

Abstract
Background
Sepsis, the most severe form of infection, involves endothelial dysfunction which contributes to organ failure. To improve therapeutic prospects, elucidation of molecular mechanisms underlying endothelial vascular failure is of essence.
Methods
Polymicrobial contamination induced sepsis mouse model and primary endothelial cells incubated with sepsis serum were used to study SHP-2 in sepsis-induced endothelial inflammation. SHP-2 activity was assessed by dephosphorylation of pNPP, ROS production was measured by DCF oxidation and protein interactions were assessed by proximity ligation assay. Vascular inflammation was studied in the mouse cremaster model and in an in vitro flow assay.
Findings
We identified ROS-dependent inactivation of the tyrosine phosphatase SHP-2 to be decisive for endothelial activation in sepsis. Using in vivo and in vitro sepsis models, we observed a significant reduction of endothelial SHP-2 activity, accompanied by enhanced adhesion molecule expression. The impaired SHP-2 activity was restored by ROS inhibitors and an IL-1 receptor antagonist. SHP-2 activity inversely correlated with the adhesive phenotype of endothelial cells exposed to IL-1β as well as sepsis serum via p38 MAPK and NF-κB. In vivo, SHP-2 inhibition accelerated IL-1β-induced leukocyte adhesion, extravasation and vascular permeability. Mechanistically, SHP-2 directly interacts with the IL-1R1 adaptor protein MyD88 via its tyrosine 257, resulting in reduced binding of p85/PI3-K to MyD88.
Interpretation
Our data show that SHP-2 inactivation by ROS in sepsis releases a protective break, resulting in endothelial activation.
Fund
German Research Foundation, LMU Mentoring excellence and FöFoLe Programme, Verein zur Förderung von Wissenschaft und Forschung, German Ministry of Education and Research.
Abbreviations: Akt, protein kinase B; AP-1, activator protein-1; BW, body weight; C, control; CREB, cAMP response element-binding protein; CRP, C-reactive protein; CS, Cys459 to Ser459 mutation (catalytically inactive SHP-2mutant); DAPI, 4′,6-Diamidin-2-phenylindol; DCF, Dichlorofluorescein; EA, Glu76 to Ala76 mutation (constitutively active SHP-2 mutant).; EC, endothelial cells; ELISA, enzyme-linked immunosorbent assay; FITC, fluorescein isothiocyanate; hSerum, human serum; Hsp27, heat shock protein 27; HUVEC, human umbilical vein endothelial cells; ICAM-1, intercellular adhesion molecule 1; IKK, IκB kinase; IL-18R, Interleukin-18 receptor; IL-1R1, Interleukin-1 receptor 1; IL-1Ra, Interleukin-1 receptor antagonist; IL-1β, interleukin-1β; IκBα, inhibitor of kappa B; MAPK, Mitogen-activated protein kinase; MyD88 Y257F, Tyr257 to Phe257 mutation of MyD88; MyD88, Myeloid differentiation primary response 88; NAC, N-acetylcysteine; NF-κB, Nuclear factor ‘kappa-light-chain-enhancer’ of activated B-cells; PCI, peritoneal contamination and infection; PI3K, Phosphoinositid-3-kinase; PLA, Proximity ligation assay; PMN, Polymorphnuclear neutrophils; PtpI IV, Protein tyrosine phosphatase Inhibitor IV (SHP-2 inhibitor); ROS, Reactive oxygen species; S, Sepsis; SH2 domain, Src homology 2 domain; SHP-2, Src homology 2 domain containing phosphatase-2; TIR domain, Toll/interleukin-1 receptor domain; TLR, Toll-like receptor; VAS-2870, NAD(P)H-oxidase inhibitor; VCAM-1, Vascular cell adhesion molecule 1; WT, Wildtype
Keywords: Endothelial cells, IL-1β, MyD88, ROS, SHP-2, Sepsis
Research in context.
Evidence before this study
Sepsis is a life-threatening condition, where vascular dysfunction caused by elevated cytokine levels plays a major role in disease progression. In sepsis, cytokines such as IL-1 activate the endothelium, leading to expression of adhesion molecules and microvascular leakage. This mediates adhesion of leukocytes and their extravasation, further promoting inflammation. Hence, the vascular inflammatory response needs to be tightly regulated to prevent tissue injury. The tyrosine phosphatase SHP-2 regulates growth factor and cytokine signalling and is known to play a role in endothelial physiology but its function in inflammatory endothelial activation in sepsis has not been investigated.
Added value of this study
Our study identifies SHP-2 as a new key player in the control of endothelial activation in sepsis. We observed a significant reduction of SHP-2 phosphatase activity in cells of septic mice and in cultured human endothelial cells treated with serum from sepsis patients, which was accompanied by enhanced adhesion molecule expression. SHP-2 activity was restored by blockage of the IL-1 receptor and inhibition of reactive oxygen species. Mechanistically, we show by overexpression of different SHP-2 activity mutants in endothelial cells that SHP-2 activity inversely correlates with the adhesive phenotype of EC exposed to IL-1β as well as serum from sepsis patients via regulation of p38 MAPK and NF-κB. Accordingly, SHP-2 inhibition accelerated IL-1β-induced leukocyte adhesion, extravasation and vascular permeability in vivo. The protective effect of SHP-2 on endothelial activation was due to interaction of SHP-2 with tyrosine 257 of the IL-1R1 adaptor protein MyD88, resulting in reduced binding of the p85 subunit of PI3-Kinase to MyD88.
Implications of all the available evidence
Our data suggest that ROS mediated SHP-2 inactivation plays a key role in endothelial activation in sepsis. Restoration of SHP-2 activity may therefore represent a promising target in preventing a hyperinflammatory state of the endothelium, thus limiting endothelial dysfunction and capillary leakage in sepsis.
Alt-text: Unlabelled Box
1. Introduction
Sepsis is a severe clinical condition caused by a deregulated systemic host response to infection leading to organ dysfunction and tissue damage [1,2]. As one of the first lines of defense against infection, cellular components of innate immunity release inflammatory cytokines such as interleukin (IL)-1β and tumor necrosis factor α [3]. IL-1β induces endothelial activation, which involves reactive oxygen species (ROS) formation and upregulation of adhesion molecules, a decisive step in the disease progression of sepsis, mediating the recruitment and adhesion of leukocytes to the endothelium [4,5]. Additionally, IL-1β promotes microvascular leakage, which is important for leukocyte extravasation [6] and the development of tissue edema during sepsis [7]. Hence, the dysregulation of the endothelium in sepsis is a pivotal component in the complex process of sepsis, involving several different factors leading to organ failure [7]. To regain control of the endothelial responses to inflammatory stress is thus essential to prevent tissue injury. However, no specific anti-inflammatory therapy for sepsis is yet available [8]. Therefore, a deeper understanding of the deregulated endothelial response is of great importance for the development of future therapeutic strategies.
