

Abstract
Background
We have recently reported that activation of cannabinoid type 2 receptors (CB2Rs) reduces dopamine (DA) neuron excitability in mouse ventral tegmental area (VTA). Here, we elucidate the underlying mechanisms.
Methods
Patch-clamp recordings were performed in mouse VTA slices and dissociated single VTA DA neurons.
Findings
Using cell-attached recording in VTA slices, bath-application of CB2R agonists (JWH133 or five other CB2R agonists) significantly reduced VTA DA neuron action potential (AP) firing rate. Under the patch-clamp whole-cell recording model, JWH133 (10 μM) mildly reduced the frequency of miniature excitatory postsynaptic currents (mEPSCs) but not miniature inhibitory postsynaptic currents (mIPSCs). JWH133 also did not alter evoked EPSCs or IPSCs. In freshly dissociated VTA DA neurons, JWH133 reduced AP firing rate, delayed AP initiation and enhanced AP after-hyperpolarization. In voltage-clamp recordings, JWH133 (1 μM) enhanced M-type K+ currents and this effect was absent in CB2−/− mice and abolished by co-administration of a selective CB2R antagonist (10 μM, AM630). CB2R-mediated inhibition in VTA DA neuron firing can be mimicked by M-current opener (10 μM retigabine) and blocked by M-current blocker (30 μM XE991). In addition, enhancement of neuronal cAMP by forskolin (10 μM) reduced M-current and increased DA neuron firing rate. Finally, pharmacological block of synaptic transmission by NBQX (10 μM), D-APV (50 μM) and picrotoxin (100 μM) in VTA slices failed to prevent CB2R-mediated inhibition, while intracellular infusion of guanosine 5'-O-2-thiodiphosphate (600 μM, GDP-β-S) through recording electrode to block postsynaptic G-protein function prevented JWH133-induced reduction in AP firing.
Interpretation
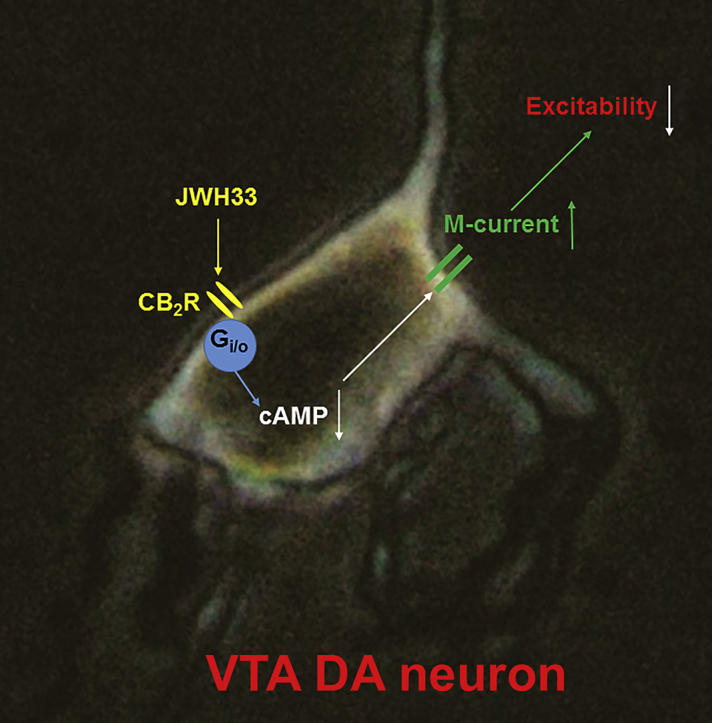
Our results suggest that CB2Rs modulate VTA DA neuron excitability mainly through an intrinsic mechanism, including a CB2R-mediated reduction of intracellular cAMP, and in turn enhancement of M-type K+ currents.
Fund
This research was supported by the Barrow Neuroscience Foundation, the BNI-BMS Seed Fund, and CNSF (81771437).
Keywords: Cannabinoid, CB2 receptor, Ventral tegmental area, Dopamine neuron
Graphical abstract
Research in context.
Evidence before this study
Cannabis sativa (marijuana) is a fibrous flowering plant that naturally produces an abundant variety of molecules, some with psychoactive effects. At least 4% of the world's adult population uses cannabis annually, making it one of the most frequently used illicit drugs in the world. The psychoactive effects of cannabis are mediated primarily through two cannabinoid receptor (CBR) subtypes, CB1R and CB2R. The prevailing view is that CB1Rs are mainly expressed in central neurons while CB2Rs are predominantly expressed in peripheral immune cells. However, this traditional view has been challenged by strong emerging evidence that shows CB2Rs are moderately expressed and function in specific brain areas. New evidence has demonstrated that brain CB2Rs mediate important modulations in animal cocaine-seeking behaviors (Xi et al., Nature of Neuroscience, Jul 24;14 [9]:1160-6, 2011), suggesting that these receptors may exist in the brain regions that regulate drug addiction. Recently, we further confirmed that functional CB2Rs are expressed in mouse ventral tegmental area (VTA) DA neurons, and activation of VTA CB2Rs reduces neuronal excitability and cocaine-seeking behavior (Zhang et al., PNAS 111 [46]:E5007-15, 2014). However, the mechanisms and the CB2R-G protein intracellular signaling that underlies these VTA CB2R-mediated modulations are unknown.
Added value of this study
In this paper, we elucidate the underlying mechanisms. Using cell-attached recording in VTA slices, bath-application of a CB2R agonist (JWH133) inhibited VTA DA neuron firing in a concentration-dependent manner. In addition to JWH133, five other CB2R agonists exhibited similar inhibition. Under the patch-clamp whole-cell recording model, JWH133 (10 μM) inhibited miniature excitatory postsynaptic currents (mEPSCs) but not miniature inhibitory postsynaptic currents (mIPSCs). However, JWH133 did not inhibit evoked EPSCs or IPSCs. In freshly dissociated VTA DA neurons, JWH133 reduced the action potential (AP) firing rate, delayed AP initiation and enhanced AP after-hyperpolarization. In voltage-clamp recordings, M-type K+ currents were enhanced by JWH133. This effect was absent in CB2−/− mice and abolished by co-administration of a selective CB2R antagonist (AM630). In addition, CB2R-mediated inhibition in VTA DA neuron firing can be mimicked by an M-current opener and blocked by an M-current blocker. Finally, CB2R-cAMP signaling plays a role in modulating DA neuron excitability since enhancement of cAMP by forskolin reduced M-current and increased DA neuron firing rate.
Implications of all the available evidence
These results suggest that CB2Rs modulate VTA DA neuron excitability through both synaptic and intrinsic mechanisms, including a reduction in presynaptic glutamate release and enhancement of postsynaptic neuronal M-currents. This work provides new insights into understanding the impact of brain CB2Rs in a variety of DA system associated disease pathogenesis and potential therapeutics.
Alt-text: Unlabelled Box
1. Introduction
The CB2R is a G-protein-coupled receptor that was cloned in 1993 [5]. Since then, the expression and function of CB2Rs in the brain has been debated. Early studies suggested that CB2Rs were absent in the brain because CB2 mRNA transcripts were not detected in brain tissues [[1], [2], [3], [4]]. Based on these findings, the CB2R has been considered as a “peripheral” cannabinoid receptor [[5], [6], [7]]. Recently, this concept has been challenged by the identification of CB2R mRNA and receptor proteins throughout the central nervous system (CNS) [8,9], including the cerebral cortex [10], hippocampus [11], striatum [12], amygdala and brainstem [[12], [13], [14], [15], [16], [17]]. Immunoblot and immunohistochemistry (IHC) assay also detect CB2R-immunoreactivity or immunostaining in various brain regions [16,[18], [19], [20], [21], [22], [23]]. Consistent with these findings, we reported that brain CB2Rs modulate cocaine self-administration, cocaine-enhanced locomotion and extracellular dopamine (DA) levels in the nucleus accumbens in mice [24]. This finding was further supported by a study where mice over-expressing CB2Rs exhibit a significantly lower level of cocaine self-administration and cocaine-enhanced locomotion compared to wild type (WT) mice [22]. Recently, we identified functional CB2R expression in mouse VTA DA neurons, and selective activation of these CB2Rs reduced DA neuronal excitability in vitro and in vivo preparations [25]. More recently, Stempel et al. reported that CB2Rs mediated a cell type-specific plasticity in the hippocampal CA3 neurons [11]. However, the cellular and molecular mechanisms underlying CB2R modulation of VTA DA neuronal excitability are poorly understood.
In the present study, we systematically investigated synaptic and intrinsic ionic channel mechanisms possibly underlying CB2R modulation of VTA DA neurons using patch-clamp recordings. We found that activation of VTA CB2Rs reduced the frequency of mEPSCs but not mIPSCs in DA neurons. By using a perforated patch-clamp recording in acutely dissociated VTA DA neurons, a simple single cell model without intact presynaptic connections, we found that JWH133 reduced DA neuron action potential (AP) firing rate, prolonged AP initiation, enhanced after-hyperpolarization, and enhanced M-type K+ channel currents. In addition, we determined whether the presynaptic (reduction of mEPSC frequency) or postsynaptic (intrinsic) or both contribute to JWH133-induced reduction in AP firing, and found that the intrinsic mechanism such as enhancement of M-current, contributes to CB2R–mediated reduction of VTA DA neuronal excitability, which may play an important role in the regulation of DA-associated behaviors and diseases.
2. Methods and materials
2.1. Animals
Male wild-type (WT) and Zimmer strain CB2−/− mice [6,7] with C57BL/6J genetic backgrounds were bred at Barrow Neurological Institute. Genotyping was performed in our laboratory before experiments were begun. All experiments were in compliance with the Guide for the Care and Use of Laboratory Animals and experimental protocol has been approved by IACUC of Barrow Neurological Institute. When we tested the effects of CB2R agonist on VTA DA neurons using CB2−/− mice, the littermate WT mice were used as the control. All experimental procedures were approved by the Institutional Animal Care and Use Committee at the Barrow Neurological Institute and by the Animal Ethics Committee of Qingdao University.
