

SUMMARY
CaMKIIα is a central mediator of bidirectional synaptic plasticity, including long-term potentiation (LTP) and long-term depression (LTD). To study how CaMKIIα movement during plasticity is affected by soluble amyloid-β peptide oligomers (Aβ), we used FingR intrabodies to simultaneously image endogenous CaMKIIα and markers for excitatory versus inhibitory synapses in live neurons. Aβ blocks LTP-stimulus-induced CaMKIIα accumulation at excitatory synapses. This block requires CaMKII activity, is dose and time dependent, and also occurs at synapses without detectable Aβ; it is specific to LTP, as CaMKIIα accumulation at inhibitory synapses during LTD is not reduced. As CaMKII movement to excitatory synapses is required for normal LTP, its impairment can mechanistically explain Aβ-induced impairment of LTP. CaMKII movement during LTP requires binding to the NMDA receptor, and Aβ induces internalization of NMDA receptors. However, surprisingly, this internalization does not cause the block in CaMKIIα movement and is observed for extrasynaptic, but not synaptic, NMDA receptors.
Graphical Abstract
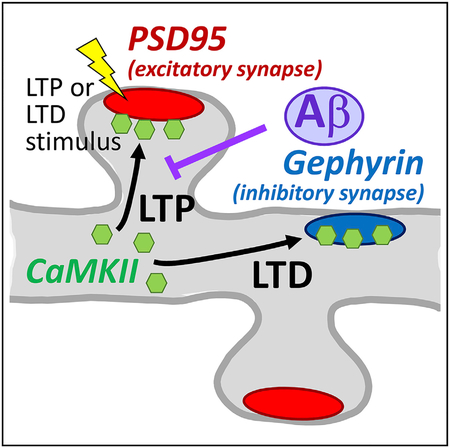
In Brief
Cook et al. used simultaneous live imaging of multiple endogenous proteins to probe LTP- versus LTD-induced CaMKII trafficking to excitatory versus inhibitory synapses. They show that Ab inhibits LTP by blocking LTP-induced CaMKII trafficking via signaling mechanisms that require CaMKII activity.
INTRODUCTION
Higher brain functions such as learning, memory, and cognition require forms of synaptic plasticity, including long-term potentiation (LTP) and long-term depression (LTD) (for review, see Lee and Silva, 2009; Malenka and Bear, 2004; Martin et al., 2000). These higher brain functions are impaired in Alzheimer’s disease, and one of the main pathological agents of the disease, soluble amyloid-b peptide oligomers (Aβ), acutely inhibits LTP and facilitates LTD in the hippocampus (Lambert et al., 1998; Li et al., 2009; O’Riordan et al., 2018; Shankar et al., 2008; Walsh et al., 2002). Thus, we decided to investigate the effects of Aβ on synaptic trafficking of the Ca2+/calmodulin (CaM)-dependent protein kinase II (CaMKII) α isoform, a central mediator of LTP and LTD at both excitatory and inhibitory synapses (Coultrap and Bayer, 2012; Coultrap et al., 2014; Giese et al., 1998; Malinow et al., 1989; Marsden et al., 2010; Silva et al., 1992).
Live imaging of fluorescent-labeled proteins is a powerful tool, but the associated protein overexpression comes with various caveats (Giepmans et al., 2006; Rodriguez et al., 2017; Tsien, 1998). For instance, excitatory or inhibitory synapses can be effectively labeled by expressing GFP-PSD95 or GFP-gephyrin, respectively, but such overexpression also directly affects synaptic functions (El-Husseini et al., 2000; Gross et al., 2013; Yu et al., 2007). Excitatory synapse function of cultured neurons is also altered by overexpression of CaMKIIα (Barcomb et al., 2014; Thiagarajan et al., 2002). Several methods for live imaging of endogenous protein (i.e., without overexpression) have been developed (Campagnola et al., 2002; Mikuni et al., 2016; Panza et al., 2015; Wakayama et al., 2017; Xue et al., 2015). Intrabodies that bind to specific proteins can be expressed in cells and imaged by fusion with a fluorescent protein. FingRs (fibronectin intrabodies generated by mRNA display) are recombinant, antibody-like proteins based on a fibronectin scaffold that bind to their target with high affinity and specificity (Gross et al., 2013) (Figure 1A). Here, we show successful simultaneous live imaging of endogenous CaMKIIα, PSD95, and gephyrin.
Figure 1. Simultaneous Live Imaging of Three Endogenous Proteins Using FingR Intrabodies.

(A) Schematic representation of FingR intrabodies. Unbound intrabody translocates to the nucleus, where it shuts down its own expression.
(B) Simultaneous imaging of three intrabodies in a live hippocampal neuron: anti-PSD95-mCherry (red; excitatory synapse marker), anti-gephyrin-mTurquois (blue; inhibitory synapse marker), and anti-CaMKIIα-YFP2 (green). Scale bars indicate 5 μm.
(C) Individual versus triple expression of our intrabodies did not change their co-localization with the corresponding antibody stain (as shown by the ratio of their Pearson correlation of localization) (n = 9, 11, 9 neurons).
(D) Percentage of antibody (ab) and intrabody (ib) punctae that are positive for the respective other staining in cultures transfected with all three intrabodies. The percentage of ib-positive ab punctae is lower, as ab-positive punctae include nearby non-transfected neurons (n = 10, 10, 9, and 10 neurons).
Our results show that excitatory LTP versus LTD stimuli cause differential trafficking of endogenous CaMKII to excitatory versus inhibitory synapses. While the LTP-induced trafficking is thought to occur specifically to the stimulated synapses (Zhang et al., 2008), the LTD-induced trafficking to inhibitory synapses represents transsynaptic communication from the stimulated synapse to an inhibitory synapse. Additionally, we show that Aβ engaged CaMKII signaling that prevented subsequent LTP-induced CaMKII movement. Importantly, because such CaMKII movement is required for normal LTP (Barria and Malinow, 2005; Halt et al., 2012), the blocking of its movement provides a mechanistic explanation for the well-established Aβ-induced inhibition of LTP (Lambert et al., 1998; Shankar et al., 2008; Walsh et al., 2002), which is thought to contribute to the cognitive impairments in Alzheimer’s disease.