In sepsis, several signalling pathways have been shown to play a role in endothelial activation and dysfunction leading to activation of transcription factors like NF-κB and AP-1 [1]. IL-1β signals through the IL-1R1 inducing binding of the adaptor protein MyD88 to the receptor. This promotes the generation of a signalling complex (Myddosome) through interaction of the MyD88 death domain [9], which transduces downstream signals [10,11]. Additionally, MyD88 has been shown to harbour a SH2-binding motif, through which it can directly interact with SH2-containing proteins [12].
The Src-homology 2 (SH2) domain containing tyrosine phosphatase SHP-2 binds to phosphorylated tyrosines within SH2-binding motifs and regulates signal transduction by dephosphorylation of tyrosine residues on receptors and regulatory adaptor proteins [13,14]. Although SHP-2 has been implicated in the control of growth factor and cytokine signalling [[15], [16], [17], [18], [19], [20]] and is known to play a role in endothelial physiology [[21], [22], [23], [24], [25], [26]], its function in inflammatory endothelial activation and the development of endothelial dysfunction in sepsis has not been investigated.
As we observed a significant reduction of SHP-2 phosphatase activity in endothelial cells exposed to serum from sepsis patients in this study, we further investigated whether this may contribute to the detrimental dysregulation of endothelial processes in sepsis, such as increased adhesion molecule upregulation, leukocyte recruitment, and vascular permeability. We show that SHP-2 becomes inactivated by cytokine-induced ROS formation in endothelial cells in sepsis, thereby releasing an inhibitory interaction between SHP-2 and MyD88 and accelerating the endothelial inflammatory response.
2. Materials and methods
2.1. Antibodies and chemicals
Rabbit phospho-Akt (Ser473) (D9E) XP™ (#4060, RRID: AB_2315049), rabbit phospho-p38 MAPK (Thr180/Tyr182) (D3F9) XP™ (#4511, RRID:AB_2139682), rabbit phospho-Hsp27 (Ser82) (#2401, RRID:AB_331644), rabbit VCAM-1 (#12367), rabbit MyD88 (#4283, RRID:AB_10547882), rabbit NF-κB p65 (D14E12) XP™ (#8242), rabbit myc-tag (#2278, RRID:AB_490778), rabbit HA-tag (C29F4) (# 3724, RRID:AB_1549585), rabbit β-Actin (13E5) (#4790) as well as mouse and rabbit IgG isotype (#5415 and #3900, RRID:AB_1550038) antibodies were from Cell Signaling Technology. Rabbit ICAM-1 (H-108) (#sc-7891, RRID:AB_647486), mouse IL-1R1 (#sc-393,998, RRID:AB_2737063), goat MyD88 (N-19) (#sc-8196, RRID:AB_2146863), mouse SHP-2 (B-1) (#sc-7384, RRID:AB_628252) and PI3-K p85α (B-9) (#sc-1637) were from Santa Cruz Biotechnology. Mouse GAPDH (#MAB374) as well as anti-mouse and rabbit horseradish peroxidase-conjugated secondary antibodies (#401253 and #401353) were from Merck Millipore. Anti-rabbit Alexa-fluor 546 labelled antibody (A11035) was from Invitrogen. Mouse monoclonal APC-labelled ICAM-1 (#559771, RRID:AB_398667), VCAM-1 (#551147, RRID:AB_398496) and IgG1 isotype control antibody (#555751, RRID:AB_398613) for flow cytometry were from BD Biosciences. Recombinant human IL-1β was purchased from PeproTech. SHP-2 (PtpI IV) inhibitor was from Merck Millipore. IL-1Ra (# 280-RA) was from R&D. Alexa-Fluor 488 labelled phalloidin was from Molecular Probes. All other chemicals were from Sigma-Aldrich.
2.2. Human umbilical vein endothelial cell (HUVEC) culture and treatments
Primary HUVEC were isolated and cultivated as previously described [27] and used up to passage five.
2.3. Lentiviral constructs and transductions
Expression cassettes for wild type (WT) SHP-2 and the catalytically inactive mutant SHP-2 CS (Cys459 to Ser459) were kindly provided by the Bennett laboratory [28]. The constitutively active mutant SHP-2 EA (Glu76 to Ala) was generated as previously described [26] Lentiviral particles were produced and physical and biological viral titers were measured and calculated as previously described [29,30]. Overexpression of SHP-2 WT and mutant constructs in HUVEC was achieved by lentiviral transduction, as described elsewhere [26]. Cells were left 72 h for gene expression before assaying.
2.4. Plasmids and transfections
Plasmids for HA-tagged MyD88 wild type (WT) and HA-tagged MyD88 YF mutant (mutation of Tyr257 to Ala) were kindly provided by Dr. Rhee and Dr. Pothoulakis [12]. HUVEC were transfected with MyD88 WT and MyD88 Y257F using jetPRIME transfection reagent (PeqLab) according to manufacturer's protocol. Cells were used for further experiments 48 h after transfection.
2.5. Flow cytometry
Detection and quantification of surface adhesion molecules (ICAM-1 and VCAM-1) was performed as previously described [27] using APC-labelled anti-ICAM-1 and VCAM-1 antibodies or IgG1 isotype antibody as control for background staining.
2.6. Immunoblotting and quantification of protein bands
Lysates were prepared and subjected to SDS-PAGE following western blotting and protein band intensities quantified as previously described [26,31].
2.7. Isolation of human polymorphonuclear neutrophils (PMN)
Human PMN were freshly isolated from human whole blood of healthy donors at the day of the experiment as described elsewhere [32]. PMN were resuspended in Hank's solution (Biochrom) supplemented with 0.25% BSA and 0.1% glucose.
2.8. PMN adhesion assay in vitro
HUVEC expressing the different SHP-2 constructs (WT, CS or EA) were grown to confluence on μ-channel slides IV0.4 (ibiTreat, IBIDI). Cells were stimulated with 10 ng/ml IL-1β for 4 h. Isolated human PMN were stained by incubation with 2 μl/ml 0.05% rhodamine 6G (R6G) for 5 min at 37 °C in the dark. Stained PMN were pelleted (300 g, 5 min), resuspended in 37 °C warm phosphate buffered saline (PBS) supplemented with Ca2+ and Mg2+ and brought to a final concentration of 1 × 105 PMN/ml. After priming confluent HUVEC by 15 min perfusion with warm PBS (37 °C) supplemented with Ca2+ and Mg2+ at 1 dyn/cm2 using a syringe pump (kdScientific) slides were flushed with R6G-stained PMN suspensions for 5 min at 1 dyn/cm2. PMN adhering to the EC layer were monitored by fluorescence microscopy using the Zeiss Axiovert 200 M microscope, and pictures from four independent areas per channel were taken at 10× magnification.