2.2. VTA slice preparation
Mice (14–31 days old) were used as previously described [[26], [27], [28]]. Briefly, mice were housed under standard conditions at 22–24 °C and 50–60% humidity under a 12 h light/dark cycle. On the experimental day, mice were anesthetized (isoflurane USP) and then sacrificed by decapitation. Horizontal (or coronal) midbrain slices (250 μm) were cut using a vibratome 1000 (Vibratome 1000 plus; Jed Pella Inc., Redding, CA) to include the VTA. Slices were prepared in an ice-cold, sucrose-based, artificial cerebrospinal fluid containing (in mM): 212.7 sucrose, 5 KCl, 1.25 NaH2PO4, 3 MgCl2, 1 CaCl2, 26 NaHCO3, and 10 glucose. The slices were then incubated for at least 1 h in a pre-incubation chamber (Warner Ins., Holliston, MA) at room temperature (22–24 °C) in conventional artificial cerebrospinal fluid containing (ACSF, in mM): 124 NaCl, 5 KCl, 2 CaCl2, 2 MgCl2, 1.25 NaH2PO4, 26 NaHCO3 and 10 glucose. The ACSF was continuously saturated with 95% O2 and 5% CO2.
2.3. Cell-attached patch-clamp recordings in VTA slices
For slice patch recordings, horizontal or coronal midbrain slices (250 μm thickness) were prepared from 14- to 31-day old mice (WT and CB2−/−) mice as described previously [28]. To maintain an intact intracellular environment, we employed a cell-attached mode of patch recording. Thus, we were able to measure stable extracellular action potential (AP) firing for hours. The recording pipette was filled with intracellular solution containing in mM: 140 potassium gluconate, 5 KCl, 10 HEPES, 0.2 EGTA, 2 MgCl2, 4 MgATP, 0.3 Na2GTP and 10 Na2-phosphocreatine (pH 7.3 with KOH). After patch recording, we converted from cell-attached mode to conventional whole-cell recording to identify the neuronal phenotype of the recorded neuron according to standard electrophysiological, pharmacological and immunostaining criteria as described previously [28,29]. Briefly, in this study, we mostly recorded DA neurons from lateral VTA, which demonstrated a low action potential (AP) firing rate (1–3 Hz), large H-current, and TH positive reaction by immunostaining (Fig. 1A-C). We have analyzed 12 VTA neurons (from 12 mice), under the cell-attached recording mode to record extracellular AP firing, the averaged AP duration was 2.34 ± 0.07 ms, the averaged firing rate was 1.65 ± 0.14 Hz, and after perfusion of 100 nM quinpirole for 10 min, the AP firing rate was reduced to 0.53 ± 0.08 Hz (paired t-test p < 0.0001). After washout of quinpirole for 10 min, AP firing rate recovered to 1.40 ± 0.12 Hz (quinpirole washout p < 0.0001). In addition, we have examined the effects of JWH133 on H-currents of DA neurons in mouse VTA slices. In 4 cells tested (from 2 mice), the amplitudes of H-currents (hyperpolarizing from −60 to −120 mV) before and after exposure of 10 μM JWH133 for 10 min were 166.0 ± 24.3 pA and 161.3 ± 11.2 pA (paired t-test, p = 0.86). These results suggest that acute application of 10 μM JWH133 to VTA slices did not alter H-current.
Fig. 1.

Identification of VTA DA neurons from patch-clamp recording in VTA slice. (a) In patch-clamp cell-attached recording mode, bath-application of 100 nM quinpirole (dopamine D2 receptor agonist) inhibited firing rate of DAergic neurons. (b) Under whole-cell recording mode, DA neurons exhibited hyperpolarizing pulse-activated currents (H-current, left traces) whereas GABAergic neurons usually did not express H-currents (Right traces). (c) After patch recording, the biocytin (2 mg/ml) was delivered into recorded neuron (left panel). Tyrosine hydroxylase (TH) staining was performed post hoc and labels DA neurons (middle and right panels).
2.4. Miniature or evoked excitatory/inhibitory postsynaptic currents (mEPSCs/mIPSCs, eEPSCs/eIPSCs) in VTA slices
To determine whether brain CB2Rs modulate presynaptic glutamate or GABA release, thereby altering postsynaptic DA neuronal activity, we recorded mEPSCs in the presence of TTX (a voltage-dependent Na+ channel blocker, 1 μM), D-APV (a NMDA receptor antagonist, 50 μM) and picrotoxin (a selective GABAA receptor antagonist, 100 μM), or mIPSCs in the presence of TTX, NBQX (an AMPA receptor antagonist, 10 μM) and D-APV (50 μM) in VTA DA neurons. In addition, we recorded electrical stimulation-evoked EPSCs (eEPSCs) or -evoked IPSCs (eIPSCs) in the absence of TTX to determine whether JWH133 alters eEPSCs or eIPSCs in VTA DA neurons.
2.5. Patch-clamp recording from single dissociated VTA DA neurons
Perforated-patch whole-cell recording techniques were employed [30,31]. Compared to performing conventional whole-cell recordings, this approach was crucial to obtain stable nicotinic responses from dissociated VTA neurons, presumably due to minimal perturbation of the intracellular environment. Pipettes (3–5 MΩ) used for perforated-patch recordings were filled with intracellular recording solution, which contained (in mM): 140 K-gluconate, 10 KCl, 5 MgCl2, and 10 HEPES; pH 7.2 (with Tris-OH) freshly supplemented before use with amphotericin B to 200 μg/mL from a 40 mg/mL DMSO stock. The liquid junction potential was 14 mV and calculated using Clamplex 9.2 (Axon Instruments, Foster City, CA), and corrections were made for junction potentials post-hoc. After tight seal (>2 GΩ) formation, about 5–20 min was required for conversion to the perforated-patch mode, and an access resistance of <60 MΩ was accepted to start the experiments. Series resistance was not compensated in this study. Data were acquired at 10 kHz, filtered at 2 kHz, digitized on-line (Digidata 1440 series A/D board, Axon Instruments), and displayed and stored on a PC computer. Rapid application of drugs was performed using a computer-controlled “U-tube” system [30]. Periods of drug exposure are specified in the text and figures or legends. All experiments were performed at room temperature (21 ± 1 °C). To enable identification of single, dissociated VTA neurons, cells were labeled with biocytin (Sigma-Aldrich, St. Louis, MO; 2.0 mg/mL in the recording electrode) in some experiments. After conversion from perforated to a conventional whole-cell recording mode, the dye was injected into the cytoplasm by a pulse (200 ms, 0.5 Hz) of depolarizing current (1.0 nA) for 3 min. Labeled cells were visualized using epifluorescence microscopy (see detail in our recent publication) [25].
2.6. Data analyses and statistics
All data are presented as means (± S.E.M.). One-way or two-way analysis of variance (ANOVA) was used to analyze the significance of the effects of JWH133 or other drugs on neuronal firing. Individual group comparisons were carried out using the Student Newman-Keuls or Tukey method. In addition, paired (or unpaired) t-tests were also used to analyze some electrophysiological data as described previously [30,31].
3. Results
3.1. Activation of CB2R reduced VTA DA neuron firing
Initial experiments were designed to examine the concentration-dependent effect of CB2R agonist (JWH133) on VTA DA neuron firing using cell-attached recording mode. Fig. 2A shows a typical trace of VTA DA firing before (A top trace) and during (A bottom trace) CB2R agonist JWH133 exposure. Bath applications of different concentrations of JWH133 (1, 10, 100 μM) reduced DA neuron firing rate in a concentration-dependent manner (Fig. 2B, C, One-way ANOVA, F [4,35] = 6.97, p < 0.001). Then, we examined the effects of JWH133 on DA neuron firing from horizontal (n = 13 from 4 mice) or coronal (b = 8 from 3 mice) VTA slices, and found a modest but significantly consistent effect on inhibition of VTA DA neuron firing in both types of slice preparations (One-way ANOVA, F [2,31] = 4.79, p < 0.05, Fig. 2D, E). Further, Tukey analysis showed that compared to baseline, JWH133 inhibited DA fairing rate in either coronal (p < 0.05, n = 8, Tukey analysis) or horizontal (p < 0.05, n = 13, Tukey analysis) slices. Furthermore, we tested whether different CB2R agonists exhibit similar inhibitory effects on VTA DA neuron firing. As shown in Fig. 2F, the CB2R agonists, GW, SER, CB65, JWH105, Hu308 and L759656 exhibited a similar reduction of DA firing rate (One-way ANOVA, F [6,31] = 2.4, p < 0.05), but Tukey analysis showed that JWH105 and L759656 did not reach statistical significance (p > 0.05, Tukey analysis). Finally, to exclude the possibility that JWH133-induced reduction of AP firing rate was mediated by AP firing rundown with time, we performed a group of control experiments, in which, VTA DA neuronal AP firing was recorded using cell-attached patch for 20 min without any drugs. In 6 slices tested (from 3 mice), there was no functional rundown of AP firing within 20 min (Fig. 2G), demonstrating that cell-attached recording to measure extracellular AP firing was stable. Considering the ages of mice used in this study were between 2 and 4 weeks, we classified mice into two age groups and compared the effects of JWH133. Group one contained 21 slices with an average of age 16.1 ± 0.3 postnatal days and group two contained 6 slices with an average of 30.5 ± 0.6 postnatal days. The difference of the ages between these two groups were statistically significant (p < 0.001, unpaired t-test, Fig. 2H). 10 μM JWH133 inhibited VTA DA AP firing rate in a similar rate (Fig. 2I), in group one, the inhibitory rate was 18 ± 4% (n = 21 from 8 mice), while in group two, the inhibitory rate was 15 ± 3% (n = 6 from 2 mice, p = 0.59, unpaired t-test), suggesting that during animal developing between P16 and P30, the CB2R-mediated inhibition in VTA DA neuronal firing rate is not changed. Together, these results are further consistent with our previous findings that activation of VTA CB2Rs reduces VTA DA neuron firing rate [25].