RESULTS
Simultaneous Live Imaging of CaMKIIα, PSD95, and Gephyrin
Two of the intrabodies used here were directed against post-synaptic markers for excitatory versus inhibitory synapses (anti-PSD95-mCherry and anti-gephyrin-mTurquois, respectively) and are thus widely applicable in general neuronal cell biology. A third intrabody was directed against CaMKIIα and was labeled with YFP2. Individual expression of the corresponding originally GFP-labeled intrabodies does not alter synaptic functions or localization of the detected endogenous proteins (Barcomb et al., 2015; Gross et al., 2013; Mora et al., 2013). Individual expression of the modified intrabodies also resulted in faithful labeling of the subcellular localization of their respective endogenous protein (Figures S1A and S1B). Two days after transfecting hippocampal neurons with all three modified intra-body constructs simultaneously, all three intrabodies localized as expected both from the distribution of their respective endogenous target proteins and from the distribution of the individually expressed intrabodies (Figures 1B and S1A–S1C). The anti-PSD95-mCherry intrabody localized almost exclusively to the dendritic spines that form the postsynaptic compartment of excitatory synapses; the anti-gephyrin-mTurquois intrabody instead localized almost exclusively to discrete punctae on the membrane of dendrite shafts, where inhibitory synapses form; and the anti-CaMKIIα-YFP2 intrabody was found throughout the neuron, but with some enrichment in dendritic spines even without stimulation of the neurons. Thus, all three intrabodies can be successfully expressed together in the same neuron and appear to faithfully label the localization of their respective target protein. Faithful labeling in neurons expressing all three intrabodies was further verified by fixation and immunostaining with the respective antibodies (Figures 1C, 1D, and S1D–S1F). The simultaneous expression of all three intrabodies did not interfere with the intrabody specificity, as the correlation of co-localization with their respective antibody stain was almost identical to the one after individual expression (Figure 1C). As expected, co-localization of each antibody stain was high with the matching intrabody and significantly lower with the other intrabodies (Figures S1D and S1E). Also as expected, co-localization of each synaptic marker was better with CaMKII than with the other synaptic marker (Figure S1E). More than 90% of excitatory or inhibitory synapses identified by intrabody labeling were verified also by antibody staining (Figure 1D). The percentage of synapses identified by antibody staining that was also positive for intrabody staining was slightly lower (Figure 1D), consistent with the fact that the antibody also labels synapses on dendrites of nearby non-transfected neurons.
CaMKII Trafficking to Excitatory versus Inhibitory Synapses after cLTP Versus cLTD Stimuli
Next, we utilized the three intrabodies in order to monitor the CaMKII trafficking to excitatory versus inhibitory synapses in response to chemical LTP versus LTD stimuli (Figures 2 and S2). In response to chemical LTP (cLTP) stimuli (100 μM gluta-mate, 10 μM glycine), endogenous CaMKII preferentially moved to excitatory over inhibitory synapses (Figures 2A and S2). By contrast, after chemical LTD (cLTD) stimuli (30 μM NMDA, 10 μM cyanquixaline [CNQX], and 10 μM glycine), endogenous CaMKII instead accumulated at inhibitory but not excitatory synapses (Figure 2B). Thus, while the LTP-induced CaMKII accumulation is restricted to the synapses that were stimulated (Zhang et al., 2008), the CaMKII accumulation at inhibitory synapses in response to excitatory LTD stimuli represents a form of transsynaptic communication (Figure 2C). While quantification of both CaMKII trafficking events was significant and conclusive, the movement to excitatory synapses was visually much more obvious compared to the movement to inhibitory synapses (see Figures 2A and 2B).
Figure 2. CaMKII Trafficking to Excitatory versus Inhibitory Synapses after Opposing Stimuli.

(A) cLTP stimuli induced CaMKII trafficking to excitatory over inhibitory synapses in hippocampal neurons, as determined by our live imaging method of endogenous protein introduced in Figure 1 (n = 28 neurons; ***p < 0.001). Scale bars indicate 5 μm in all panels.
(B) By contrast, cLTD stimuli instead induce CaMKII trafficking to inhibitory but not excitatory synapses (n = 30 neurons; ***p < 0.001). The statistical analysis (by two-way ANOVA comparing data in A and B, followed by Bonferroni post hoc tests) additionally indicated that the translocation for each synapse type is significantly different in LTP compared to LTD (p < 0.001).
(C) Schematic of the homo-synaptic CaMKII trafficking to excitatory synapses after LTP versus hetero-synaptic trafficking to inhibitory synapses after LTD.
Aβ Blocks cLTP-Induced CaMKII Trafficking to Excitatory Synapses
It has been shown recently that application of Ab caused activation of CaMKII (Opazo et al., 2018). As CaMKII activation can trigger its movement to excitatory or inhibitory synapses (see Figure 2), we decided to determine the effect of Aβ on synaptic CaMKII targeting (Figure 3). Incubation with Aβ (500 nM for 45 min) had no effect on basal localization of CaMKII (Figures 3A and 3B). However, such Aβ incubation dramatically reduced subsequent cLTP-induced movement to excitatory synapses (Figures 3A and 3C). A control peptide with scrambled Aβ sequence had no effect (Figure 3C), and Aβ did not inhibit the cLTD-induced movement of CaMKII to inhibitory synapses (Figure 3D). The cLTP-induced movement was also significantly impaired by 50 nM Aβ, but not by 5 nM Aβ (Figure 3E). Next, we tested the effect of different incubation times with 500 nM Aβ (Figure 3F). CaMKII movement was completely blocked after 45 min and significantly reduced after 20 min but remained largely unaffected after 5 min. CaMKII translocation to excitatory synapses was also induced by treatment with the Ca2+ ionophore ionomycin, and this translocation was also blocked by Aβ (Figure 3G).
Figure 3. Ab Blocks LTP-Induced CaMKII Accumulation at Excitatory Synapses.

(A) Representative images of CaMKIIα in hippocampal neuron before and after treatment with Aβ peptide (500 nM; labeled with Alexa Fluor 647) and cLTP stimulation. Scale bar indicates 5 μm.
(B) Aβ application (500 nM for 45 min, unless indicated otherwise) does not affect the basal localization of endogenous CaMKIIα in hippocampal neurons, as detected with an intrabody (n = 13 neurons; unpaired Student’s t test).
(C) Preincubation with Aβ blocks subsequent cLTP-induced CaMKII movement to excitatory synapses. A control peptide with scrambled Ab sequence did not affect the cLTP-induced CaMKII movement (n = 19, 11, 11 neurons; one-way ANOVA, Tukey’s post hoc test, ***p < 0.001).
(D) Aβ does not affect the cLTD-induced CaMKII movement to inhibitory synapses (n = 15, 12 neurons; unpaired Student’s t test).
(E and F) cLTP-induced CaMKII movement to excitatory synapses is inhibited by 500 nM and 50 nM Aβ, but not 5 nM Aβ (E; n = 14, 16, 16, 17 neurons; one-way ANOVA, Tukey’s post hoc test, ***p < 0.001) and by 45-min and 20-min, but not 5-min, preincubation with Aβ (F; n = 9, 17, 21, 11 neurons; one-way ANOVA, Tukey’s post hoc test, ***p < 0.001).
(G) Ionomycin (5 μM) induces CaMKIIα movement to excitatory synapses, which is also blocked by Aβ (n = 10, 13 neurons; unpaired Student’s t test, *p < 0.05).
Aβ Has Homo- and Hetero-synaptic Effects on cLTP-Induced CaMKII Trafficking
Using fluorescently labeled Aβ at different concentrations, Aβ labeling was detected at varying percentages within 1 μm proximity of excitatory synapses (Figure 4A), at ~20% (5 nM Aβ), ~70% (50 nM Aβ), and > 70% (500 nM Aβ) of synapses, respectively. No significant labeling of inhibitory synapses was observed at either Aβ concentration. Interestingly, Aβ had both homo- and hetero-synaptic effects regarding suppression of LTP-induced CaMKII translocation to excitatory synapses. While the suppression was strongest for synapses with bound Aβ, incubation with 500 nM Aβ also caused significant suppression of CaMKII translocation to synapses with no detectable bound Aβ (Figures 4B, and 4C). After incubation with 5 nM Aβ, LTP-induced CaMKII translocation appeared slightly reduced for the synapses with detectable Aβ; however, this apparent reduction was not statistically significant (Figure 4B). This suggests that the Aβ-mediated suppression of CaMKII translocation requires a threshold number of synapses with bound Aβ.
