
Figure 2.
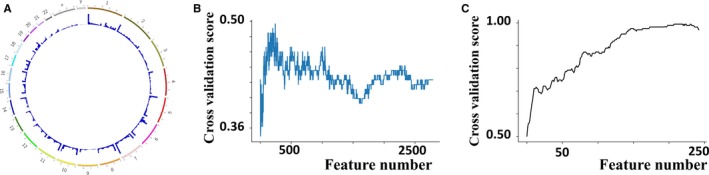
(A) Using ChAMP, we identified 47 099 differentially methylated sites in the sample of 377 HCC samples and 50 adjacent normal tissues. (B) Results of applying the SVM‐RFE algorithm to 2785 methylation sites significantly associated with overall survival based on Cox regression, and the best 243 were selected based on the 10‐fold cross‐validation score for the number of recursive features at each level. The corresponding 10‐fold cross‐validation score was 0.50.C. Results of applying the FW‐SVM algorithm to 243 methylation sites obtained with the SVM‐RFE method, and we finally built a predictive model containing the best 134 features, which gave a mean 10‐fold cross‐validation score of 0.95