2.9. Leukocyte-endothelium interaction and transmigration in vivo
Leukocyte-endothelium-interaction in vivo was assessed in post capillary venules of the cremaster muscle of 16–18 week old male C57BL/6J WT mice (Charles River Laboratories). In detail, mice were treated with PtpI IV or sham solution (DMSO) diluted in 150 μl 0.9% NaCl for 30 min followed by 4 h of IL-1β stimulation (10 ng/ml) by intrascrotal injection, respectively. Mice were anesthetized (5 mg/kg midazolam, 0.5 mg/kg medetomidin, 0.05 mg/kg fentanyl) and the cremaster muscle was prepared for intravital microscopy as originally described by Baez et al. [33]. The cremaster muscle was superfused with 35 °C warm buffered saline and intravital microscopy was performed using a Zeiss Axiotech Vario microscope at 20-fold magnification. Leukocyte-endothelium-interaction was recorded in three post capillary venules per mouse. Mean leukocyte rolling velocities as well as microvascular parameters (venular diameter, venular vessel segment length) were determined using the Fiji software. No statistical difference in the blood flow velocities in vessels of the two treatment groups could be detected, as assessed by the beam length of green fluorescent microspheres injected via the tail vein (1 mm diameter; Polysciences) under 20 ms exposure time. Extravasated leukocytes were detected in 4% formalin-fixed cremaster muscles and stained (Hemacolor, Millipore) immediately after sacrificing the animals. Microscopy images were taken from 6 representative areas with 10× magnification.
2.10. Permeability assay in vivo
IL-1β-induced vascular permeability of microvessels was assessed in the cremaster muscle of 20–25 week old male and female C57BL/6 WT mice (Charles River Laboratories) by detection of extravasation of the fluorescent dye FITC-dextran, as previously described [27]. 150 μl of PtpI IV (2 μM) or sham solution were injected into the scrotum 30 min before preparation of the cremaster muscle. After FITC-dextran injection and obtaining the first picture, 50 μl of 10 ng/ml IL-1β supplemented with 2 μM PtpI IV or sham were applied on the cremaster muscle tissue.
2.11. Induction of polymicrobial sepsis
Animals were held with ad libitum access to water and a standard nutritionally complete rodent chow diet and on a 12 h (h) dark-light cycle in a conventional barrier facility. Polymicrobial sepsis was induced by peritoneal contamination and infection (PCI) following the protocol established by Gonnert et al. [34] and as previously performed [35]. Briefly, to induce polymicrobial sepsis, 20–25 week old male and female C57BL/6J WT mice (originally obtained from Charles River Laboratories) were intraperitoneally injected with a standardized and microbiologically validated human faeces solution (2 μl/g body weight (BW), 650 g-positive colony forming units/μl, 3200 g-negative colony forming units/μl). The resulting systemic inflammatory reaction reflects several characteristics of a human sepsis. Sham-treated animals were injected with 0.9% sodium chloride solution (2 μl/g BW). All sepsis-induced animals received subcutaneous volume substitution (0.9% sodium chloride, 25 μl/g BW) every 4 h and antibiotic treatment (Meropenem 25 ml/kg BW) twice daily. At 24 h and 48 h after PCI the sepsis-induced animals had lost 8.0 ± 0.6% and 13.6 ± 1.0% body weight, had −4.8 ± 0.6 °C and − 2.6 ± 0.8 °C decreased body temperature and had a disease severity (clinical sepsis score, CSS, according to Gonnert et al. [34]) of 12.6 ± 0.26 and 12.5 ± 0.65 (CSS at 0 h: 4.0 ± 0), respectively (n = 12). For the CSS maximal 16 points can be reached (Grade IV). For the definition of the illness severity depending on the points, see Table S1.
2.12. NF-κB (p65) nuclear translocation and DNA binding activity
HUVEC expressing the SHP-2 constructs were stimulated with 10 ng/ml IL-1β for 30 min or left non treated. Nuclear translocation was assessed by immunofluorescence staining of p65 in cells cultured in 8-well μ-slides (ibiTreat, IBIDI), as previously described [36]. Nuclear translocation of p65 was quantified by number of stained nuclei normalized to the total number of nuclei per field of view. The specific DNA binding activity of the NF-κB subunit p65 in nuclear extracts of HUVEC (1 × 106 cells) was measured in triplicates using the Transcription Factor Assay Kit (#KA1341) from Abnova following the manufacturer's instructions.
2.13. Proximity ligation assay
HUVEC were cultivated on cover slips coated with 0.1% gelatine in PBS, treated with 10 ng/ml IL-1β and fixed with 1% formalin. Cells were permeabilized with 0.1% Triton-X in PBS and slides were bordered using a liquid blocker PAP-Pen (Sigma-Aldrich). DuoLink proximity ligation assay (Sigma-Aldrich) was performed according to manufacturer's protocol. In detail, slides were blocked with Duolink® blocking solution for 1 h following incubation with primary antibodies (1:100) targeting the interacting proteins and AlexaFluor 488 labelled phalloidin (1:300, Molecular Probes) diluted in Duolink® AB diluent for 1 h. Slides were washed three times with Duolink® washing buffer A for 5 min and appropriate Duolink® PLUS and MINUS PLA probes were incubated for 1 h. Ligation and amplification of antibody-conjugated DNA-oligos was performed as described by the manufacturer. To visualize nuclei, cells were incubated with DAPI for 5 min before the final washing step. Slides were air dried at room temperature and mounted with Dako fluorescence mounting medium. Image acquisition was performed using a Zeiss Axiovert 200 M fluorescence microscope at 40-fold magnification. Data are expressed as number of interaction spots/cell.
2.14. RNA isolation and qRT-PCR
RNA isolation and quantitative reverse-transcriptase PCR was performed as described before [37]. The probes (Applied Biosystems) used in the experiments included human ICAM-1 NM_000201.2 (#Hs00164932_m1), VCAM-1 NM_001078.3 (#Hs00365486_m1), and GAPDH NM_002046.3 (#4310884E).
2.15. SHP-2 phosphatase activity assay
Phosphatase activity in SHP-2 pull downs was measured as previously described [26]. To inhibit basal phosphatase activity of SHP-2, the phosphatase inhibitor Na3VO4 (25 mM) or the specific SHP-2 inhibitor PtpI IV (2 μM) were added to the substrate solution previous to incubation with precipitated SHP-2. After incubation for 1 h at 37 °C in the dark, supernatants were transferred to multiwell plates, and extinction was measured at 405 nm (SpectraFluor, Tecan).
2.16. ROS measurement
Intracellular ROS was measured by ROS-dependent oxidation of H2–2′,7′-Dichlorofluorescein (H2-DCF) to fluorescent DCF as previously described [37].
2.17. Measurement of IL-1β concentration in serum
Levels of IL-1β in serum of sepsis patients and healthy participants were assessed using the human IL-1β/IL-1F12 Quantikine® ELISA (#DLB50) from R&D according to the manufacturer's instructions. Levels of IL-1β in serum of PCI mice and healthy littermates were detected using the Mouse IL-1β ELISA Kit (#RAB0275) from Sigma-Aldrich according to the manufacturer's instructions.