Figure 4. Mechanisms of the Effects of Aβ on CaMKII Movement.

(A) The fraction of excitatory synapses (identified by PSD95 intrabody staining; red) with juxtaposed Aβ (labeled with Alexa Fluor 647; purple) detected within 1 μm is concentration-dependent. Scale bar indicates 1 μm (n = 12, 10, 10 neurons; one-way ANOVA, Tukey’s post hoc test, ***p < 0.001).
(B) The Aβ -mediated blockade of cLTP-induced CaMKII movement has both homo- and hetero-synaptic components. At all Aβ concentrations, CaMKII movement was less for synapses that are in in proximity to Aβ (n = 13, 14, 9 neurons; paired t tests, # p < 0.05, ### p < 0.001). However, at higher Aβ concentrations, CaMKII movement is significantly reduced compared to control without Aβ (dotted line) for synapses with and without proximal Aβ (one-way ANOVA, Tukey’s post hoc test, *p < 0.05, **p < 0.01, ***p < 0.001).
(C) Schematic illustration of the homo- and hetero-synaptic inhibition of CaMKII movement by Aβ.
(D) CaMKII inhibition (with either 10 μM KN93 or 5 μM tatCN21) during Aβ application alleviates the blockade of cLTP-induced CaMKII movement. Note that inhibitors were washed out (using artificial cerebral spinal fluid [ACSF]) prior to cLTP stimulation (n = 11, 7, 8, 9 neurons; one-way ANOVA, Tukey’s post hoc test, ***p < 0.001).
(E) Aβ application does not alter GluN2B phosphorylation at S1303 either directly (left; n = 9) or indirectly in combination with subsequent additional LTP stimulation (right; n = 4), as shown by western analysis with specific antibodies (unpaired Student’s t test).
(F) Surface biotinylation followed by western analysis shows that 500 nM Aβ induces removal of surface NMDA receptor (GluN1) with the same time course as seen for block of CaMKII translocation. At the initial 5-min time point, the surface NMDA receptor was even increased (n = 9; one-way ANOVA, Tukey’s post hoc test, ***p < 0.001).
(G) Surface NMDA-receptor internalization after 45 min of 500 nM Aβ is unaffected by the CaMKII inhibition (with either KN93 or tatCN21) that restored CaMKII translocation, showing that receptor internalization is not responsible for the block of CaMKII translocation (n = 9; one-way ANOVA, Tukey’s post hoc test, ***p < 0.001).
(H) Fractionation experiments show that 45 min of 500 nM Aβ does not affect the synaptic NMDA-receptor pool that is found in the PSD fraction (n = 3 experiments; unpaired Student’s t test).
Aβ Effects on LTP and CaMKII Trafficking Are Mediated by CaMKII Activity
CaMKII movement to excitatory synapses is required for normal LTP (Barria and Malinow, 2005; Halt et al., 2012); thus, the Aβ-induced block of this movement found here could explain the well-documented Aβ-induced inhibition of LTP (Lambert et al., 1998; Shankar et al., 2008; Walsh et al., 2002). We have recently shown that this Aβ -induced inhibition of LTP is prevented by CaMKII inhibition during Aβ application (Opazo et al., 2018). Thus, we decided to test if CaMKII inhibition also abolishes the Aβ-induced block of CaMKII movement after a cLTP stimulus. In presence of either of two mechanistically distinct CaMKII inhibitors, KN93 or tatCN21 (Deng et al., 2017; Sumi et al., 1991; Vest et al., 2007), Aβ no longer blocked the cLTP-induced CaMKII movement (Figure 4D). Notably, either of the two inhibitors is also sufficient to prevent the Aβ-induced LTP impairment (Opazo et al., 2018). (In both cases, the inhibitor was present during Ab application but washed out prior to LTP induction.)
How does Aβ-induced CaMKII activity mediate the Aβ-induced inhibition of CaMKII movement and LTP? One potential mechanism is phosphorylation of the NMDA-receptor subunit GluN2B at S1303, which impairs the CaMKII binding to GluN2B (O’Leary et al., 2011; Strack et al., 2000) that mediates the CaMKII movement to excitatory synapses during LTP (Bayer et al., 2001; Halt et al., 2012). We have previously shown that we can detect changes in GluN2B S1303 phosphorylation, both in vitro and in response to LTD stimuli (Coultrap et al., 2010; Goodell et al., 2017). However, Aβ did not alter GluN2B S1303 phosphorylation (neither directly nor indirectly in response to subsequent LTP stimulation; Figure 4E), suggesting that a CaMKII-mediated mechanism other than S1303 phosphorylation underlies the Aβ effect.
Aβ Causes Internalization of the Extrasynaptic, but Not Synaptic, NMDA Receptor
It is well described that Ab causes the internalization of NMDA receptors (Jo et al., 2011; Kurup et al., 2010; Shankar et al., 2008; Sinnen et al., 2016; Snyder et al., 2005), a mechanism that would be expected to inhibit the synaptic translocation of CaMKII that is mediated by binding to the receptor. Indeed, block of CaMKII translocation and induction of NMDA-receptor internalization have the same dependence on the Aβ dose, with both effects seen only in response to concentrations higher than 5 nM (see Figure 3E and Sinnen et al., 2016). Additionally, in experiments using surface receptor biotinylation, both effects are seen only after incubation times longer than 5 min (Figures 4F and S3A; compare to Figure 3F). Notably, at the initial 5-min time point, we detected a significant increase in surface receptor in our hippocampal neurons (Figure 4F). While this was somewhat surprising, a similar initial surface receptor increase has been observed previously in cortical neurons (Um et al., 2012).
If NMDA-receptor internalization causes the Aβ-induced block of CaMKII translocation, then the receptor internalization should be alleviated by the CaMKII inhibitor treatment that restores the translocation (see Figure 4D). However, surprisingly, neither of the CaMKII inhibitors (KN93 and tatCN21) suppressed the Aβ-induced NMDA-receptor internalization (Figures 4G and S3B). Thus, CaMKII translocation can be restored by treatments that leave the NMDA-receptor internalization unaffected (even though the CaMKII translocation requires binding to the receptor). This apparent conundrum could be explained if Aβ induces internalization of the extrasynaptic, but not synaptic, NMDA receptor. In order to directly test this possibility, we isolated a post-synaptic density (PSD) fraction that contains synaptic receptors, synaptic scaffolding proteins, and associated signaling proteins (by biochemical fraction, as we have described previously (Coultrap et al., 2014). Indeed, while 45 min of 500 nM Aβ treatment caused significant removal of NMDA receptors from the overall cell surface (Figure 4G), it did not affect the synaptic NMDA-receptor content in the PSD fraction (Figure 4H). Thus, Aβ induces the removal of extrasynaptic, but not synaptic, NMDA receptors.