Fig. 2.

Activation of VTA CB2Rs reduced VTA DA neuron firing rate. In VTA slices, cell-attached recording showed the typical traces of extracellular action potential (AP) firing before and after JWH133 exposure (a). A time-frequency histograph (b) and Bar graph from the normalized data (c) showed that JWH133 inhibited VTA DA neuron firing in a concentration-dependent manner. (d) Comparison of the effects of JWH133 on AP firing rate between horizontal and coronal VTA slices, and showed no significant difference between these two types of slices (e). (f) In addition to JWH133, other 6 CB2R agonists tested showed 4 of them exhibited inhibitory effects on VTA DA neurons. * p < 0.05, ** p < 0.01. (g) In control experiments (without any drugs), cell-attached patch recording showed a stable AP firing of VTA DA neurons for 20 min. (h) Mice were classified into two age groups (postnatal 16 and 30). (i) Statistical comparison of JWH133 (10 μM)-induced inhibition of VTA DA neuron firing rate between these two age-groups, and showed no statistical significance (N.S.).
3.2. Effects of CB2R activation on excitatory synaptic transmission in VTA DA neurons
VTA DA neuron firing is controlled by both presynaptic and postsynaptic mechanisms. To elucidate the mechanisms underlying CB2R-mediated reduction of VTA DA neuronal firing, we tested the effects of CB2R agonist on excitatory synaptic transmission measured by miniature excitatory postsynaptic currents (mEPSCs) in DA neurons of VTA slices. The identification of VTA DA neurons was based on their electrophysiological characteristic features and the TH staining after patch-clamp recording as described previously [25,28] and shown in Fig. 1A-C. In 18 VTA slices tested prepared from 9 WT mice, bath perfusion of 10 μM JWH133 for 10 min altered mEPSCs (Fig. 3A,B), represented as reduced frequency in 14 slices (Fig. 3C,E, left panel) and enhanced frequency in 4 slices. Averaged data showed that JWH133 reduced mEPSC frequency from 0.91 ± 0.19 to 0.75 ± 0.14 Hz (p = 0.03, paired t-test, n = 18, Fig. 3F, left panel). Further cumulative distribution of mEPSC inter-event interval showed significantly shift (K—S test, p = 0.002). For mEPSC amplitude, JWH133 reduced amplitude in 3 slices and enhanced amplitude in 14 slices; and no change in 1 slice (Fig. 3D,E, right panel), and together, there was no significant change of amplitude (from 11.0 ± 0.5 to 10.6 ± 0.4 pA, p = 0.10, paired t-test, n = 18, Fig. 3F, right panel). These results suggest that acute activation of CB2Rs shows a modest, but statistically significant, reduction in glutamatergic synaptic transmission in VTA DA neurons, including reduced presynaptic glutamate release probability.
Fig. 3.

Effects of JWH133 on excitatory synaptic transmission in VTA DA neurons. (a) Typical traces of mEPSCs in the presence of TTX (1 μM) and picrotoxin (100 μM) before (top trace) and after (bottom trace) JWH133 (10 μM) exposure using whole-cell patch-clamp recording in a VTA slice. In 19 slices, cumulative probability analysis for mEPSC inter-event interval (b) and amplitude (c) under control conditions (black curve) and JWH133 exposure (red curve). (d) Comparison of mEPSC frequency (left panel) and amplitude (right panel) before and after JWH133 exposure from 19 VTA slices tested. (e) Bar graph data illustrating JWH133 significantly reduced mEPSC frequency, but not amplitude. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.3. Effects of CB2R activation on inhibitory synaptic transmission in VTA DA neurons
We then examined the effects of JWH133 on miniature inhibitory postsynaptic currents (mIPSCs) in the presence of TTX (1 μM), NBQX (10 μM) and D-APV (50 μM). In 23 VTA slices prepared from 11 WT mice, bath perfusion of 10 μM JWH133 for 10 min altered mIPSC frequency and amplitude, represented as reduced frequency in 9 slices and enhanced frequency in 14 slices; reduced amplitude in 8 slices and enhancement amplitude in 14 slices; and no change in one slice. After averaged, these data showed that after JWH133 exposure, mIPSC frequency was 109% (109 ± 5%, p = 0.09, paired t-test, n = 23) and amplitude was 101% (101 ± 2%, p = 0.73, paired t-test, n = 23) compared to the baseline level (Fig. 4A, E). Neither inter-event interval nor amplitude cumulative distribution showed shift (Fig. 4B, C). These results suggest that activation of CB2Rs exhibits little effects on either presynaptic GABA release or postsynaptic GABAA receptor function.
Fig. 4.

Effects of JWH133 on inhibitory synaptic transmission in VTA DA neurons. (a) Typical traces of mIPSCs in the presence of NBQX (10 μM) and D-APV (50 μM) before (top trace) and after (bottom trace) JWH133 (10 μM) exposure using the whole-cell patch-clamp recording in a VTA slice. (b) and (c) In 23 VTA slices, cumulative probability analysis for mIPSC inter-event interval and amplitude under control conditions (baseline) and JWH133 exposure. K—S Test showed that JWH133 did not significantly alter both mIPSC frequency (p = 0.432, b) and amplitude (p = 0.384, c). (d) Comparison of mIPSC frequency (left panel) and amplitude (right panel) before and after JWH133 exposure from 23 VTA slices tested. (e) Normalized bar graph data illustrating JWH133 did not alter mIPSC frequency and amplitude.
3.4. Effects of CB2R activation on evoked EPSCs and IPSCs in VTA DA neurons
We also examined the effects of JWH133 on evoked EPSCs (eEPSCs) and IPSCs (eIPSCs). In these experiments, 100 μM PTX (for eEPSCs) or NBQX(10 μM) + APV (50 μM) (for eIPSCs) was added into the bath solution. A bipolar tungsten stimulation electrode was placed at ~150 μm rostral to the recording electrode and the paired stimulation pulses with 50 ms interval were applied (Fig. 5A). The results showed that bath perfusion of 10 μM JWH133 for 10 min failed to alter the current amplitude and the P2/P1 ratio of paired-pulse-induced eEPSCs (Fig. 5A-C) and eIPSCs (Fig. 5D-F). These results suggest that acute activation of VTA CB2Rs does not affect evoked synaptic neurotransmitter release.
Fig. 5.

Effects of JWH133 on evoked EPSCs and IPSCs in VTA DA neurons. (a) Representative typical traces of paired-pulse (50 ms interval) stimulation-induced eEPSCs before (left trace pair) and after (right trace pair) exposure to JWH133. Bar graph illustrating that JWH133 (10 μM) altered neither eEPSC amplitude (b) nor P2/P1 ratio (c) in 10 VTA slices tested. (d) Representative typical traces of paired-pulse (50 ms interval) stimulation-induced eIPSCs before (left trace pair) and after (right trace pair) exposure to JWH133. Bar graph illustrating that JWH133 (10 μM) did not change eIPSC amplitude (e) and P2/P1 ratio (f) in 10 VTA slices tested.
3.5. Characteristic features of CB2R-mediated modulation in VTA DA neuron action potentials
The data presented thus far demonstrated that a mild reduction of presynaptic glutamatergic by acute CB2R activation. We further elucidated possible intrinsic mechanisms of the CB2R-mediated modulation in postsynaptic DA neurons. To accomplish this, we used freshly dissociated single VTA DA neurons, which is a pure postsynaptic cell model since the enzymatic treatment and mechanical dissociation remove all presynaptic terminals/boutons from these cells [25,30,31]. Perforated (amphotericin B) patch-clamp recording from the single dissociated VTA DA neurons showed that bath application of 1 μM JWH133 for 1–3 min reduced AP firing rate (Fig. 6A-C), prolonged AP initiation (Fig. 6D) and enhanced the amplitude of after-hyperpolarization current (Fig. 6E). Fig. 6F summarizes these AP alterations before, during and after exposure to JWH133. These results suggest that, in a single neuron model, the activation of somatodendritic CB2Rs in VTA DA neurons reduces neuronal excitability likely via alterations of intrinsic ion channels such as the M-type K+ channels.
Fig. 6.

Analysis of CB2R-mediated changes of action potential (AP) in dissociated VTA DA neurons. Representative typical traces of AP of VTA DA neurons before (a), during (b) and after (c) exposure of 1 μM JWH133 under perforated current-clamp recording conditions. AP firing was elicited by injection of 10 pA current through recording electrode. Superimposed the first AP from trace (a)-(c) to show the AP initiation (d) and AP after-hyperpolarization (e) with expended time scale. (f) Normalized data showing the effect of JWH133 on AP firing parameters. * p < 0.05, ** p < 0.01.