2.18. Statistical analysis
All data are presented as means ± SEM. All statistical analyses were performed using Sigma Plot 10.0. For comparisons between two groups of normal distributed data, the student's t-test was used. For comparisons between two groups of data lacking normal distribution, a Mann-Whitney rank sum test was performed. For multiple comparisons of normal distributed data, the one-way analysis of variance (1-way ANOVA), followed by Student Newman-Keuls post-hoc test was performed. Differences were considered significant at an error probability level of p < 0.05.
2.19. Study approvals
All animal studies were conducted in accordance with the German animal protection law and approved by the district government of upper Bavaria (Regierung von Oberbayern, approval reference number AZ55.2–1-54-2532-172-13) as well as government of Thuringia (Thüringer Landesamt für Lebensmittelsicherheit und Verbraucherschutz, approval reference number AZ. 02–033/15). The investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85–23, revised 1996). Serum from patients, who were diagnosed with sepsis at the intensive care unit of the university hospital of Munich, was used for this study (for more information see Table 1). Control serum was drawn from healthy donors. Studies involving patient material were approved by the clinical ethics committee at the University of Munich, and written informed consent was received from all participants.
Table 1.
Clinical parameters of sepsis patients, whose blood was used in this study.
| Parameter sepsis patientsa | Mean ± SEM (range) |
|---|---|
| Sex (female / male) | n = 5 / n = 15 |
| Age (years) | 66 ± 2 (41–80) |
| C-reactive protein (mg/dl) | 19.9 ± 2.7 (0.9–34.0) |
| Leukocyte count (*109/l) | 17.1 ± 1.7 (8.5–33.6) |
Parameters were considered within the normal range with CRP values ≤0.5 mg/dl and leukocyte counts between 4 and 10*10 [9]/l.
3. Results
3.1. SHP-2 phosphatase activity is repressed in sepsis and causes an excessive inflammatory activation of EC
During systemic infection the endothelium is exposed to a complex composition of inflammatory factors contributing to the induction of adhesion molecules and disease progression [4,38]. To study if endothelial SHP-2 activity is affected by the inflammatory conditions in sepsis, we first performed experiments in mice suffering from polymicrobial sepsis induced by peritoneal contamination and infection (PCI) [34]. Interestingly, we detected significantly reduced SHP-2 phosphatase activity in lungs, which are highly vascularized organs, of septic mice compared to healthy littermates at all measured time points (6 h, 24 h and 48 h) (Fig. 1A). The downregulated SHP-2 activity was accompanied by significantly enhanced levels of the cytokine Interleukin (IL)-1β in serum of septic mice (Fig. 1B). Similarly, analysis of lungs from mice suffering from sepsis revealed significantly increased Vcam1 and Icam1 expression already 6 h after PCI, which was sustained over 48 h, as compared to healthy littermates (Fig. 1C). Next, we studied SHP-2 in primary EC incubated with serum obtained from sepsis patients (Table 1). Similar to septic mice, SHP-2 activity was significantly impaired in primary EC incubated with sepsis serum (Fig. 1D), as well as in blood cells of sepsis patients compared to blood cells of healthy donors (Fig. S1A). Additionally, the reduced SHP-2 activity was associated with strongly induced adhesion molecule expression (ICAM-1 and VCAM-1) in EC compared to cells treated with control serum (Fig. 1E and Fig. S1B). To investigate if the observed reduction of SHP-2 phosphatase activity is directly linked to the increased inflammatory endothelial response seen in sepsis, we treated EC transduced with different SHP-2 mutant constructs with serum from sepsis patients. The increased ICAM-1 and VCAM-1 expression induced by sepsis serum was even more pronounced in EC expressing a dominant negative, and thus inactive, SHP-2 (CS), as compared to SHP-2 wild-type (WT) expressing cells (Fig. 1F). In contrast, induction of SHP-2 activity by expression of a constitutively active SHP-2 (EA) prevented the increase of ICAM-1 and VCAM-1 induced by sepsis serum in comparison to SHP-2 WT cells (Fig. 1F).
Fig. 1.

Impaired SHP-2 activity in sepsis leads to increased expression of endothelial adhesion molecules.
A) SHP-2 activity was impaired in lungs of animals with polymicrobial sepsis (6 h, 24 h and 48 h upon PCI) compared to sham-treated animals (*p < 0.05, 1-way ANOVA, n = 5 animals/group). B) IL-1β levels in serum of mice were increased after PCI induction (*p < 0.05, 1-way ANOVA, n = 10 animals/group), as measured by ELISA. Numbers below dots show mean IL-1β concentration in pg/ml ± SEM at baseline and 48 h after PCI. C) Expression of Icam1 and Vcam1 was induced in PCI mice (*p < 0.05, 1-way ANOVA, n = 4 animals/group), as assessed by qRT-PCR of whole lungs. D) Incubation of EC with serum from sepsis patients reduced endothelial SHP-2 phosphatase activity (*p < 0.05, student's t-test, n = 7 patients). E) ICAM-1 and VCAM-1 surface expression was increased upon incubation with serum from sepsis patients (S) compared to serum from healthy donors (C) (*p < 0.05, student's t-test, n = serum from 4 to 5 patients measured in duplicates). F) Incubation of inactive SHP-2 CS expressing EC with sepsis serum further enhanced expression of ICAM-1 and VCAM-1 compared to SHP-2 WT expressing cells, whereas constitutively active SHP-2 EA expressing cells impaired this, as assessed by flow cytometry (*p < 0.05, 1-way ANOVA, n = serum from 4 to 5 patients measured in duplicates).
3.2. SHP-2 phosphatase activity is suppressed by ROS production in sepsis
ROS are known to inactivate phosphatases [39] and are strongly induced in the endothelium in sepsis [40]. Therefore, we next studied if inflammatory ROS production may be the cause of the observed SHP-2 inactivation in septic conditions. Indeed, treatment of EC with serum from sepsis patients led to a significant increase in ROS release (Fig. 2A). SHP-2 had a high basal phosphatase activity in EC, since incubation with the general phosphatase inhibitor sodium orthovanadate as well as with the pharmacological SHP-2 inhibitor PtpI IV reduced the phosphatase activity by 60% ± 16% and 68% ± 11% below basal levels, respectively (Fig. S2A), similar to sepsis serum. Importantly, the decrease in endothelial SHP-2 activity observed after exposure to serum from sepsis patients (in Fig. 1D) was restored upon selective inhibition of the NAD(P)H-oxidase (VAS-2870) (Fig. 2B), indicating a ROS-dependent mechanism. To investigate if cytokines released in sepsis induces ROS formation and thus are the upstream source of SHP-2 inactivation, we measured IL-1β levels in serum from sepsis patients. Similar to septic mice, significantly enhanced IL-1β concentrations were detected in human sepsis serum (Fig. 2C) and IL-1β exposure induced ROS production in EC (Fig. 2D). Interestingly, IL-1β treatment reduced endothelial SHP-2 activity, similar to sepsis serum (Fig. 2E). Furthermore, the reduced SHP-2 activity observed upon IL-1β stimulation was completely rescued when cells were incubated with the general anti-oxidant N-acetylcysteine (NAC) as well as with the NAD(P)H-oxidase inhibitor VAS-2870 (Fig. 2F). Of note, treatment of EC with IL-1 receptor-antagonist (IL-1Ra) prior to exposure to sepsis serum prevented SHP-2 inactivation (Fig. 2G). As IL-1α also signals through the IL-1R [10] and is released during sepsis [41], we measured SHP-2 activity upon IL-1α stimulation. In contrast to IL-1β stimulation, IL-1α failed to inactivate SHP-2 (Fig. S2B), indicating a major contribution of IL-1β in sepsis induced SHP-2 inactivation.