DISCUSSION
Here, we demonstrate a molecular mechanism for the Aβ-induced impairment of hippocampal LTP: CaMKII-mediated inhibition of the LTP-induced CaMKII movement to excitatory synapses. Indeed, this CaMKII trafficking is required for normal LTP (Barria and Malinow, 2005; Halt et al., 2012). Importantly, the impairment of LTP by Aβ is thought to contribute to the cognitive impairments in Alzheimer’s disease (Lambert et al., 1998; Shankar et al., 2008; Walsh et al., 2002). Interestingly, Aβ had both homo- and hetero-synaptic effects on CaMKII movement. While the LTP-induced CaMKII movement was most strongly inhibited to excitatory synapses that had Aβ associated with them (here defined by nearby binding, i.e., within 1 μm), movement to neighboring synapses without bound Aβ was also significantly inhibited. Additionally, at low Aβ concentrations that resulted in Aβ binding to only a minority of synapses, no signifi-cant inhibition of CaMKII movement was detected at all, i.e., not even to synapses that contained associated Aβ. This suggest that a cumulative hetero-synaptic effect of Aβ binding may cause the most significant functional detriment to the processes required for normal LTP. By contrast, the cLTD-induced hetero-synaptic CaMKII movement to inhibitory synapses was not inhibited.
The LTP-induced CaMKII movement to excitatory synapses is mediated by the regulated CaMKII binding to the NMDA-receptor subunit GluN2B (Bayer et al., 2001, 2006; Halt et al., 2012; O’Leary et al., 2011). Thus, Aβ-induced signaling must somehow prevent this CaMKII-GluN2B interaction, and our results indicate that the underlying mechanism requires CaMKII activity. Indeed, recent results indicate that Aβ can activate CaMKII (although without inducing T286 autophosphorylation; Opazo et al., 2018). However, while CaMKII activity could prevent CaMKIIGluN2B binding by inducing phosphorylation of GluN2B at S1303 ((Barcomb et al., 2014; O’Leary et al., 2011; Strack et al., 2000), our results show that Aβ application did not increase this S1303 phosphorylation either basally or in response to LTP stimuli. CaMKII binding to GluN2B is induced by Ca2+/ CaM-stimulation (Bayer et al., 2001; O’Leary et al., 2011), which is provided during LTP-stimuli by NMDA-receptor activation. Recent results indicate that Aβ can reduce the Ca2 currents through the NMDA receptor (Sinnen et al., 2016), which in turn could reduce the Ca2+-induced CaMKII movement. However, our results show that Aβ also inhibits CaMKII movement that is triggered when the Ca2+-stimulus is instead directly induced by ionomycin.
Another obvious potential mechanism for the Aβ-induced block of CaMKII trafficking was through the well-described acute Aβ-induced internalization of surface NMDA receptors. Initially, this mechanism appeared to be supported by the similar dependence on Aβ dose and incubation time for both block of CaMKII trafficking and induction of NMDA-receptor internalization. However, CaMKII inhibitors rescued the block of CaMKII trafficking without affecting NMDA-receptor internalization. Thus, the Aβ-induced receptor internalization is not what causes the block of CaMKII trafficking. But why would NMDA-receptor internalization not block the LTP-induced synaptic CaMKII trafficking that depends on CaMKII binding to this receptor? The answer was provided by our biochemical fractionation experiments: Aβ caused internalization only of the extrasynaptic NMDA receptor, while the synaptic receptor pool in the PSD fraction remained entirely unaffected. It will be interesting to elucidate the long-term functional consequences of the internalization of extrasynaptic receptors. However, acutely, the Aβ-induced internalization of only extrasynaptic NMDA receptors cannot explain the Aβ-mediated suppression of LTP, which instead depends on synaptic NMDA receptors. By contrast, as normal LTP requires CaMKII trafficking to excitatory synapses (Barria and Malinow, 2005; Halt et al., 2012), the Aβ-mediated inhibition of LTP is instead readily explained by the suppression of this CaMKII trafficking event. Indeed, the CaMKII inhibition shown here to rescue CaMKII trafficking was recently shown to also rescue LTP from the impairment by Aβ (Opazo et al., 2018).
Our findings were made by live imaging of endogenous CaMKII (rather than by live imaging of overexpressed GFPCaMKII or post hoc immunostaining of CaMKII in fixed neurons). Importantly, we demonstrated that FingR intrabodies can be utilized to enable simultaneous live imaging of multiple endogenous proteins. Two of the three intrabodies used here simultaneously (against PSD95 and gephyrin) enable live labeling of excitatory versus inhibitory synapses (without affecting basal synaptic functions, in contrast to overexpression of labeled PSD95 or gephyrin; El-Husseini et al., 2000; Gross et al., 2013; Yu et al., 2007); this simultaneous live labeling of both synapse types should be of wide general utility in neuronal cell biology. Notably, although endogenous CaMKII and overexpressed GFP-CaMKII show similar trafficking, there are also some important differences. While endogenous CaMKII traffics to excitatory synapses only after LTP-type stimuli (Goodell et al., 2017; Mars-den et al., 2010), GFP-CaMKII can traffic to excitatory synapses also after LTD-type stimuli (Bayer et al., 2006; Marsden et al., 2010). This difference may be explained by the fact that the lack of CaMKII trafficking to excitatory synapses after LTD stimuli requires an active suppression mechanism (Goodell et al., 2017) that may be overwhelmed by GFP-CaMKII overexpression. This highlights the significant drive on CaMKII to move to excitatory synapses even in response to weaker neuronal stimuli and further supports that the observed block by Aβ requires specific signaling as opposed to merely a general reduction in NMDA-receptor mediated Ca2+ signals.