3.6. Effects of CB2R agonist on voltage-dependent M-type K+ currents
M-channel is a voltage-gated, non-inactivating K+ channel. It is expressed in VTA DA neurons [32] and functionally modulates neuronal excitability [33,34]. It has been reported that M-channels are modulated by multiple G-protein-coupled receptors, including CB1Rs [35]. Therefore, we explored the possibility that CB2Rs may also modulate M-currents in VTA DA neurons. M-currents were induced with a standard deactivation voltage protocol [32,36] as shown in Fig. 7A. M-current is measured as the inward relaxation current caused by M-current deactivation during the voltage steps. Bath-applied JWH133 (1 μM) significantly enhanced M-currents elicited by three hyperpolarizing holding potentials in dissociated VTA DA neurons in WT mice (Fig. 7B), and this effect was reversible after JWH133 washout (Fig. 7C). Fig. 7D shows the superimposed traces of M-currents before and during JWH133 exposure, showing a JWH133-induced M-current augmentation. This effect was blocked by AM630 (1 μM) and absent in VTA DA neurons prepared from CB2−/− mice. To confirm the identity of the recorded M-current, we examined the effects of retigabine (a selective M-current enhancer, 10 μM) and XE991 (a selective M-current blocker, 30 μM) on the recorded current. Like JWH133, retigabine enhanced (n = 5 from 3 mice, p = 0.02, paired t-test), but XE991 reduced (n = 8 from 4 mice, p = 0.003, paired t-test) the recorded currents produced by the M-current-inducing protocol, supporting the conclusion that the recorded currents were, indeed, M-currents. Fig. 7E shows pooled data, illustrating the effects of each individual group tested, in which JWH133 (1 μM) enhanced M-currents (n = 19 from 8 mice, p = 0.0002, paired t-test), AM630 + JWH133 abolished JWH133's effect, retigabine enhanced but XE991 reduced M-currents in WT mice. While in CB2−/− mice, JWH133 failed to enhance M-current but retigabine still enhances and XE991 still reduced M-currents. These results suggest that the inhibitory effect of JWH133 on M-current is mediated through the CB2Rs, while the effects of retigabine and XE991 on M-current are CB2R-independent.
Fig. 7.

Effects of JWH133 on M-type K+ currents in single dissociated VTA DA neurons. (a)-(c) Representative typical traces of M-currents before, during and after exposure of JWH133. M-currents were induced by a pre-depolarization from holding potential of −60 mV to −30 mV, then three steps back to −60 mV in 10 mV increments. Fig. 6b illustrates that JWH133 increased M-current amplitude in VTA DA neurons. (d) Superimposed M-current before (Black trace) and during (red trace) JWH133 exposure with expanded time scale. (e) Bar graph illustrating that JWH133 (1 μM) enhanced M-currents, which was blocked by AM630 or absent in CB2−/− mice. Retigabine enhanced, while XE991 attenuated M-currents in both WT and CB2−/− mice. The above experiments were performed separately by each paired (before and after tested drug exposure) group. In statistical analyses, the baseline (before drug exposure) was normalized as 1, and the changed value after drug perfusion was compared to the baseline of each individual group, and analyzed using paired t-test. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.7. Roles of CB2R-mediated enhancement of M-current in altered VTA DA neuron firing
To further determine whether JWH133-enhanced M-currents contribute to JWH133-induced VTA DA neuronal inhibition, we examined the effects of retigabine and XE991 on VTA DA neuronal firing. In single dissociated VTA DA neurons, we found that retigabine (10 μM) increased M-currents, in an amplitude similar to JWH133, causing a reduction in VTA DA neuronal firing, while XE991 (30 μM) attenuated M-currents, producing an increase in neuronal firing (Fig. 8A, B). Fig. 8C and D show the summarized results after statistical analyses that retigabine reduced AP firing rate and prolonged AP initial time, and XE991 increased AP firing rate and shorted AP initial time. These results suggest that M-current in VTA DA neurons play an important role in modulation of neuronal firing rate. In VTA slices, cell-attached recordings showed that, in the presence of XE991, JWH133 failed to inhibit DA neuron firing (Fig. 8E, F). These results suggest that JWH133-enhanced M-currents play a role in the JWH133-induced reduction in VTA DA neuronal firing.
Fig. 8.

Roles of M-type K+ currents in JWH133-modulated VTA DA neuron firing. (a) Representative typical traces of M-type K+ currents in control (top trace), retigabine (middle trace) and XE991 (bottom trace) from the same acutely-dissociated neuron, and showed that retigabine enhanced and XE991 reduced M-current. (b) Representative typical traces of AP firing in control (top trace), retigabine (middle trace) and XE991 (bottom trace) from the same recorded neuron, and showed that retigabine decreased and XE991 increased AP firing rate. (c) Statistical analysis of the effects of retigabine on VTA DA neuronal AP firing including firing rate (FR), resting membrane potential (RP) and AP initial time. AP firing was evoked by an injection of depolarizing current (20 pA). (d) Statistical analysis of the effects of XE991 on VTA DA neuronal AP firing including firing rate (FR), resting membrane potential (RP) and AP initial time. AP firing was evoked by an injection of depolarizing current (20 pA). (e) In VTA slices using cell-attached recording, the time-frequency histograph showed no detectable change of AP firing rate after exposure to XE991 plus JWH133. (f) Statistical analysis showed JWH133 failed to reduce VTA DA firing rate in the presence of XE991.
3.8. Intracellular signal pathway of JWH133-induced enhancement of M-current
In these experiments, we determined which second messenger molecule mediates the CB2R-G-protein signaling to M-type K+ channels. It is known that CB2R activation primarily initiates two signal pathways, MAPK/ERK and cAMP/PKA, producing an increase in the former and decrease in the latter [[37], [38], [39]]. Since cAMP, but not MAPK, is associated to M-type K+ channels [37,40], we examined the effect of enhanced intracellular cAMP by forskolin on JWH133-induced enhancement of M-current. As shown in Fig. 9, bath-perfusion of 10 μM forskolin to dissociated single VTA DA neuron reduced M-current by 19.2 ± 4.9% and in the presence of forskolin, JWH133 (1 μM) potentiated M-current by 8.1 ± 3.7% (n = 10 from 5 mice, One-Way ANOVA, F [3,27]=14.9, p < 0.0001). Further analysis showed that forskolin-induced reduction of M-current was significant (p < 0.01, One-way ANOVA, Tukey), while in the presence of forskolin, JWH133-induced change of AP firing was not significant (p > 0.05, Fig. 9A,B, One-way ANOVA, Tukey). These results suggest that CB2R-mediated reduction of intracellular cAMP likely contributes to the enhancement of M-current by JWH133. Finally, we determined whether forskolin can prevent the JWH133-induced DA firing reduction. As shown in Fig. 9C and D, using cell-attached recording in VTA slices, bath-application of forskolin (10 μM) increased DA firing rate and prevented JWH133 (10 μM)-mediated reduction of DA firing rate (n = 11 from 5 mice, One-way ANOVA, F [2,20] = 4.63, p = 0.018, Fig. 9C, D). Further analysis showed that forskolin increased AP firing rate by 8.2 ± 2.0% (p < 0.05, paired t-test), while in the presence of forskolin, JWH133 altered AP firing by 1.2 ± 3.0% (p > 0.05, paired t-test). Together, these results suggest that JWH133 activates CB2R-G-protein signaling, thereby reducing intracellular cAMP levels, in turn enhancing M-type K+ channel function, and ultimately reducing VTA DA neuronal excitability.
Fig. 9.

Effects of forskolin on JWH133 modulated M-current and AP firing in VTA DA neurons. (a) Representative the typical traces of M-current under four different conditions: control (control, red trace), forskolin (Forsk, green trace), forskolin plus JWH133 (blue trace) and washout of drugs (light blue trace). The enhanced inset showed a reduction of M-current by forskolin. (b) Statistical analysis showed that forskolin significantly reduced M-current and in the presence of forskolin, JWH133 failed to enhance M-current. (c) In VTA slices using cell-attached recording, a time-frequency histograph showed AP firing rate's change during exposure to forskolin (10 μM) and forskolin plus JWH133 (10 μM). (d) Statistical analysis showed that forskolin increased AP firing rate, and in the presence of forskolin, JWH133 failed to reduce AP firing rate. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.9. Do both synaptic and intrinsic mechanisms contribute to CB2R-mediated reduction of VTA DA neuron firing?
To further determine the above findings that presynaptic mechanism (reduced mEPSC frequency) and intrinsic mechanism (enhanced M-current) contribute to CB2R-mediated inhibition in VTA DA neuronal firing, we designed two additional experiments. First, we examined the effects of JWH133 on neuronal firing in the presence of NBQX (10 μM), APV (50 μM) and picrotoxin (100 μM) to block excitatory and inhibitory presynaptic transmissions. We found that under cell-attached recording mode, JWH133 (10 μM) still significantly reduced DA neuron AP firing rate (inhibitory rate was 11 ± 2%, n = 21 from 6 mice, p = 0.0001, paired t-test, Fig. 10A), while without this pharmacological cocktail, JWH133-induced reduction in AP firing rate was 18 ± 4% (n = 21 from 8 mice, p = 0.0002, Fig. 10B). Although the block of synaptic transmission did reduce the inhibitory rate by JWH133 from 18 ± 4% (without synaptic block) to 11 ± 2% (with synaptic block), the difference of the inhibitory rate between these two groups (18 ± 4% vs. 11 ± 2%) was not statistical significant (p = 0.165, unpaired t-test). These results suggest that JWH133-induced mild reduction of presynaptic glutamate release seems not to play an important role in the mediation of JWH133-induced reduction in DA neuron firing. Second, we selectively blocked postsynaptic CB2R-G protein function by addition of 600 μM GDP-β-S, an intracellular G-protein blocker, into the recording electrode, thus allowing GDP-β-S to infuse into the recorded neuron after converted into whole-cell recording mode. As shown in Fig. 10, in the presence of intracellular GDP-βS, bath application of 10 μM JWH133 failed to alter AP firing rate in VTA DA neurons (from 1.97 ± 0.39 to 2.03 ± 0.44 Hz, p = 0.44, paired t-test, n = 7 from 3 mice, Fig. 10C). However, in the absence of intracellular GDP-βS, bath application of 10 μM JWH133 reduced VTA DA neuronal rate (from 1.99 ± 0.23 to 1.70 ± 0.22 Hz, p = 0.004, paired t-test, n = 14 from 4 mice, Fig. 10D). Statistical analysis showed that under whole-cell recording conditions (within 5 min after converted into whole-cell configuration), JWH133 failed to reduce AP firing rate in the presence of intracellular GDP-βS (Fig. 10E), while JWH133 reduced AP firing in the group neurons that did not contain intracellular GDP-βS (Fig. 10F). These results suggest that the intrinsic mechanism (CB2R-mediated enhancement of M-current) predominantly underlies the JWH133-induced reduction of VTA DA neuron excitability.