Fig. 2.

SHP-2 activity in endothelial cells is inhibited via ROS in inflammation and sepsis.
A) Treatment of EC with serum from sepsis patients (hSerum) induced ROS production, as assessed by DCF assay (*p < 0.05 vs control serum, student's t-test, n = serum from 6 patients). B) Incubation with the NAD(P)H-oxidase inhibitor VAS-2870 (5 μM) prior to stimulation with human sepsis serum prevented the decrease of SHP-2 activity in EC (*p < 0.05, 1-way ANOVA, n = serum from 4 to 7 patients). C) Analysis (ELISA) of serum from sepsis patients showed enhanced IL-1β concentrations compared to healthy donors (*p < 0.05, student's t-test, n = 7 patients measured in duplicates). D) IL-1β stimulation (10 ng/ml) induced ROS production in EC, as measured by the DCF assay (***p < 0.001, Mann-Whitney rank sum test, n = 3 in octuplicates). E) Stimulation with IL-1β (10 ng/ml, 4 h) repressed SHP-2 phosphatase activity in EC (*p < 0.05, 1-way ANOVA, n = 4). As a control, cells were incubated with an IL-1R antagonist (40 ng/ml) prior to incubation with IL-1β. F) Incubation with ROS inhibitors NAC (10 mM) or VAS-2870 (5 μM) prior to IL-1β stimulation restored the diminished SHP-2 activity in EC (*p < 0.05, 1-way ANOVA, n = 6–7). G) Treatment of EC with an IL-1R antagonist (40 ng/ml) prior to exposure to serum from sepsis patients restored SHP-2 activity (*p < 0.05, 1-way ANOVA on ranks, n = 12–15).
3.3. Loss of SHP-2 activity enhances IL-1β-induced endothelial inflammation in vivo
To further study the physiological impact of SHP-2 inactivation in an inflammatory setting and having observed that SHP-2 activity can be restored by IL-1R blockage in sepsis, we investigated the influence of SHP-2 activity on IL-1β-induced endothelial activation in vivo. Leukocyte recruitment was studied in postcapillary venules of the cremaster muscle of mice 4 h after treatment with IL-1β. To mimic the in vivo situation with reduced SHP2 activity in sepsis (Fig. 1A, D), mice were treated with the pharmacological SHP-2 inhibitor PtpI IV, which also enhanced IL-1β-induced endothelial ICAM-1 and VCAM-1 expression (Fig. S3A). The number of rolling leukocytes as well as the mean leukocyte rolling velocity was significantly decreased in animals receiving PtpI IV compared to sham treated animals (Fig. 3A). As a consequence, PtpI IV treatment significantly increased leukocyte adhesion (Fig. 3B). As EC permeability is a further hallmark of inflammation during sepsis and also determines leukocyte transmigration into the tissue, we assessed this parameter in the mouse cremaster muscle measuring FITC-dextran extravasation. Treatment with the SHP-2 inhibitor PtpI IV further augmented the IL-1β-induced vascular permeability of microvessels in vivo compared to sham treatment (Fig. 3C). In accordance with that, PtpI IV treatment enhanced leukocyte extravasation 4 h after IL-1β application compared to IL-1β treatment alone (Fig. 3D).
Fig. 3.
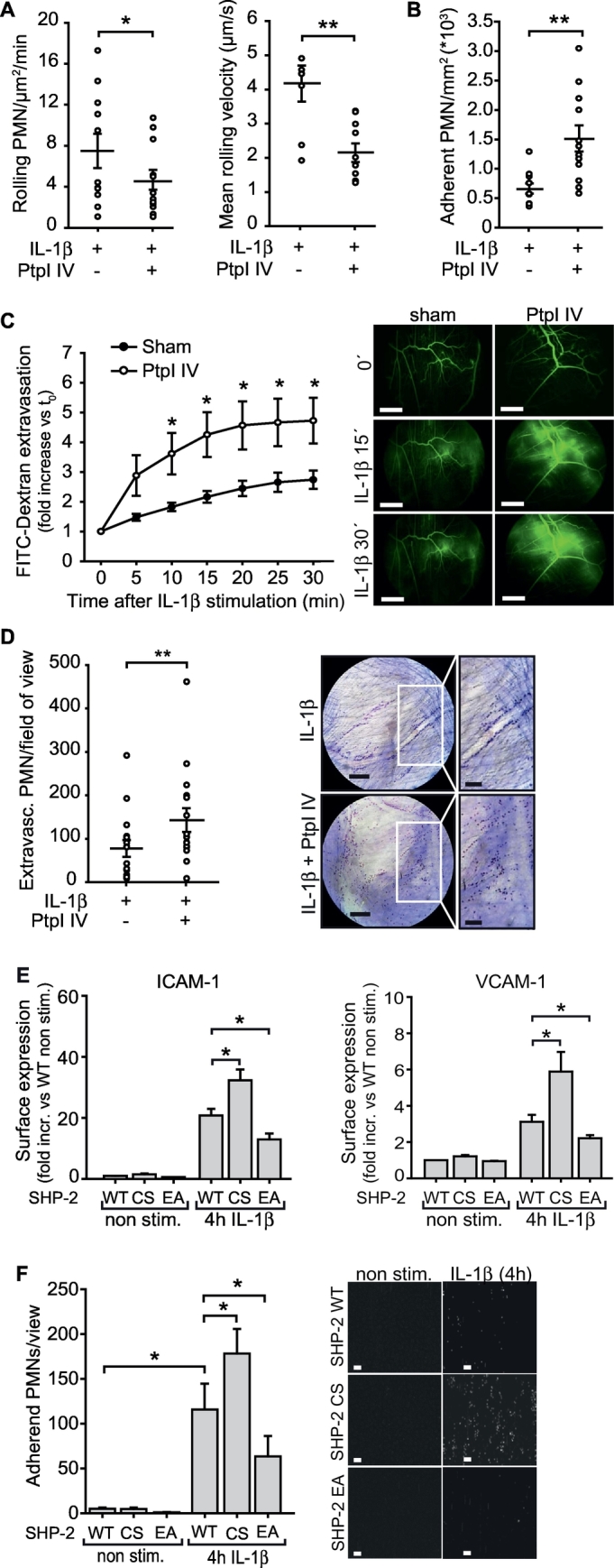
Inhibition of SHP-2 phosphatase activity augments IL-1β-induced vascular inflammation in vivo.