STAR★METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| PSD-95 | Chemicon | MAB1596; RRID: AB_2092365 |
| GABAAR 2/3 | Millipore | 05–474; RRID: AB_309747 |
| Gephyrin | Abcam | ab25784; RRID: AB_1209349 |
| CaMKIIα | Made in House | CBα2 |
| GluN2B | PhosphoSolutions | AB_2492176; RRID: AB_2492176 |
| GluN2B pS1303 | Millipore | 07–398; RRID: AB_310582 |
| GluN1 | BD Biosciences | 556308; RRID: AB_396353 |
| Beta (III) Tubulin | Millipore | 05–559; RRID: AB_309804 |
| Rabbit | GE Healthcare | NA934V; RRID: AB_772206 |
| Mouse | GE Healthcare | NA931V; RRID: AB_772210 |
| Alexa-647 Anti-Rabbit | Molecular Probes | A21245; RRID: AB_2535813 |
| Alexa-647 Anti-Mouse | Molecular Probes | A21236; RRID: AB_141725 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| HiLyte Fluor 647-labeled Aβ1–42 peptide | AnaSpec | AS-64161 |
| Unlabeled Aβ1–42 peptide | AnaSpec | AS-20276 |
| Scrambled Aβ1–42 peptide | AnaSpec | AS-25382 |
| Papain | Worthington | LS 03126 |
| Lipofectamine 2000 | Invitrogen | 11668027 |
| B-27 supplement | GIBCO | 17504044 |
| KN-93 inhibitor | Tocris | 139298–40-1 |
| tatCN21 inhibitor | Millipore | 5.32385.0001 |
| cOmplete protease inhibitor cocktail (EDTA-free) | Roche | 1187380001 |
| Critical Commercial Assays | ||
| Pierce BCA protein assay | Thermo-Fisher | 23225 |
| Recombinant DNA | ||
| CaMKIIα-FingR-GFP | Dr. Donald Arnold (USC) Mora et al., 2013 | N/A |
| PSD-95-FingR-GFP | Addgene | 46295 |
| Gephyrin-FingR-GFP | Addgene | 46296 |
| Software and Algorithms | ||
| Slidebook 6.0 | Intelligent Imaging Innovations (3i) | N/A |
| Prism 7.0 | Graphpad | RRID: SCR_002798 |
| AlphaEase FC 4.0 | Alpha Innotech | N/A |
CONTACT FOR REAGENT AND RESOURCE SHARING
Requests for resources, reagents, or questions about methods should be directed to Lead Contact, K. Ulrich Bayer (ulli.bayer@ucdenver.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Primary rat hippocampal cultures
Primary hippocampal neurons were cultured as described previously (Vest et al., 2010) and imaged after 14–17 days in vitro (DIV14–17). Pregnant Sprague-Dawley rats were supplied by Charles River Labs for preparation of primary hippocampal cultures from P0–P1 neonatal rat pups of both sexes. All animal treatment was in accordance with the University of Colorado Denver Institutional Animal Care and Use Committee. Pups were decapitated and hippocampi were dissected and incubated in dissociation solution (7 mL HBSS buffered saline, 150 μL 100mM CaCl2, 10 μL 1M NaOH, 10 μL 500mM EDTA, 200 units Papain [Worthington]) at 25°C for 1 h. Hippocampi were then washed 5x with plating media (DMEM, FBS, 50 units/ml Penn/strep, 2 mM L-glutamine, filter sterilized) and manually dissociated and counted using a hemocytometer. Dissociated neurons were plated on poly-D-lysine (0.1mg/mL in 1M Borate Buffer: 3.1g boric Acid, 4.75 g borax, in 1 L deionized H20, filter sterilized) and laminin (0.01mg/mL in PBS)-coated 18 mm glass coverslips in 12 well plates at a density of 75,000–100,000 neurons per well in plating media and maintained at 37°C with 5% CO2. After 1 day in vitro (DIV 1), media was switched to 100% feeding media (Neurobasal-A, B27 supplements, and 2 mM L-gluta-mine, filter sterilized). At DIV 4, neurons were treated with FDU (70 μM 5-fluoro-2ʹ-deoxyuridine/140 μM uridine) to suppress glial growth by halting mitosis. The following day (DIV 5), 50% of media was replaced with fresh neuron feeding media.
METHOD DETAILS
Material and DNA constructs
Material was obtained from Sigma, unless noted otherwise. The expression vectors for the GFP-labeled FingR intrabodies targeting CaMKIIα, PSD-95, and gephyrin were kindly provided by Dr. Donald Arnold (University of Southern California, Los Angeles, CA, USA) as described previously (Gross et al., 2013; Mora et al., 2013). The FingRs against PSD95 and gephyrin contained the CCR5TC repressor (Gross et al., 2013); the FingR against CaMKIIα did not contain a repressor (Mora et al., 2013). The fluorophore label was exchanged to contain the following tags in place of GFP: CaMKIIa-FingR-YFP2, PSD-95-FingR-mCh, and gephyrin-FingR-mTurquois. These modifications were performed using Gibson Assembly (Gibson et al., 2009).
Transfection of primary hippocampal cultures
At 12–14 DIV, neurons were transfected with the intrabodies using a 1:1:1 ratio, at a concentration of 1 μg total DNA/well, using Lipofectamine 2000 (2.5 μL/well, Invitrogen) according to the manufacturer’s recommendations.
Live imaging of primary hippocampal cultures
Two to three days following transfection (DIV 14–17), neurons were imaged using a Axio Observer microscope (Carl Zeiss) fitted with a 63x Plan-Apo/1.4 numerical aperture (NA) objective, using 405, 488, 561, and 638 nm laser excitation and a CSU-XI spinning disk confocal scan head (Yokogawa) coupled to an Evolve 512 EM-CCD camera (Photometrics) and controlled using Slidebook 6.0 software (Intelligent Imaging Innovations [3i]), as described previously (Woolfrey et al., 2018). During image acquisition, neurons were maintained in a climate controlled chamber at 34°C in a HEPES buffered ACSF solution containing the following (in mM): 130 NaCl, 5 KCl, 10 HEPES pH 7.4, 20 glucose, 2 CaCl2, and 1 MgCl2 (adjusted to proper osmolarity with sucrose). cLTP was induced using 100 μM glutamate and 10 μM glycine and cLTD was induced with 30 μM NMDA, 10 μM glycine, and 10 μM CNQX. After 1 min of either cLTP or cLTD application, 5 volumes of fresh ACSF was perfused through the imaging chamber to wash out the stimulation. The cLTD stimuli used does not induce CaMKII accumulation at dendritic spines (shown here and in Marsden et al., 2010; Goodell et al., 2017), whereas the cLTP stimulation has been shown to induce robust CaMKII accumulation in spines (also shown here and in Barcomb et al., 2013; O’Leary et al., 2011). For Aβ experiments, control wells were incubated for 45 min in ACSF + 1X PBS/0.004% DMSO, while experimental wells were incubated in ACSF + 5, 50, or 500 nM of Fluor-647 labeled Aβ for 45 min (and 500 nM for 5 and 20 min for time-response experiments) before being transferred to a Ludin chamber for imaging. CaMKIIα inhibitor experiments were perform by incubating neurons with 10 μM KN-93 or 5 μM tatCN21 for 20 min during the end of a 45 min 500 nM Aβ application. Neurons were washed 2X with ACSF before being transferred to the imaging chamber and imaged in fresh ACSF. Hippocampal neurons were selected based on pyramidal shaped soma and presence of spiny apical dendrites, and tertiary dendritic branches were selected for analysis to maintain consistency. Images were analyzed at 1 min before stimulation and 1 min or 5 mins after wash out for cLTP and cLTD, respectively. 2D maximum intensity projection images were then generated and analyzed by an experimenter blinded to condition using Slidebook 6.0 software (3i). The mean YFP intensity (CaMKIIα) at excitatory and inhibitory synapses was quantified using threshold masks of PSD-95 and gephyrin puncta. PSD-95 and gephyrin puncta masks were defined using the mean intensity of mCh or mTurquois + 2 standard deviations. Synaptic CaMKIIα was calculated using the mean YFP intensity of PSD-95 or gephyrin puncta masks divided by the mean intensity of a line drawn in the dendritic shaft. Changes in CaMKIIα synaptic accumulation were determined by dividing the net change in YFP at PSD-95 or gephyrin puncta-to-shaft ratio by the pre-stimulation YFP puncta-to-shaft ratio. Excitatory synapses in proximity to Aβ were characterized as dendritic spines containing PSD-95 (as labeled by intrabody) within 1 mm of 647-labeled Ab punctae.
Immunocytochemistry
Neurons were plated and transfected as described above and fixed in a solution containing 4% paraformaldehyde and 4% sucrose in PBS for 15 min then permeabilized and blocked in 0.1% Triton/5% BSA/PBS at 25°C for 1 h. Fixed neurons were stained in 5% BSA/PBS with anti-CaMKIIα (1:1000; CBα2, made in house), anti-PSD-95 (1:2000, Chemicon) or anti-GABAAR b2/3 (1:1000, Millipore) primary antibodies overnight at 4°C. Neurons were then washed 3X with PBS and incubated with Alexa-647 labeled species-specific secondary antibodies (1:500, Thermo Fisher) for 1–2 h at 25°C. After washing 4X with ACSF, coverslips were embedded using ProLong gold anti-fade reagent (Thermo-Fisher) for imaging. Images were acquired on a Zeiss Axiovert 200M inverted microscope (Carl Zeiss) fitted with a 63X objective (1.4 NA; plan-Apo). Focal plane z stacks (0.3-μm steps; over 1.8–2.4 μm) were acquired and deconvolved to eliminate out-of-focus light. 2D maximum intensity projection images were generated and analyzed by an experimenter blinded to condition using Slidebook 6.0 software (3i). Pearson’s correlation of fluorescence overlap was used to quantify co-localization between the three intrabodies and their respective antibodies.