Fig. 10.

Determination of the role of synaptic and intrinsic mechanisms in JWH133-induced reduction of VTA DA neuronal excitability. (a) Bar graph summarizes data that showed the alteration of AP firing rate before and after JWH133 exposure. The VTA DA neuron AP firing was recorded using cell-attach recording mode in the presence (a) and absence (b) of and of cocktail pharmacology (NBQX+APV + PTX) to block synaptic transmission. (c) Representative typical traces showing the AP firing measured by whole-cell recordings in current-clamp mode before (left) and after (right) JWH133 exposure. In these experiments, the GDP-βS (600 μM) was added into recording electrode, and converted to whole-cell recording mode allowed GDP-βS infusion into recorded cells, and data showed that JWH133 failed to alter AP firing rate. (d) Representative typical traces in control group showing the AP firing measured by whole-cell recordings in current-clamp mode before (left) and after (right) JWH133 exposure without GDP-βS in the pipette solution and showed that JWH133 reduced AP firing rate. (e)/(f) Bar graph summarizes pooled data from (c) and (d) demonstrating that JWH133 significantly reduced AP firing rate in control (d), but not GDP-βS-treated neurons (c). ** p < 0.01, *** p < 0.001, compared to AP firing before JWH133 exposure.
4. Discussion
In this study, we find that acute exposure of the CB2R agonist, JWH133, reduces DA neuronal excitability in the VTA, which is mainly mediated through an intrinsic mechanism. Although JWH133 mildly reduces presynaptic glutamate release probability and results in a decreased glutamatergic synaptic transmission in VTA DA neurons, this may play little role in JWH133-induced reduction of VTA DA neuron firing because pharmacological block of synaptic transmission fails to prevent JWH133induced inhibitory effect. JWH133 activates a postsynaptic CB2R-Gi/o signaling cascade, reduces intracellular cAMP levels, in turn enhances M-type K+ channel function, and leading to the reduction of VTA DA neuronal excitability. Through this intrinsic mechanism, CB2Rs significantly reduce VTA DA neuronal excitability, which may underlie CB2R-mediated modulations in mesolimbic DA system function and DA-related behavior and diseases.
VTA DA neuronal firing is controlled by excitatory and inhibitory synaptic innervations. Modulations of these synaptic inputs alter neuronal excitability. Numerous G-protein-coupled receptors such as opioid, 5-HT2, metabolic glutamate, GABAB and CB1 receptors modulate synaptic transmission by alteration of presynaptic neurotransmitter release. Emerging evidence demonstrates that activation of CB2Rs modulates synaptic transmission in the entorhinal cortex [41], transfected hippocampal Duald neurons [42] and hippocampal pyramidal neurons from hippocampal slice cultures [43]. Thus, it is reasonable that CB2Rs are expressed in presynaptic terminals/boutons and modulate neurotransmitter release. Indeed, in this study, we found a mild, but statistically significant, reduction in the frequency of mEPSC after exposure to JWH133 (10 μM), suggesting that JWH133-induced reduction in VTA DA neuronal excitability may be mediated, at least in part, through a reduction of glutamatergic synaptic transmission onto DA neurons. However, additional experiments showed that pharmacological block of glutamatergic and GABAergic transmissions by perfusion of NBQX, APV and picrotoxin to VTA slice failed to prevent JWH133 (10 μM)-induced reduction in DA neuron firing rate. These results suggest that JWH133-induced the mild reduction of presynaptic glutamate release does not contribute to the JWH133-induced reduction in DA neuron firing under our experimental conditions. Previous report has shown that the activation of CB2Rs reduced inhibitory synaptic transmission in the entorhinal cortex [41], but this study did not identify the cellular location of the CB2Rs involved [41]. Thus, the pharmacological effects reported may be caused by CB2Rs expressed in either cortical neurons or microglia [44] since microglia are capable of modulating synaptic plasticity [45]. Another report demonstrated that selective activation of neuronal CB2Rs reduced inhibitory synaptic transmission via a presynaptic mechanism in transfected hippocampal Duald neurons prepared from CB1R−/− mice [42]. Compared to these two reports, we found that CB2Rs mildly reduced glutamatergic, but not GABAergic transmission in mouse VTA DA neurons. This discrepancy may be due to several reasons. First, different brain regions or neuronal phenotypes may have varied levels of CB2R expression [46]. This is supported by our recent finding that CB2Rs are highly expressed in the midbrain and moderately expressed in the prefrontal cortex (PFC), while the expression in the striatum is very low [25]. Second, brain CB2R expression is inducible and dynamic. For example, repeated cocaine administration or self-administration significantly up-regulates brain CB2R expression in the PFC, striatum and midbrain, as well as VTA neurons in rats [47]. Similarly, chronic, rather than acute, treatment of the slices with JWH133 for 7 days significantly enhanced excitatory synaptic transmission, suggesting that chronic treatment of JWH133 may up-regulate CB2R expression in the hippocampus [43]. Third, different CB2R agonists, with varying pharmacological actions, were used in separate studies. These pharmacological variances, resulting in the activation of diverse intracellular signal pathways [48], may explain these inconsistent electrophysiological findings. Collectively, our data suggest that glutamatergic presynaptic terminal/boutons may express very low level of CB2Rs, where they mildly modulate glutamate release probability, but contribute little to the alteration of DA neuron firing rate. Interestingly, we did not see a significant effect of JWH133 on mIPSCs, eEPSCs and eIPSCs in VTA DA neurons, which is consistent with previous report that activation of CB2Rs modulated glutamatergic, but not GABAergic synaptic, transmission in hippocampal slices [43]. The reason remains unknown, it may suggest that CB2Rs are mainly distributed on the neuron somato, and only limited CB2Rs are expressed on glutamatergic presynaptic terminals/boutons in VTA DA neurons. Our previous work supports this idea [25] as we showed predominant expression of CB2Rs on the VTA DAergic cell body.
In addition to presynaptic effect, VTA DA neuronal excitability is also profoundly modulated by intrinsic postsynaptic mechanisms, including the linkage of G-protein-coupled receptors to ion channels located on the cell membrane. CB2Rs are G-protein-coupled receptors that, when activated, alter G-protein (Gi/Go)-coupled intracellular signaling cascades [49]. Emerging lines of evidence suggest that in VTA DA neurons, G-protein-coupled receptor signaling modulates neuronal excitability through several types of ion channels, e.g., G-protein-coupled inwardly rectifying K+ channels (GIRK) [[50], [51], [52]], M-type K+ channels [32,53,54], A-type of K+ channels [55], and L-type voltage-gated Ca2+ channels [56]. In the present study, we found that JWH133 reduced neuron firing in acutely-dissociated VTA DA neurons by prolonging AP initiation, enhancing AHP amplitude and hyperpolarizing membrane potential, suggesting a potentiation of M-type K+ current. Under voltage-clamp mode, we measured M-current in dissociated VTA DA neurons, and found a CB2R-dependent enhancement of M-current amplitude by JWH133, confirming that CB2Rs modulate M-current. M-channels are voltage-gated K+ channels that strongly activate on depolarization to repolarize the cell, and potently regulate neuronal excitability [57,58]. Since M-type channels are activated at a membrane potential near the threshold for action potential initiation, it has a strong influence on neuronal excitability and responsiveness to synaptic inputs [59,60], and plays a critical role in terminating bursting activity in hippocampal CA1 neurons [61]. Importantly, under resting physiological conditions, M-channels are not fully open. Single channel analysis of both native [53,59] and recombinant [62,63] Kv7.2/Kv7.3 channels demonstrates that the maximal channel open probability is <1.0, suggesting that there is a considerable range to fully activate M-channels, and therefore, regulate neuronal activity. It was reported that VTA DA neurons express M-type K+ channels [32] and these channels play an important role in controlling VTA DA neuronal excitability [64]. It has been reported that activation of G-protein-coupled receptors increases M-currents in hippocampal neurons, by administration of somatostatin [65], corticostatin [66], or dynorphin [67]. This G-protein-mediated enhancement of M-current is important since it reduces neuronal excitability and counteracts convulsions. Indeed, it was also reported that the M-current enhancer, retigabine, shifts the current–voltage curve to the left (so that the channels open at more hyperpolarized membrane potentials) and increases the maximal opening probability [68,69], therefore, producing a strong suppression of neuronal firing [68] and exhibiting a potent anticonvulsant effect [70]. We also compared the effects of JWH133 and retigabine on M-current and VTA DA neuronal firing. We found that JWH133 and retigabine enhanced M-current amplitude and reduced neuronal firing rate. However, in the presence of M-current blocker (XE991), JWH133 failed to inhibit VTA DA neuron firing. These results suggest that CB2R-mediated enhancement of M-current is an important intrinsic mechanism underlying the CB2R-mediated reduction in VTA DA neuronal excitability. Then, we further examined the signaling between CB2R activation and M-current enhancement. Based on the previous studies, activation of CB2Rs primarily initiates two signal pathways, MAPK/ERK and cAMP/PKA [[37], [38], [39],48]. Considering cAMP plays an important role in modulation of M-current [40] and neuronal firing [71,72], we tested whether or not CB2R-induced reduction of cAMP contributes to the enhanced effect of M-current. To reach this goal, we applied forskolin, a cAMP enhancer to the recorded cell to increase intracellular cAMP levels [73]. We tried to address 3 questions: [1] Does forskolin suppress M-current? [2] Does forskolin increase VTA DA neuron firing? [3] Does forskolin prevent JWH133-induced inhibition of VTA DA neuron firing? Our data clearly showed that forskolin (10 μM) reduced M-current and increased VTA DA neuron firing. In the presence of forskolin, JWH133 failed to potentiate M-current and reduce AP firing. These results suggest that CB2R-G-protein-cAMP pathway likely plays a critical role in modulation of M-current and consequently neuronal excitability. Thus, under our experimental conditions (in vitro preparations), the activation of somatodendritic CB2Rs on VTA DA neurons reduces intracellular cAMP levels and enhances M-current amplitude, leading to a reduction in neuronal firing and excitability. However, in vivo conditions may be more complex, for example CB2Rs can modulate neuronal excitability by acting on glial cells [74,75], by modulating endogenous cannabinoid substances or even by acting on peripheral cells.