Leukocyte recruitment was assessed in post capillary venules of the mouse cremaster muscle after treatment with IL-1β (10 ng/ml) in combination with the pharmacological SHP-2 inhibitor PtpI IV (2 μM) or sham solution for 4 h by intravital microscopy. A) Number of rolling leukocytes and leukocyte rolling velocity was reduced upon SHP-2 inhibition (*p < 0.05, **p < 0.01, student's t-test, n = 4–5 animals/group, 2–3 venules/animal). B) SHP-2 inhibition further enhanced leukocyte adherence to the vessel wall compared to sole IL-1β treatment (**p < 0.01, student's t-test, n = 4–5 animals/group, 2–3 venules/animal). C) Stimulation of the mouse cremaster muscle with IL-1β (10 ng/ml) time dependently increased vascular permeability, which was further enhanced by pretreatment with PtpI IV (2 μM, 4 h) (*p < 0.05, two-tailed t-test between the respective time points, n = 5 animals/group), as assessed by FITC-dextran extravasation. Representative images of FITC-dextran (green) extravasation in the mouse cremaster muscle before (0 min) and after IL-1β stimulation and PtpI IV treatment, respectively, are shown next to the graph. Scale bar: 500 μm. D) Number of extravasated leukocytes under IL-1β stimulation was enhanced after SHP-2 inhibition (2 μM PtpI IV) compared to sham treatment (**p < 0.01, student's t-test, n = 3 animals/group, each 6 fields of view). Photos show representative microscopy images of stained cremaster tissues. Scale bar: 100 μm. White frames: Magnification of selected areas shown to the right; scale bar: 50 μm. E) Overexpression of SHP-2 CS enhanced IL-1β-induced (10 ng/ml) ICAM-1 and VCAM-1 surface expression, whereas SHP-2 EA expression prevented this induction, as measured by flow cytometry (*p < 0.05, 1-way ANOVA, n = 6 in duplicates). F) Expression of SHP-2 CS in human EC increased IL-1β-dependent (10 ng/ml, 4 h) human leukocyte adhesion under flow (1 dyn/cm2), whereas SHP-2 EA expression impaired adhesion compared to SHP-2 WT expressing cells (*p < 0.05, 1-way ANOVA, n = 3 each 4–5 visual fields). Representative images of human PMN adhesion to human EC expressing SHP-2 mutants are shown next to the graph. Scale bar: 50 μm.
To further verify the functional relevance of SHP-2 inhibition in the endothelium, we analyzed adhesion molecule expression and functional adhesion of isolated human leukocytes to EC expressing the different SHP-2 mutants (WT, CS and EA) under flow (1 dyn/cm2). This revealed that the increased number of adherent leukocytes after 4 h of IL-1β stimulation to SHP-2 WT expressing EC was even more pronounced in dominant negative SHP-2 CS expressing cells due to enhanced surface expression of VCAM-1 and ICAM-1 in these cells (Fig. 3E, F and S3B), similarly to pharmacological SHP-2 inhibition in vivo. In contrast, expression of constitutively active SHP-2 EA in EC significantly impaired adhesion molecule expression and accordingly leukocyte adhesion (Fig. 3E, F and S3B).
3.4. SHP-2 controls IL-1β-induced downstream signalling by a regulatory interaction with MyD88
Next, we sought to identify the specific signalling pathway affected by SHP-2 phosphatase activity during IL-1β-induced inflammation in sepsis. Firstly, pharmacological inhibition of the transcription factors AP-1 (c-jun), CREB and NF-κB showed that only inhibition of NF-κB affects IL-1β-induced ICAM-1 or VCAM-1 expression (Fig. S4A). Overexpression of SHP-2 CS in EC enhanced IL-1β-induced NF-κB activation compared to SHP-2 WT expression. In contrast, SHP-2 EA impaired this, as assessed by immunofluorescence staining of nuclear translocation of the NF-κB subunit p65 (Fig. 4A, B), p65 DNA-binding activity (Fig. 4C), and phosphorylation of IKK and IκBα (Fig. S4B). Finally, NF-κB inhibition in SHP-2 CS cells prevented the observed enhancement of ICAM-1 and VCAM-1 surface expression and protein levels in these cells (Fig. 4D).
Fig. 4.

SHP-2 activity controls IL-1β-induced NF-κB activation.
A) IL-1β-dependent (10 ng/ml, 30 min) nuclear translocation of the NF-κB subunit p65 in SHP-2 mutant EC was assessed by immunofluorescence staining (red: p65, blue: DAPI). B) SHP-2 EA expressing cells showed less p65 nuclear translocation upon IL-1β stimulation (*p < 0.05, 1-way ANOVA, n = 3 in duplicates) compared to SHP-2 WT cells. Graph shows quantification of NF-κB-positive nuclei. C) As no enhancement of p65 nuclear translocation was detected with SHP-2 CS, potentially due to already maximal response in SHP-2 WT expressing cells, p65 DNA-binding activity was measured. IL-1β induced p65 DNA binding activity in nuclear extracts of SHP-2 WT expressing EC, which was further enhanced by SHP-2 CS expression (*p < 0.05, 1-way ANOVA, n = 3). D) NF-κB inhibition (Ro 106–9920, 10 μM) prevented enhancement of IL-1β-dependent ICAM-1 and VCAM-1 upregulation in SHP-2 CS expressing cells (*p < 0.05, 1-way ANOVA, n = 6 in duplicates), as measured by flow cytometry (graph) and subsequent western blot of the same samples (lower panels). E and F) IL-1β-induced activation of downstream signalling molecules was analyzed by western blot. SHP-2 CS expression enhanced IL-1β-dependent Akt (n = 12), p38 MAPK (n = 4) and Hsp27 phosphorylation (n = 11) in EC compared to SHP-2 WT (*p < 0.05, 1-way ANOVA). Graphs show quantitative analysis of protein band intensities.
Secondly, to identify further signalling molecules upstream of NF-κB transcriptional activation, pharmacological inhibition of PI3K, Akt, ERK, and p38 MAPK prior to IL-1β stimulation was performed. Only p38 MAPK inhibition impaired IL-1β-induced ICAM-1 surface expression and protein levels (Fig. S4C). VCAM-1 expression was reduced after inhibition of p38 MAPK, PI3K, and Akt (Fig. S4D). Furthermore, Akt was activated by p38 MAPK and PI3K (Fig. S4E). Heat shock protein 27 (Hsp27), however, was only activated by p38 (Fig. S4F), indicating a splitting of the p38-mediated signalling cascade. Moreover, expression of SHP-2 CS in EC further enhanced the IL-1β-induced phosphorylation of p38 MAPK and Akt (Fig. 4E) as well as Hsp27 compared to SHP-2 WT (Fig. 4F).