Western analysis
Hippocampal neurons were plated on poly-D-lysine (0.1mg/mL in 1M Borate Buffer) coated 6 well plates at a density of 300,000 cells/ per well and maintained as described above. At DIV 14, neurons were incubated in various treatments and harvested via sonication in buffer containing 1% SDS, 1 mM EDTA, and 10 mM Tris pH 8 and boiled for 10 mins. Protein content was determined using the Pierce BCA protein assay (Thermo-Fisher). 4 to 10 μg of total protein was subjected to SDS-PAGE on 10% polyacrylamide gels and transferred to polyvinylidene fluoride (PVDF) membrane for 1–2 h at 4°C using Idea Scientific transfer apparatus in western transfer buffer (1200 mL methanol, 67.5g glycine, 14.54 g Tris Base, volume to 6 L in deionized H20, pH 8.3). Immuno-detection was then preformed using anti-CaMKIIα 1:5000 (CBα2, available at Invitrogen, but made in house), anti-GluN2B 1:3000 (Phospho-Solutions), or anti-pS1303 GluN2B 1:1000 (Millipore) followed by Amersham ECL anti-mouse IgG, horseradish peroxidase-linked secondary 1:5000 (GE Healthcare) or goat anti-rabbit IgG horseradish peroxidase conjugate 1:4000 (Bio-Rad). The dilution buffer was TBS-T (20 mM Tris HCl pH 7.4, 150 mM NaCl and 0.1% Tween-20). Blots using CBα2 or anti-pS1303 were blocked in 5% milk in TBS-T with antibody and secondary dilutions in 1% milk in TBS-T. Anti-GluN2B blots were blocked in 5% BSA in TBS-T and antibody and secondary dilutions were in 1% BSA in TBS-T. Blots were developed using chemi-luminescence (Super Signal West Femto, Thermo-Fisher) and imaged using the Chemi-Imager 4400 system (Alpha-Innotech). Densitometry was calculated in AlphaEaseFC (Alpha-Innotech). Three technical replicates per condition were loaded per gel, and the relative immuno-detection value (IDV) was normalized as a percent of the average of all control conditions for the same blot, which was set at a value of one to allow comparison between multiple experiments.
Soluble Aβ peptide preparation
HiLyte Fluor 647-labeled and unlabeled Aβ1–42 (AnaSpec) were prepared in aliquots as a dried 1,1,1,3,3,3-hexafluoro-2-propanol film and stored at 80 C, as described previously (Klein, 2002), however, the peptides were diluted in PBS instead of F12 medium (Sinnen et al., 2016). The peptide film was dissolved in 4.4 μL of anhydrous DMSO and diluted to 50 μM with 1X PBS and allowed to oligomerize at 4°C overnight. The preparation was centrifuged at 14,000 × g for 10 min at 4°C to remove insoluble aggregates. The peptide was then diluted in 600 μL of PBS to 10 μM and kept on ice until use. Scrambled Ab1–42 peptide (AnaSpec) was prepared in the same manner. 647-labeled and scrambled Ab1–42 peptide were used for live-imaging experiments, while unlabeled Ab1–42 peptide was used in biochemical assays.
Surface biotinylation assay
Surface biotinylation assays were performed as described previously with slight modifications (Mammen et al., 1997; Sinnen et al., 2016). After Aβ treatment, neurons were washed 3X in fresh ACSF and then incubated in ACSF containing 1 mg/ml EZ-link-sulfo-NHS-LC-biotin (Thermo Fisher) for 10 min at 25°C. Neurons were rinsed 3X in ACSF + 0.1% BSA and then scraped and lysed in 200 μl warm precipitation buffer (PB) containing the following (in mM): 10 NaPO4, pH 7.4, 5 EDTA, 5 EGTA, 100 NaCl, 1 Na3VO4, 10 sodium pyrophosphate, and 50 NaF with cOmplete protease inhibitor cocktail (EDTA free, Roche), 1% SDS, and 1% Triton X-100. Lysates were sonicated and centrifuged for 20 min at 18,000 X g. Before pull-down of biotinylated proteins, 10% of the cell lysate was reserved for total input quantification. The remaining sample was combined with NeutrAvidin agarose beads (30 μl; Thermo Fisher) and incubated overnight at 4°C. The beads were washed 2X each with PB containing 0.1% Triton X-100, 0.1% Triton X-100 plus 600 mM NaCl, and PB alone. To elute captured proteins, 1X SDS-PAGE buffer was added to the beads and the samples were boiled for 5 min before loading onto 10% SDS-PAGE gels. Immunoblotting was performed with GluN1 antibody (BD Biosciences) and β-tubulin loading control (Millipore). Blots were quantified by comparing the ratio of biotinylated GluN1 with the total GluN1 for a given sample. Values were normalized to control samples treated in an identical manner but not exposed to Ab. Variations in sample loading concentrations were accessed using β-tubulin (Millipore) as a loading control.
Subcellular fractionation assay
Experiments were performed as previously described (Coultrap et al., 2014; Deng et al., 2017), however, dissociated rat hippocampal neurons (plated in 10 cm dishes, 2,500,000–3,000,000 neurons/dish) were used instead of whole hippocampi. Neurons were maintained as described and were treated in the same conditions as used for imaging then washed 3X in fresh ACSF before fractionation. Briefly, hippocampal neurons were scraped and homogenized using a pestle homogenizer in 1 mL sucrose buffer (SB) containing (in mM): 10 tris (pH 7.4), 320 sucrose, 2.5 NaF, 1 EDTA, 1 EGTA, cOmplete protease inhibitor cocktail (EDTA free, Roche), microcysin-LR, 2.5 β-glycerophosphate, 2.5 NaPPi, brought to 5 mL deionized H20. A sample was taken for the homogenate fraction. The remaining homogenate was spun for 10 mins at 1000 g. Supernatant (S1) was decanted to a fresh tube and spun for 15 mins at 10,000 g. The pellet containing the synaptosomal plasma fraction was rinsed with SB then resuspended in 400 μL Triton buffer containing 0.5% Triton X-100 and (in mM): 10 tris (pH 7.4), 2.5 NaF, 1 EDTA, 1 EGTA, cOmplete protease inhibitor cocktail (EDTA free, Roche), microcysin-LR, 2.5 β-glycerophosphate, 2.5 NaPPi, brought to 5 mL deionized H20. The resuspension was spun at for 20 mins at 32,000 g. The pellet containing the post-synaptic density fraction was resuspended in 60 μL storage buffer and boiled for 5 mins. Protein content was determined using the Pierce BCA protein assay (Thermo-Fisher) and 2 μg protein per condition was subjected to SDS-PAGE and western analysis. Blots were probed for GluN1 (BD Biosciences) and GluN2B (PhosphoSolutions) and values were normalized to control samples. Variations in sample loading concentrations were accessed and normalized using β-tubulin (Millipore) as a loading control.