Our findings that the CB2R-mediated reduction of VTA DA neuronal excitability, the underlying intracellular signaling (cAMP) and target (M-channels) have translational significance. Accumulating lines of evidence demonstrate that CB2Rs mediate a variety of important modulations in DA-associated behaviors [76] including food intake, body weight [[77], [78], [79], [80]], depression [81], anxiety [14,82], and schizophrenia-like behavior [15,83]. Recent reports emerging from several labs, including ours, have shown brain CB2Rs play a pivotal role in the elimination of cocaine, alcohol and nicotine addiction [[84], [85], [86]]. These lines of evidence strongly suggest an important impact of CB2Rs in the mesocorticolimbic system as well as critical roles in various brain functions including psychiatric, cognitive and neurobiological activity. The finding of the CB2R-mediated reduction of midbrain DA neuronal excitability provides an insight into understanding of the roles played by CB2Rs in the above mentioned DA-associated function and diseases. When compared to CB1Rs, brain CB2Rs exhibit highly translational potential to be used in clinical practices since the following unique features: [1] Lower expression levels, suggesting that these receptors may not mediate the effect of cannabis under normal physiological conditions; [2] Highly inducible, meaning that under some pathological conditions (e.g., addiction, inflammation, stroke, schizophrenia, stress, anxiety, etc.), CB2R expression is enhanced in the brain [87], suggesting a close relationship between the alteration of CB2R expression/function and various psychiatric and neurological diseases; and [3] Exhibit special distribution, given that CB2Rs are chiefly expressed in neuronal somatodendritic areas [8] (postsynaptic) but CB1Rs are predominantly expressed on neuronal terminals, especially on GABAergic terminals (presynaptic), which leads to some opposing effects after activation by these two receptor subtypes [88]. Considering these characteristic features, the mesolimbic CB2R appears to be an important substrate for neuroprotection [89], and targeting CB2Rs or manipulations of their signaling (cAMP/PKA) and target (M-channels) will likely offer a novel therapeutic strategy for treating neuropsychiatric and neurological diseases without typical CB1R-mediated side-effects.
The following are the supplementary data related to this article.
Supplementary fig. 1.

A signalling pathway of CB2R-mediated reduction of VTA DA neuronal excitability.
Conflicts of interest
All authors have nothing to disclose.
Author contributions
Z.M., F.G., B.L., M.G., Z.L., D.C., X.M., S.Q., Y.Z., Z.X. conducted the experiments, analyzed data, prepared original figures, and revised the manuscript. Z.X. and J.X. participated in the experimental designs, analyzed the data, and revised the manuscript. J.W. designed and conducted the experiments, analyzed data, finalized figures, and wrote the manuscript.
Acknowledgments
This research was supported by the Barrow Neuroscience Foundation, the BNI-BMS Seed Fund, and CNSF (81771437). These funders have no role in study design, data collection, data analysis, interpretation, writing of the report.
References
- 1.Galiegue S., Mary S., Marchand J., Dussossoy D., Carriere D., Carayon P. Expression of central and peripheral cannabinoid receptors in human immune tissues and leukocyte subpopulations. Eur J Biochem. 1995;232(1):54–61. doi: 10.1111/j.1432-1033.1995.tb20780.x. [DOI] [PubMed] [Google Scholar]
- 2.Schatz A.R., Lee M., Condie R.B., Pulaski J.T., Kaminski N.E. Cannabinoid receptors CB1 and CB2: a characterization of expression and adenylate cyclase modulation within the immune system. Toxicol Appl Pharmacol. 1997;142(2):278–287. doi: 10.1006/taap.1996.8034. [DOI] [PubMed] [Google Scholar]
- 3.McCoy K.L., Matveyeva M., Carlisle S.J., Cabral G.A. Cannabinoid inhibition of the processing of intact lysozyme by macrophages: evidence for CB2 receptor participation. J Pharmacol Exp Ther. 1999;289(3):1620–1625. [PubMed] [Google Scholar]
- 4.Burdyga G., Lal S., Varro A., Dimaline R., Thompson D.G., Dockray G.J. Expression of cannabinoid CB1 receptors by vagal afferent neurons is inhibited by cholecystokinin. J Neurosci. 2004;24(11):2708–2715. doi: 10.1523/JNEUROSCI.5404-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Munro S., Thomas K.L., Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature. 1993;365(6441):61–65. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- 6.Buckley N.E., McCoy K.L., Mezey E., Bonner T., Zimmer A., Felder C.C. Immunomodulation by cannabinoids is absent in mice deficient for the cannabinoid CB(2) receptor. Eur J Pharmacol. 2000;396(2–3):141–149. doi: 10.1016/s0014-2999(00)00211-9. [DOI] [PubMed] [Google Scholar]
- 7.Buckley N.E. The peripheral cannabinoid receptor knockout mice: an update. Br J Pharmacol. 2008;153(2):309–318. doi: 10.1038/sj.bjp.0707527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Onaivi E.S., Ishiguro H., Gong J.P., Patel S., Meozzi P.A., Myers L. Functional expression of brain neuronal CB2 cannabinoid receptors are involved in the effects of drugs of abuse and in depression. Ann N Y Acad Sci. 2008;1139:434–449. doi: 10.1196/annals.1432.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Onaivi E.S., Ishiguro H., Gong J.P., Patel S., Perchuk A., Meozzi P.A. Discovery of the presence and functional expression of cannabinoid CB2 receptors in brain. Ann N Y Acad Sci. 2006;1074:514–536. doi: 10.1196/annals.1369.052. [DOI] [PubMed] [Google Scholar]
- 10.Skaper S.D., Buriani A., Dal Toso R., Petrelli L., Romanello S., Facci L. The ALIAmide palmitoylethanolamide and cannabinoids, but not anandamide, are protective in a delayed postglutamate paradigm of excitotoxic death in cerebellar granule neurons. Proc Natl Acad Sci U S A. 1996;93(9):3984–3989. doi: 10.1073/pnas.93.9.3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stempel A.V., Stumpf A., Zhang H.Y., Ozdogan T., Pannasch U., Theis A.K. Cannabinoid type 2 receptors mediate a cell type-specific plasticity in the Hippocampus. Neuron. 2016;90(4):795–809. doi: 10.1016/j.neuron.2016.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lanciego J.L., Barroso-Chinea P., Rico A.J., Conte-Perales L., Callen L., Roda E. Expression of the mRNA coding the cannabinoid receptor 2 in the pallidal complex of Macaca fascicularis. J Psychopharmacol. 2011;25(1):97–104. doi: 10.1177/0269881110367732. [DOI] [PubMed] [Google Scholar]
- 13.Liu Q.R., Pan C.H., Hishimoto A., Li C.Y., Xi Z.X., Llorente-Berzal A. Species differences in cannabinoid receptor 2 (CNR2 gene): identification of novel human and rodent CB2 isoforms, differential tissue expression and regulation by cannabinoid receptor ligands. Genes Brain Behav. 2009;8(5):519–530. doi: 10.1111/j.1601-183X.2009.00498.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garcia-Gutierrez M.S., Garcia-Bueno B., Zoppi S., Leza J.C., Manzanares J. Chronic blockade of cannabinoid CB(2) receptors induces anxiolytic-like actions associated to alterations in GABA(A) receptors. Br J Pharmacol. 2012 Feb;165(4):951–964. doi: 10.1111/j.1476-5381.2011.01625.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Navarrete F., Perez-Ortiz J.M., Manzanares J. Cannabinoid CB(2) receptor-mediated regulation of impulsive-like behaviour in DBA/2 mice. Br J Pharmacol. 2012;165(1):260–273. doi: 10.1111/j.1476-5381.2011.01542.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Van Sickle M.D., Duncan M., Kingsley P.J., Mouihate A., Urbani P., Mackie K. Identification and functional characterization of brainstem cannabinoid CB2 receptors. Science. 2005;310(5746):329–332. doi: 10.1126/science.1115740. [DOI] [PubMed] [Google Scholar]
- 17.Viscomi M.T., Oddi S., Latini L., Pasquariello N., Florenzano F., Bernardi G. Selective CB2 receptor agonism protects central neurons from remote axotomy-induced apoptosis through the PI3K/Akt pathway. J Neurosci. 2009;29(14):4564–4570. doi: 10.1523/JNEUROSCI.0786-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ashton J.C., Friberg D., Darlington C.L., Smith P.F. Expression of the cannabinoid CB2 receptor in the rat cerebellum: an immunohistochemical study. Neurosci Lett. 2006;396(2):113–116. doi: 10.1016/j.neulet.2005.11.038. [DOI] [PubMed] [Google Scholar]
- 19.Baek J.H., Zheng Y., Darlington C.L., Smith P.F. Cannabinoid CB2 receptor expression in the rat brainstem cochlear and vestibular nuclei. Acta Otolaryngol. 2008;128(9):961–967. doi: 10.1080/00016480701796944. [DOI] [PubMed] [Google Scholar]
- 20.Brusco A., Tagliaferro P., Saez T., Onaivi E.S. Postsynaptic localization of CB2 cannabinoid receptors in the rat hippocampus. Synapse (New York NY). 2008;62(12):944–949. doi: 10.1002/syn.20569. [DOI] [PubMed] [Google Scholar]
