
Figure 1.
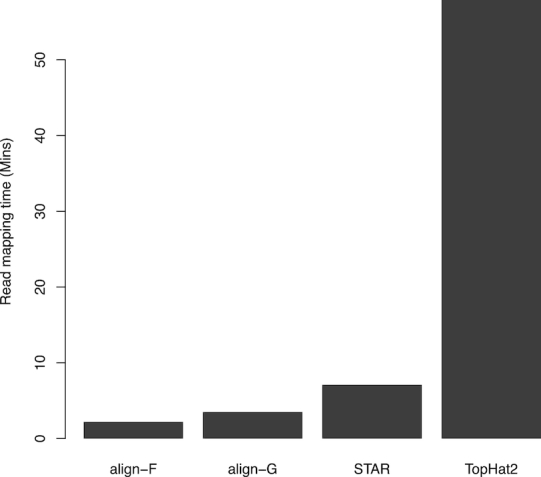
Run times of read aligners. Each aligner used ten threads to map 15 million 100 bp read-pairs from the SEQC UHRR sample to the human reference genome GRCh38. Rsubread::align is faster than STAR or TopHat2 regardless of whether the full index (align-F) or a gapped index (align-G) is used.