QUANTIFICATION AND STATISTICAL ANALYSIS
All data are shown as mean ± SEM. Statistical significance and sample size (n) are indicated in the figure legends. Data obtained from the imaging experiments were obtained using SlideBook 6.0 software (3i) and analyzed using Prism (GraphPad) software. All data met parametric conditions, as evaluated by a Shapiro-Wilk test for normal distribution and a Brown-Forsythe test (3 or more groups) or an F-test (2 groups) to determine equal variance. Comparisons between two groups were analyzed using unpaired, two-way Student’s t tests. Comparisons between pre- and post-treatment images at the same synapse type from the same neurons were analyzed using paired, two-way Student’s t tests. Comparisons between three or more groups were done by one-way ANOVA with Tukey’s post hoc analysis. Comparisons between three or more groups with two independent variables were accessed using a two-way ANOVA to determine whether there is an interaction and/or main effect between the variables.
Supplementary Material
Highlights.
FingR intrabodies enabled simultaneous live imaging of three endogenous proteins
Aβ inhibits CaMKII trafficking after LTP-, but not LTD-inducing, stimuli
The Aβ-induced block of CaMKII trafficking requires CaMKII activity
Aβ induced internalization of the extrasynaptic, but not synaptic, NMDA receptor
ACKNOWLEDGMENTS
We thank Drs. Kevin Woolfrey, Brooke Sinnen, and Matt Kennedy for helpful discussions and for critical reading of our manuscript. The research was funded by NIH grants T32GM007635 (UCD pharmacology training grant supporting S.G.C.), F31NS092265 (to D.J.G.), P30NS048154 (UCD neuroscience center grant) and R01NS081678 (to D.B.A.), and R01NS081248, R01NS110383, and R01NS080851 (to K.U.B.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.03.041.
DECLARATION OF INTERESTS
The University of Colorado holds the patent rights for tatCN21, its derivatives, and its uses (PCT/US08/077934 “Compositions and methods for improved CaMKII inhibitors and uses thereof”). K.U.B. is founder of Neurexus Therapeutics, LLC.
REFERENCES
- Barcomb K, Buard I, Coultrap SJ, Kulbe JR, O’Leary H, Benke TA, and Bayer KU (2014). Autonomous CaMKII requires further stimulation by Ca2+/calmodulin for enhancing synaptic strength. FASEB J. 28, 3810–3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barcomb K, Coultrap SJ, and Bayer KU (2013). Enzymatic activity of CaMKII is not requires for its interaction with the glutamate receptor subunit GluN2B. Mol. Pharmacol 84, 834–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barcomb K, Goodell DJ, Arnold DB, and Bayer KU (2015). Live imaging of endogenous Ca2+/calmodulin-dependent protein kinase II in neurons reveals that ischemia-related aggregation does not require kinase activity. J. Neurochem 135, 666–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barria A, and Malinow R (2005). NMDA receptor subunit composition controls synaptic plasticity by regulating binding to CaMKII. Neuron 48, 289–301. [DOI] [PubMed] [Google Scholar]
- Bayer KU, De Koninck P, Leonard AS, Hell JW, and Schulman H (2001). Interaction with the NMDA receptor locks CaMKII in an active conformation. Nature 411, 801–805. [DOI] [PubMed] [Google Scholar]
- Bayer KU, LeBel E, McDonald GL, O’Leary H, Schulman H, and De Koninck P (2006). Transition from reversible to persistent binding of CaMKII to postsynaptic sites and NR2B. J. Neurosci 26, 1164–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campagnola PJ, Millard AC, Terasaki M, Hoppe PE, Malone CJ, and Mohler WA (2002). Three-dimensional high-resolution second-harmonic generation imaging of endogenous structural proteins in biological tissues. Biophys. J 82, 493–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coultrap SJ, and Bayer KU (2012). CaMKII regulation in information processing and storage. Trends Neurosci 35, 607–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coultrap SJ, Buard I, Kulbe JR, Dell’Acqua ML, and Bayer KU (2010). CaMKII autonomy is substrate-dependent and further stimulated by Ca2+/ calmodulin. J. Biol. Chem 285, 17930–17937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coultrap SJ, Freund RK, O’Leary H, Sanderson JL, Roche KW, Dell’Acqua ML, and Bayer KU (2014). Autonomous CaMKII mediates both LTP and LTD using a mechanism for differential substrate site selection. Cell Rep 6, 431–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng G, Orfila JE, Dietz RM, Moreno-Garcia M, Rodgers KM, Coul-trap SJ, Quillinan N, Traystman RJ, Bayer KU, and Herson PS (2017). Autonomous CaMKII activity as a drug target for histological and functional neuroprotection after resuscitation from cardiac arrest. Cell Rep 18, 1109–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Husseini AE, Schnell E, Chetkovich DM, Nicoll RA, and Bredt DS (2000). PSD-95 involvement in maturation of excitatory synapses. Science 290, 1364–1368. [PubMed] [Google Scholar]
- Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA 3rd, and Smith HO (2009). Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345. [DOI] [PubMed] [Google Scholar]
- Giepmans BN, Adams SR, Ellisman MH, and Tsien RY (2006). The fluorescent toolbox for assessing protein location and function. Science 312, 217–224. [DOI] [PubMed] [Google Scholar]
- Giese KP, Fedorov NB, Filipkowski RK, and Silva AJ (1998). Autophosphorylation at Thr286 of the alpha calcium-calmodulin kinase II in LTP and learning. Science 279, 870–873. [DOI] [PubMed] [Google Scholar]
- Goodell DJ, Zaegel V, Coultrap SJ, Hell JW, and Bayer KU (2017). DAPK1 mediates LTD by making CaMKII/GluN2B binding LTP specific. Cell Rep 19, 2231–2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross GG, Junge JA, Mora RJ, Kwon HB, Olson CA, Takahashi TT, Liman ER, Ellis-Davies GC, McGee AW, Sabatini BL, et al. (2013). Recombinant probes for visualizing endogenous synaptic proteins in living neurons. Neuron 78, 971–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halt AR, Dallapiazza RF, Zhou Y, Stein IS, Qian H, Juntti S, Wojcik S, Brose N, Silva AJ, and Hell JW (2012). CaMKII binding to GluN2B is critical during memory consolidation. EMBO J 31, 1203–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo J, Whitcomb DJ, Olsen KM, Kerrigan TL, Lo SC, Bru-Mercier G, Dickinson B, Scullion S, Sheng M, Collingridge G, and Cho K (2011). Aβ(1–42) inhibition of LTP is mediated by a signaling pathway involving caspase-3, Akt1 and GSK-3β. Nat. Neurosci 14, 545–547. [DOI] [PubMed] [Google Scholar]
- Klein WL (2002). Abeta toxicity in Alzheimer’s disease: globular oligomers (ADDLs) as new vaccine and drug targets. Neurochem. Int 41, 345–352. [DOI] [PubMed] [Google Scholar]