- 21.Gong J.P., Onaivi E.S., Ishiguro H., Liu Q.R., Tagliaferro P.A., Brusco A. Cannabinoid CB2 receptors: immunohistochemical localization in rat brain. Brain Res. 2006;1071(1):10–23. doi: 10.1016/j.brainres.2005.11.035. [DOI] [PubMed] [Google Scholar]
- 22.Aracil-Fernandez A., Trigo J.M., Garcia-Gutierrez M.S., Ortega-Alvaro A., Ternianov A., Navarro D. Decreased cocaine motor sensitization and self-administration in mice overexpressing cannabinoid CB(2) receptors. Neuropsychopharmacology. 2012;37(7):1749–1763. doi: 10.1038/npp.2012.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schmidt W., Schafer F., Striggow V., Frohlich K., Striggow F. Cannabinoid receptor subtypes 1 and 2 mediate long-lasting neuroprotection and improve motor behavior deficits after transient focal cerebral ischemia. Neuroscience. 2012;227:313–326. doi: 10.1016/j.neuroscience.2012.09.080. [DOI] [PubMed] [Google Scholar]
- 24.Xi Z.X., Peng X.Q., Li X., Song R., Zhang H.Y., Liu Q.R. Brain cannabinoid CB receptors modulate cocaine's actions in mice. Nat Neurosci. 2011;14(9):1160–1166. doi: 10.1038/nn.2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang H.Y., Gao M., Liu Q.R., Bi G.H., Li X., Yang H.J. Cannabinoid CB2 receptors modulate midbrain dopamine neuronal activity and dopamine-related behavior in mice. Proc Natl Acad Sci U S A. 2014;111(46):E5007–E5015. doi: 10.1073/pnas.1413210111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Q-s Liu, Pu L., M-m Poo. Repeated cocaine exposure in vivo facilitates LTP induction in midbrain dopamine neurons. Nature. 2005;437(7061):1027–1031. doi: 10.1038/nature04050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ungless M.A., Whistler J.L., Malenka R.C., Bonci A. Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature. 2001;411(6837):583–587. doi: 10.1038/35079077. [DOI] [PubMed] [Google Scholar]
- 28.Gao M., Jin Y., Yang K., Zhang D., Lukas R.J., Wu J. Mechanisms involved in systemic nicotine-induced glutamatergic synaptic plasticity on dopamine neurons in the ventral tegmental area. J Neurosci. 2010;30(41):13814–13825. doi: 10.1523/JNEUROSCI.1943-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang K., Buhlman L., Khan G.M., Nichols R.A., Jin G., McIntosh J.M. Functional nicotinic acetylcholine receptors containing alpha6 subunits are on GABAergic neuronal boutons adherent to ventral tegmental area dopamine neurons. J Neurosci. 2011;31(7):2537–2548. doi: 10.1523/JNEUROSCI.3003-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu J., George A.A., Schroeder K.M., Xu L., Marxer-Miller S., Lucero L. Electrophysiological, pharmacological, and molecular evidence for alpha7-nicotinic acetylcholine receptors in rat midbrain dopamine neurons. J Pharmacol Exp Ther. 2004;311(1):80–91. doi: 10.1124/jpet.104.070417. [DOI] [PubMed] [Google Scholar]
- 31.Yang K., Hu J., Lucero L., Liu Q., Zheng C., Zhen X. Distinctive nicotinic acetylcholine receptor functional phenotypes of rat ventral tegmental area dopaminergic neurons. J Physiol. 2009;587(Pt 2):345–361. doi: 10.1113/jphysiol.2008.162743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koyama S., Appel S.B. Characterization of M-current in ventral tegmental area dopamine neurons. J Neurophysiol. 2006;96(2):535–543. doi: 10.1152/jn.00574.2005. [DOI] [PubMed] [Google Scholar]
- 33.Brown D.A., Passmore G.M. Neural KCNQ (Kv7) channels. Br J Pharmacol. 2009;156(8):1185–1195. doi: 10.1111/j.1476-5381.2009.00111.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee S., Kwag J. M-channels modulate the intrinsic excitability and synaptic responses of layer 2/3 pyramidal neurons in auditory cortex. Biochem Biophys Res Commun. 2012;426(4):448–453. doi: 10.1016/j.bbrc.2012.08.057. [DOI] [PubMed] [Google Scholar]
- 35.Schweitzer P. Cannabinoids decrease the K(+) M-current in hippocampal CA1 neurons. J Neurosci. 2000;20(1):51–58. doi: 10.1523/JNEUROSCI.20-01-00051.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brown D.A., Adams P.R. Muscarinic suppression of a novel voltage-sensitive K+ current in a vertebrate neurone. Nature. 1980;283(5748):673–676. doi: 10.1038/283673a0. [DOI] [PubMed] [Google Scholar]
- 37.Childers S.R., Deadwyler S.A. Role of cyclic AMP in the actions of cannabinoid receptors. Biochem Pharmacol. 1996;52(6):819–827. doi: 10.1016/0006-2952(96)00419-4. [DOI] [PubMed] [Google Scholar]
- 38.Pertwee R.G. Pharmacology of cannabinoid CB1 and CB2 receptors. Pharmacol Ther. 1997;74(2):129–180. doi: 10.1016/s0163-7258(97)82001-3. [DOI] [PubMed] [Google Scholar]
- 39.Elphick M.R., Egertova M. The neurobiology and evolution of cannabinoid signalling. Philos Trans R Soc Lond B Biol Sci. 2001;356(1407):381–408. doi: 10.1098/rstb.2000.0787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stott J.B., Povstyan O.V., Carr G., Barrese V., Greenwood I.A. G-protein betagamma subunits are positive regulators of Kv7.4 and native vascular Kv7 channel activity. Proc Natl Acad Sci U S A. 2015;112(20):6497–6502. doi: 10.1073/pnas.1418605112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Morgan N.H., Stanford I.M., Woodhall G.L. Functional CB2 type cannabinoid receptors at CNS synapses. Neuropharmacology. 2009;57(4):356–368. doi: 10.1016/j.neuropharm.2009.07.017. [DOI] [PubMed] [Google Scholar]
- 42.Atwood B.K., Straiker A., Mackie K. CB(2) cannabinoid receptors inhibit synaptic transmission when expressed in cultured autaptic neurons. Neuropharmacology. 2012;63(4):514–523. doi: 10.1016/j.neuropharm.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim J., Li Y. Chronic activation of CB2 cannabinoid receptors in the hippocampus increases excitatory synaptic transmission. J Physiol. 2015;593(4):871–886. doi: 10.1113/jphysiol.2014.286633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ashton J.C., Glass M. The cannabinoid CB2 receptor as a target for inflammation-dependent neurodegeneration. Curr Neuropharmacol. 2007;5(2):73–80. doi: 10.2174/157015907780866884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ben Achour S., Pascual O. Glia: the many ways to modulate synaptic plasticity. Neurochem Int. 2010;57(4):440–445. doi: 10.1016/j.neuint.2010.02.013. [DOI] [PubMed] [Google Scholar]
- 46.Ellert-Miklaszewska A., Grajkowska W., Gabrusiewicz K., Kaminska B., Konarska L. Distinctive pattern of cannabinoid receptor type II (CB2) expression in adult and pediatric brain tumors. Brain Res. 2007;1137(1):161–169. doi: 10.1016/j.brainres.2006.12.060. [DOI] [PubMed] [Google Scholar]
- 47.Zhang H.Y., Gao M., Shen H., Bi G.H., Yang H.J., Liu Q.R. Expression of functional cannabinoid CB receptor in VTA dopamine neurons in rats. Addict Biol. 2017 May;22(3):752–765. doi: 10.1111/adb.12367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pertwee R.G. Handbook of Experimental Pharmacology. Vol. 168. 2005. Pharmacological actions of cannabinoids; pp. 1–51. [DOI] [PubMed] [Google Scholar]
- 49.Hille B. Modulation of ion-channel function by G-protein-coupled receptors. Trends Neurosci. 1994;17(12):531–536. doi: 10.1016/0166-2236(94)90157-0. [DOI] [PubMed] [Google Scholar]
- 50.Lomazzi M., Slesinger P.A., Luscher C. Addictive drugs modulate GIRK-channel signaling by regulating RGS proteins. Trends Pharmacol Sci. 2008;29(11):544–549. doi: 10.1016/j.tips.2008.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hearing M.C., Zink A.N., Wickman K. Cocaine-induced adaptations in metabotropic inhibitory signaling in the mesocorticolimbic system. Rev Neurosci. 2012;23(4):325–351. doi: 10.1515/revneuro-2012-0045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lalive A.L., Munoz M.B., Bellone C., Slesinger P.A., Luscher C., Tan K.R. Firing modes of dopamine neurons drive bidirectional GIRK channel plasticity. J Neurosci. 2014;34(15):5107–5114. doi: 10.1523/JNEUROSCI.5203-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Appel S.B., Liu Z., McElvain M.A., Brodie M.S. Ethanol excitation of dopaminergic ventral tegmental area neurons is blocked by quinidine. J Pharmacol Exp Ther. 2003;306(2):437–446. doi: 10.1124/jpet.103.050963. [DOI] [PubMed] [Google Scholar]
- 54.Hamasaki R., Shirasaki T., Soeda F., Takahama K. Tipepidine activates VTA dopamine neuron via inhibiting dopamine D(2) receptor-mediated inward rectifying K(+) current. Neuroscience. 2013;252:24–34. doi: 10.1016/j.neuroscience.2013.07.044. [DOI] [PubMed] [Google Scholar]
- 55.Koyama S., Appel S.B. A-type K+ current of dopamine and GABA neurons in the ventral tegmental area. J Neurophysiol. 2006;96(2):544–554. doi: 10.1152/jn.01318.2005. [DOI] [PubMed] [Google Scholar]
- 56.Hoddah H., Marcantoni A., Comunanza V., Carabelli V., Carbone E. L-type channel inhibition by CB1 cannabinoid receptors is mediated by PTX-sensitive G proteins and cAMP/PKA in GT1-7 hypothalamic neurons. Cell Calcium. 2009;46(5–6):303–312. doi: 10.1016/j.ceca.2009.08.007. [DOI] [PubMed] [Google Scholar]