Lastly, we investigated potential direct targets of SHP-2 through which the inflammatory response during sepsis may be influenced. The IL-1R1 adaptor protein MyD88 contains a SH-2 binding motif (YKAM between amino acids 257–260) shown to influence MyD88 downstream signalling [12]. As MyD88 was IL-1β-dependently phosphorylated on tyrosine 257 (Fig. S5), we explored if SHP-2 interacts with MyD88. As detected by a proximity ligation assay, SHP-2 was found to interact with MyD88 upon IL-1β stimulation (Fig. 5A). Importantly, mutation of the tyrosine 257 in the SH-2 binding motif of MyD88 (YF) abrogated the IL-1β-mediated interaction with endogenous SHP-2 (Fig. 5B). SHP-2/MyD88-interaction was also studied in EC expressing myc-tagged SHP-2 (WT, CS and EA). Interestingly, the previously observed IL-1β-induced interaction between SHP-2 and MyD88 (Fig. 5A) was even more pronounced between MyD88 and the inactive mutant construct SHP-2 CS, confirming its substrate trapping characteristic [42] (Fig. 5C). In contrast, SHP-2 EA exhibited a reduced interaction with MyD88, indicating a quick release of SHP-2 from its target upon dephoshorylation (Fig. 5C). We furthermore analyzed recruitment of the regulatory subunit p85 of PI3K to MyD88. Indeed, IL-1β stimulation resulted in association between MyD88 and the p85 subunit of PI3-K (Fig. 5D). Although the inactive SHP-2 CS was shown to be trapped at phospho-Y257 of MyD88 upon IL-1β stimulation, p85 recruitment to MyD88 was not impaired but even enhanced in these cells compared to SHP-2 WT (Fig. 5E). In contrast, the IL-1β-induced interaction between MyD88 and p85 was prevented in cells expressing constitutively active SHP-2 EA (Fig. 5E). These data indicate that SHP-2 binds to phospho-Y257 on MyD88 and may dephosphorylate a further binding site for p85. An illustration summarizing our findings regarding the regulatory role of SHP-2 in sepsis is shown in Fig. 6.
Fig. 5.

SHP-2 interacts with the SH-2 binding motif of MyD88.
A) IL-1β stimulation (10 ng/ml, 2 min) of EC induced MyD88 association with SHP-2 (*p < 0.05, student's t-test, n = 6 each 4–5 fields of view), as measured by a proximity ligation assay (PLA). B) HA-tagged MyD88 WT interacted with endogenous SHP-2 upon IL-1β stimulation, whereas HA-tagged MyD88 YF (mutation of Y257 in the SH-2 binding motif) did not associate with SHP-2 (*p < 0.05, 1-way ANOVA, n = 7 each 4 fields of view). C) PLA with myc-tagged SHP-2 WT, CS and EA revealed interaction between MyD88 and SHP-2 WT upon IL-1β. This interaction was increased between SHP-2 CS and MyD88, but impaired between SHP-2 EA and MyD88 (p < 0.05, 1-way ANOVA, n = 6 each 4–5 fields of view). D) IL-1β stimulation induced interaction between MyD88 and the p85 subunit of the PI3-K (*p < 0.05, student's t-test, n = 6 each 4 fields of view). E) IL-1β-mediated MyD88/p85 interaction was enhanced in EC expressing the substrate trapping and inactive mutant SHP-2 CS compared to SHP-2 WT. In contrast, interaction of MyD88 and p85 was prevented in cells expressing SHP-2 EA (*p > 0.05, 1-way ANOVA, n = 6 each 4 fields of view).
Representative images of all PLA experiments are shown to the right of the respective graph. Specific interaction spots are shown in the left panel. Merge images (right): PLA spots (red), nuclear staining with DAPI (blue) and actin staining with phalloidin AF488 (green). Scale bar: 20 μm. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
4. Discussion
In this study we unveil a novel and important regulatory role of the tyrosine phosphatase SHP-2 in protecting the endothelium from inflammation-induced activation. Of note, we found SHP-2 activity in the endothelium to be repressed by ROS during sepsis, which may constitute a critical step in the disease progression.
In mice suffering from polymicrobial sepsis, exhibiting strongly enhanced endothelial adhesion molecule expression, we detected a persistent suppression of SHP-2 activity. This corresponds to our findings in blood cells from sepsis patients, where SHP-2 phosphatase activity was found to be significantly reduced. A similar reduction of SHP-2 activity with a directly linked increase in adhesion molecule expression was observed in primary human endothelial cells (EC) exposed to serum from sepsis patients, supporting the relevance of SHP-2 activity in the control of inflammatory endothelial activation. The finding that inhibition of IL-1 receptor-mediated signalling was sufficient to reinstall SHP-2 activity in sepsis serum treated cells suggests that IL-1β is mainly responsible for SHP-2 inactivation in this condition. This is supported by the finding that IL-1α, which is also released in sepsis [41] and signals through the IL-1R [10], did not reduce SHP-2 activity in EC. The mechanism behind this selective inhibition needs to be further investigated. However, even if IL-1α does not seem to affect SHP-2 activity in EC it is still possible that SHP-2 inactivation by IL-1β influences IL-1α mediated signalling, as they exploit the same signalling molecules, such as MyD88 and p38MAPK. Whether SHP-2 is involved in the regulation of the cellular responses mediated by other pro-inflammatory factors during inflammation largely remains to be elucidated. Only a limited number of studies have investigated the role of SHP-2 in signalling mediated by other receptors involved in inflammatory responses in sepsis. For instance, Xu et al. showed that SHP-2 suppresses TLR dependent signalling in macrophages by negatively regulating TRAF-6 [43] and a study from An et al. demonstrated a negative regulation of TLR-3 and -4 mediated signalling by SHP-2 at the level of TRIF [44].
The protective role of SHP-2 in inflammatory endothelial activation is fostered by the prevention of sepsis serum- or IL-1β-induced expression of endothelial adhesion molecules and leukocyte adhesion by introduction of a constitutively active SHP-2 mutant. In contrast, expression of an inactive SHP-2 mutant, mimicking the SHP-2 inactivation in sepsis, worsened the inflammatory phenotype of EC and enhanced leukocyte adhesion. This illustrates that changes in SHP-2 activity in sepsis are functionally relevant. Accordingly, pharmacological inactivation of SHP-2 in mouse microvessels in vivo resulted in significantly enhanced leukocyte adhesion, transmigration and vascular permeability upon IL-1β stimulation. Our data are in line with the work of Coulombe et al., which found increased leukocyte infiltration into the colon in an ulcerative colitis model using mice with an SHP-2 deletion in intestinal epithelial cells [45]. The SHP-2 activity state may therefore determine the extent of vascular inflammation seen in sepsis and other systemic inflammatory diseases.