- Kurup P, Zhang Y, Xu J, Venkitaramani DV, Haroutunian V, Greengard P, Nairn AC, and Lombroso PJ (2010). Abeta-mediated NMDA receptor endocytosis in Alzheimer’s disease involves ubiquitination of the tyrosine phosphatase STEP61. J. Neurosci 30, 5948–5957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, et al. (1998). Diffusible, nonfibrillar ligands derived from Abeta1–42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. USA 95, 6448–6453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YS, and Silva AJ (2009). The molecular and cellular biology of enhanced cognition. Nat. Rev. Neurosci 10, 126–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Hong S, Shepardson NE, Walsh DM, Shankar GM, and Selkoe D (2009). Soluble oligomers of amyloid Beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron 62, 788–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, and Bear MF (2004). LTP and LTD: an embarrassment of riches. Neuron 44, 5–21. [DOI] [PubMed] [Google Scholar]
- Malinow R, Schulman H, and Tsien RW (1989). Inhibition of postsynaptic PKC or CaMKII blocks induction but not expression of LTP. Science 245, 862–866. [DOI] [PubMed] [Google Scholar]
- Mammen AL, Huganir RL, and O’Brien RJ (1997). Redistribution and stabilization of cell surface glutamate receptors during synapse formation. J. Neurosci 17, 7351–7358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsden KC, Shemesh A, Bayer KU, and Carroll RC (2010). Selective translocation of Ca2+/calmodulin protein kinase IIalpha (CaMKIIalpha) to inhibitory synapses. Proc. Natl. Acad. Sci. USA 107, 20559–20564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin SJ, Grimwood PD, and Morris RG (2000). Synaptic plasticity and memory: an evaluation of the hypothesis. Annu. Rev. Neurosci 23, 649–711. [DOI] [PubMed] [Google Scholar]
- Mikuni T, Nishiyama J, Sun Y, Kamasawa N, and Yasuda R (2016). High-throughput, high-resolution mapping of protein localization in mammalian brain by in vivo genome editing. Cell 165, 1803–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mora RJ, Roberts RW, and Arnold DB (2013). Recombinant probes reveal dynamic localization of CaMKIIα within somata of cortical neurons. J. Neurosci 33, 14579–14590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Leary H, Liu WH, Rorabaugh JM, Coultrap SJ, and Bayer KU (2011). Nucleotides and phosphorylation bi-directionally modulate Ca2+/ calmodulin-dependent protein kinase II (CaMKII) binding to the N-methyl-D-aspartate (NMDA) receptor subunit GluN2B. J. Biol. Chem 286, 31272–31281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Riordan KJ, Hu NW, and Rowan MJ (2018). Aß facilitates LTD at Schaffer collateral synapses preferentially in the left hippocampus. Cell Rep 22, 2053–2065. [DOI] [PubMed] [Google Scholar]
- Opazo P, Viana da Silva S, Carta M, Breillat C, Coultrap SJ, Grillo-Bosch D, Sainlos M, Coussen F, Bayer KU, Mulle C, and Choquet D (2018). CaMKII metaplasticity drives Aβ oligomer-mediated synaptotoxicity. Cell Rep 23, 3137–3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panza P, Maier J, Schmees C, Rothbauer U, and Söllner C (2015). Live imaging of endogenous protein dynamics in zebrafish using chromobodies. Development 142, 1879–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez EA, Campbell RE, Lin JY, Lin MZ, Miyawaki A, Palmer AE, Shu X, Zhang J, and Tsien RY (2017). The growing and glowing toolbox of fluorescent and photoactive proteins. Trends Biochem. Sci 42, 111–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, et al. (2008). Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med 14, 837–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva AJ, Stevens CF, Tonegawa S, and Wang Y (1992). Deficient hippo-campal long-term potentiation in alpha-calcium-calmodulin kinase II mutant mice. Science 257, 201–206. [DOI] [PubMed] [Google Scholar]
- Sinnen BL, Bowen AB, Gibson ES, and Kennedy MJ (2016). Local and use-dependent effects of β-amyloid oligomers on NMDA receptor function revealed by optical quantal analysis. J. Neurosci 36, 11532–11543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, Nairn AC, Salter MW, Lombroso PJ, Gouras GK, and Greengard P (2005). Regulation of NMDA receptor trafficking by amyloid-beta. Nat. Neurosci 8, 1051–1058. [DOI] [PubMed] [Google Scholar]
- Strack S, McNeill RB, and Colbran RJ (2000). Mechanism and regulation of calcium/calmodulin-dependent protein kinase II targeting to the NR2B subunit of the N-methyl-D-aspartate receptor. J. Biol. Chem 275, 23798–23806. [DOI] [PubMed] [Google Scholar]
- Sumi M, Kiuchi K, Ishikawa T, Ishii A, Hagiwara M, Nagatsu T, and Hidaka H (1991). The newly synthesized selective Ca2+/calmodulin dependent protein kinase II inhibitor KN-93 reduces dopamine contents in PC12h cells. Biochem. Biophys. Res. Commun 181, 968–975. [DOI] [PubMed] [Google Scholar]
- Thiagarajan TC, Piedras-Renteria ES, and Tsien RW (2002). alpha- and betaCaMKII. Inverse regulation by neuronal activity and opposing effects on synaptic strength. Neuron 36, 1103–1114. [DOI] [PubMed] [Google Scholar]
- Tsien RY (1998). The green fluorescent protein. Annu. Rev. Biochem 67, 509–544. [DOI] [PubMed] [Google Scholar]
- Um JW, Nygaard HB, Heiss JK, Kostylev MA, Stagi M, Vortmeyer A, Wisniewski T, Gunther EC, and Strittmatter SM (2012). Alzheimer amyloid-β oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat. Neurosci 15, 1227–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vest RS, Davies KD, O’Leary H, Port JD, and Bayer KU (2007). Dual mechanism of a natural CaMKII inhibitor. Mol. Biol. Cell 18, 5024–5033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vest RS, O’Leary H, Coultrap SJ, Kindy MS, and Bayer KU (2010). Effective post-insult neuroprotection by a novel Ca(2+)/ calmodulin-dependent protein kinase II (CaMKII) inhibitor. J. Biol. Chem 285, 20675–20682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakayama S, Kiyonaka S, Arai I, Kakegawa W, Matsuda S, Ibata K, Nemoto YL, Kusumi A, Yuzaki M, and Hamachi I (2017). Chemical labelling for visualizing native AMPA receptors in live neurons. Nat. Commun 8, 14850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, and Selkoe DJ (2002). Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416, 535–539. [DOI] [PubMed] [Google Scholar]
- Woolfrey KM, O’Leary H, Goodell DJ, Robertson HR, Horne EA, Coul-trap SJ, Dell’Acqua ML, and Bayer KU (2018). CaMKII regulates the depalmitoylation and synaptic removal of the scaffold protein AKAP79/150 to mediate structural long-term depression. J. Biol. Chem 293, 1551–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue L, Karpenko IA, Hiblot J, and Johnsson K (2015). Imaging and manipulating proteins in live cells through covalent labeling. Nat. Chem. Biol 11, 917–923. [DOI] [PubMed] [Google Scholar]
- Yu W, Jiang M, Miralles CP, Li RW, Chen G, and de Blas AL (2007). Gephyrin clustering is required for the stability of GABAergic synapses. Mol. Cell. Neurosci 36, 484–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YP, Holbro N, and Oertner TG (2008). Optical induction of plasticity at single synapses reveals input-specific accumulation of alphaCaMKII. Proc. Natl. Acad. Sci. USA 105, 12039–12044. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.