- 57.Brown D.A., Higashida H. Voltage- and calcium-activated potassium currents in mouse neuroblastoma x rat glioma hybrid cells. J Physiol. 1988;397:149–165. doi: 10.1113/jphysiol.1988.sp016993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jentsch T.J. Neuronal KCNQ potassium channels: physiology and role in disease. Nat Rev Neurosci. 2000;1(1):21–30. doi: 10.1038/35036198. [DOI] [PubMed] [Google Scholar]
- 59.Marrion N.V. Control of M-current. Annu Rev Physiol. 1997;59:483–504. doi: 10.1146/annurev.physiol.59.1.483. [DOI] [PubMed] [Google Scholar]
- 60.Aiken S.P., Lampe B.J., Murphy P.A., Brown B.S. Reduction of spike frequency adaptation and blockade of M-current in rat CA1 pyramidal neurones by linopirdine (DuP 996), a neurotransmitter release enhancer. Br J Pharmacol. 1995;115(7):1163–1168. doi: 10.1111/j.1476-5381.1995.tb15019.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Azouz R., Jensen M.S., Yaari Y. Ionic basis of spike after-depolarization and burst generation in adult rat hippocampal CA1 pyramidal cells. J Physiol. 1996;492:211–223. doi: 10.1113/jphysiol.1996.sp021302. Pt 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Selyanko A.A., Hadley J.K., Brown D.A. Properties of single M-type KCNQ2/KCNQ3 potassium channels expressed in mammalian cells. J Physiol. 2001;534:15–24. doi: 10.1111/j.1469-7793.2001.00015.x. Pt 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li Y., Gamper N., Shapiro M.S. Single-channel analysis of KCNQ K+ channels reveals the mechanism of augmentation by a cysteine-modifying reagent. J Neurosci. 2004;24(22):5079–5090. doi: 10.1523/JNEUROSCI.0882-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Drion G., Bonjean M., Waroux O., Scuvee-Moreau J., Liegeois J.F., Sejnowski T.J. M-type channels selectively control bursting in rat dopaminergic neurons. Eur J Neurosci. 2010;31(5):827–835. doi: 10.1111/j.1460-9568.2010.07107.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Moore S.D., Madamba S.G., Joels M., Siggins G.R. Somatostatin augments the M-current in hippocampal neurons. Science. 1988;239(4837):278–280. doi: 10.1126/science.2892268. [DOI] [PubMed] [Google Scholar]
- 66.de Lecea L., Criado J.R., Prospero-Garcia O., Gautvik K.M., Schweitzer P., Danielson P.E. A cortical neuropeptide with neuronal depressant and sleep-modulating properties. Nature. 1996;381(6579):242–245. doi: 10.1038/381242a0. [DOI] [PubMed] [Google Scholar]
- 67.Madamba S.G., Schweitzer P., Siggins G.R. Dynorphin selectively augments the M-current in hippocampal CA1 neurons by an opiate receptor mechanism. J Neurophysiol. 1999;82(4):1768–1775. doi: 10.1152/jn.1999.82.4.1768. [DOI] [PubMed] [Google Scholar]
- 68.Tatulian L., Delmas P., Abogadie F.C., Brown D.A. Activation of expressed KCNQ potassium currents and native neuronal M-type potassium currents by the anti-convulsant drug retigabine. J Neurosci. 2001;21(15):5535–5545. doi: 10.1523/JNEUROSCI.21-15-05535.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tatulian L., Brown D.A. Effect of the KCNQ potassium channel opener retigabine on single KCNQ2/3 channels expressed in CHO cells. J Physiol. 2003;549:57–63. doi: 10.1113/jphysiol.2003.039842. Pt 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Arora D., Hearing M., Haluk D.M., Mirkovic K., Fajardo-Serrano A., Wessendorf M.W. Acute cocaine exposure weakens GABA(B) receptor-dependent G-protein-gated inwardly rectifying K+ signaling in dopamine neurons of the ventral tegmental area. J Neurosci. 2011;31(34):12251–12257. doi: 10.1523/JNEUROSCI.0494-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gruart A., Benito E., Delgado-Garcia J.M., Barco A. Enhanced cAMP response element-binding protein activity increases neuronal excitability, hippocampal long-term potentiation, and classical eyeblink conditioning in alert behaving mice. J Neurosci. 2012;32(48):17431–17441. doi: 10.1523/JNEUROSCI.4339-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jancic D., Lopez de Armentia M., Valor L.M., Olivares R., Barco A. Inhibition of cAMP response element-binding protein reduces neuronal excitability and plasticity, and triggers neurodegeneration. Cereb Cortex. 2009;19(11):2535–2547. doi: 10.1093/cercor/bhp004. [DOI] [PubMed] [Google Scholar]
- 73.Alasbahi R.H., Melzig M.F. Forskolin and derivatives as tools for studying the role of cAMP. Pharmazie. 2012;67(1):5–13. [PubMed] [Google Scholar]
- 74.Guida F., Luongo L., Boccella S., Giordano M.E., Romano R., Bellini G. Palmitoylethanolamide induces microglia changes associated with increased migration and phagocytic activity: involvement of the CB2 receptor. Sci Rep. 2017;7(1):375. doi: 10.1038/s41598-017-00342-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Luchicchi A., Lecca S., Carta S., Pillolla G., Muntoni A.L., Yasar S. Effects of fatty acid amide hydrolase inhibition on neuronal responses to nicotine, cocaine and morphine in the nucleus accumbens shell and ventral tegmental area: involvement of PPAR-alpha nuclear receptors. Addict Biol. 2010;15(3):277–288. doi: 10.1111/j.1369-1600.2010.00222.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Vlachou S., Panagis G. Regulation of brain reward by the Endocannabinoid system: a critical review of Behavioral studies in animals. Curr Pharm Des. 2014;20(13):2072–2088. doi: 10.2174/13816128113199990433. [DOI] [PubMed] [Google Scholar]
- 77.Agudo J., Martin M., Roca C., Molas M., Bura A.S., Zimmer A. Deficiency of CB2 cannabinoid receptor in mice improves insulin sensitivity but increases food intake and obesity with age. Diabetologia. 2010;53(12):2629–2640. doi: 10.1007/s00125-010-1894-6. [DOI] [PubMed] [Google Scholar]
- 78.Ignatowska-Jankowska B., Jankowski M.M., Swiergiel A.H. Cannabidiol decreases body weight gain in rats: involvement of CB2 receptors. Neurosci Lett. 2011;490(1):82–84. doi: 10.1016/j.neulet.2010.12.031. [DOI] [PubMed] [Google Scholar]
- 79.Emadi L., Jonaidi H., Hosseini Amir Abad E. The role of central CB2 cannabinoid receptors on food intake in neonatal chicks. J Comp Physiol A Neuroethol Sens Neural Behav Physiol. 2011;197(12):1143–1147. doi: 10.1007/s00359-011-0676-z. [DOI] [PubMed] [Google Scholar]
- 80.Flake N.M., Zweifel L.S. Behavioral effects of pulp exposure in mice lacking cannabinoid receptor 2. J Endod. 2012;38(1):86–90. doi: 10.1016/j.joen.2011.09.015. [DOI] [PubMed] [Google Scholar]
- 81.Garcia-Gutierrez M.S., Perez-Ortiz J.M., Gutierrez-Adan A., Manzanares J. Depression-resistant endophenotype in mice overexpressing cannabinoid CB(2) receptors. Br J Pharmacol. 2010;160(7):1773–1784. doi: 10.1111/j.1476-5381.2010.00819.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Garcia-Gutierrez M.S., Manzanares J. Overexpression of CB2 cannabinoid receptors decreased vulnerability to anxiety and impaired anxiolytic action of alprazolam in mice. J Psychopharmacol. 2011;25(1):111–120. doi: 10.1177/0269881110379507. [DOI] [PubMed] [Google Scholar]
- 83.Ortega-Alvaro A., Aracil-Fernandez A., Garcia-Gutierrez M.S., Navarrete F., Manzanares J. Deletion of CB2 cannabinoid receptor induces schizophrenia-related behaviors in mice. Neuropsychopharmacology. 2011;36(7):1489–1504. doi: 10.1038/npp.2011.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Xi Z.X., Peng X.Q., Li X., Song R., Zhang H.Y., Liu Q.R. Brain cannabinoid CB(2) receptors modulate cocaine's actions in mice. Nat Neurosci. 2011;14(9):1160–1166. doi: 10.1038/nn.2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Navarrete F., Rodriguez-Arias M., Martin-Garcia E., Navarro D., Garcia-Gutierrez M.S., Aguilar M.A. Role of CB2 cannabinoid receptors in the rewarding, reinforcing, and physical effects of nicotine. Neuropsychopharmacology. 2013 Nov;38(12):2515–2524. doi: 10.1038/npp.2013.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ortega-Alvaro A., Ternianov A., Aracil-Fernandez A., Navarrete F., Garcia-Gutierrez M.S., Manzanares J. Role of cannabinoid CB receptor in the reinforcing actions of ethanol. Addict Biol. 2015 Jan;20(1):43–55. doi: 10.1111/adb.12076. [DOI] [PubMed] [Google Scholar]
- 87.Miller L.K., Devi L.A. The highs and lows of cannabinoid receptor expression in disease: mechanisms and their therapeutic implications. Pharmacol Rev. 2011;63(3):461–470. doi: 10.1124/pr.110.003491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Onaivi E.S., Ishiguro H., Gu S., Liu Q.R. CNS effects of CB2 cannabinoid receptors: beyond neuro-immuno-cannabinoid activity. J Psychopharmacol. 2012;26(1):92–103. doi: 10.1177/0269881111400652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pacher P., Mechoulam R. Is lipid signaling through cannabinoid 2 receptors part of a protective system? Prog Lipid Res. 2011;50(2):193–211. doi: 10.1016/j.plipres.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]