In the course of inflammation, both paracrine as well as local autocrine effects of IL-1β may come into play [46]. The observed continuous increase in IL-1β serum levels in septic mice may thus be due to increased secretion of IL-1β from recruited inflammatory cells or an autocrine positive feedback mechanism, as EC has been demonstrated to produce and release IL-1β and IL-1β can induce its own expression [46,47]. Although not investigated here, it may even be possible that SHP-2 inhibition leads to more IL-1β production in EC, as we showed that SHP-2 affects NF-κB activity, which is involved not only in the expression of adhesion molecules but also in the expression of inflammatory cytokines [48]. Thus, the consequences of SHP-2 inactivation during inflammation may be manifold and may be enhanced by positive feedback loops.
SHP-2 is known to associate with a multitude of receptors and their adaptor molecules and in this way regulates their downstream signalling [49,50]. MyD88 is an adaptor protein binding to the IL-1R, IL-18R and several Toll-like receptors (TLRs) [51,52]. Our findings that SHP-2 inhibition enhanced IL-1β-induced adhesion molecule expression and vascular leakage, both processes shown to depend on MyD88 [6], tempted us to investigate a possible link between SHP-2 phosphatase activity and MyD88. SHP-2 binds via its SH2-domains to phosphorylated tyrosines on target molecules [53]. As MyD88 was shown by us and others to become tyrosine phosphorylated [54,55], we hypothesized that SHP-2 may be recruited to this adaptor protein upon IL-1β stimulation. In fact, we found that SHP-2 interacts directly with the SH2-domain binding motif (YKAM) of MyD88 upon IL-1β stimulation, as the MyD88 Y257F mutant, lacking this binding motif, failed to bind SHP-2. It has been postulated that this motif is crucial for p85/PI3K recruitment to MyD88 and corresponding downstream signalling [12,55,56], which would imply competitive binding of SHP-2 and p85. However, we did not find evidence for a competitive binding of these molecules at this site. Both SHP-2 and p85 showed an increased association to MyD88 to a similar extent upon IL-1β stimulation, indicating that they are using different binding sites. This conclusion is supported by the observation that occupation of the Y257 within the YKAM motif of MyD88 by the substrate trapping mutant SHP-2 CS did not hamper p85 binding to MyD88 but even enhanced p85/MyD88 interaction. Therefore, based on these findings we propose an alternative model in which SHP-2 binds to the phosphorylated tyrosine in the SH-2 binding motif of MyD88 upon IL-1β stimulation, probably enabling the dephosphorylation of a different p85 binding site and subsequent inhibition of downstream signalling. This model is further highlighted by our finding that the interaction between MyD88 and p85 was reduced in cells expressing the constitutively active SHP-2 EA. Our model is in accordance with the study of Laird et al. [57], which showed that p85 binding to MyD88 occurs even if the SH-2 binding motif is mutated. In fact, screening of the PhosphoSitePlus® database revealed that MyD88 features two more tyrosine residues (Y187 and Y276) within its Toll/interleukin-1 receptor (TIR) domain, the functions of which remain to be studied, however. This mechanism may represent a vascular protective axis, limiting endothelial inflammatory responses and thus preventing vascular injury (see Fig. 6 for a summary). The high basal activity of SHP-2 is a further indication of its importance in maintaining endothelial quiescence.
Fig. 6.

Proposed model for the role of SHP-2 in the regulation of IL-1β-dependent EC signalling and vascular inflammation. A) The tyrosine phosphatase SHP-2 possesses a basal activity under physiological conditions. Thus, SHP-2 may preserve endothelial quiescence by keeping MyD88 phospho-tyrosine sites in a dephosphorylated state, thereby limiting pro-inflammatory downstream signalling via interaction partners of MyD88 such as PI3-K. B) Upon activation of the IL-1R1 by IL-1β, MyD88 is phosphorylated on Y257 enabling binding of SHP-2. However, inflammation-induced generation of ROS results in inactivation of SHP-2 by oxidation of the critical cysteine residue in the phosphatase domain suspending the protective effect of SHP-2. Hence, a different SH-2 binding site is phosphorylated, resulting in recruitment of p85/PI3-K to MyD88 and subsequent activation of inflammatory downstream signalling including p38 MAPK, AKT, Hsp27 and NF-κB. Consequently, expression of ICAM-1 and VCAM-1 at the endothelial surface is induced followed by leukocyte recruitment as well as enhanced vascular leakage (not shown). The reactivation of SHP-2 activity by IL-1Ra or ROS inhibition may be a promising therapeutic approach to prevent the extensive inflammatory responses mediated by MyD88 during inflammatory diseases such as sepsis.
Most interestingly, the suppressed SHP-2 activity in EC upon exposure to IL-1β and sepsis serum could be completely prevented by inhibition of ROS production. Indeed, ROS is a well-known mediator of endothelial inflammation [58], and SHP-2 activity has been shown to be inhibited by ROS-dependent oxidation of the critical cysteine residue in the catalytic pocket of SHP-2 [59,60]. Of note, sepsis patients were found to exhibit an impaired SHP-2 activity. In addition, the observation that IL-1β was enhanced in serum from septic mice for at least 48 h may explain the persistent SHP-2 inhibition observed in these animals. However, we cannot exclude a possible disinhibition of NAD(P)H-oxidase upon SHP-2 inactivation, which may additionally contribute to the long-term inactivation of SHP-2 in EC. On the basis of our data we thus propose that an inactivation of SHP-2 caused by cytokine-induced excessive ROS production is a crucial event in the induction of the endothelial adhesive phenotype in sepsis. This notion is further endorsed by the finding that restoration of SHP-2 activity by introduction of constitutively active SHP-2 prevented the inflammatory phenotype. This strengthens our initial belief that SHP-2 has a protective effect in the endothelium and most importantly demonstrates that SHP-2 activity can be restored during inflammatory conditions and thus may represent a putative future therapeutic target in sepsis. In inflammatory states, such as sepsis, we thus suggest that SHP-2 interacts with MyD88 upon phosphorylation of tyrosine 257. However, excessive ROS formation inhibits the phosphatase activity of SHP-2 and prevents it from dephosphorylating other phosphorylation sites on MyD88 important for binding of signalling molecules, such as the p85 subunit of the PI3-K, resulting in activation of signalling pathways, such as p38 MAPK, Akt, Hsp27, and NF-κB, and thus endothelial activation. Therapeutic application of ROS-inhibitors in the context of sepsis may reinstall endothelial SHP-2 activity and accordingly improve endothelial physiology. If this holds true in a clinical setting, needs further investigation.
In summary, our data provide novel evidence that SHP-2 is critical in the regulation of IL-1β-induced vascular dysfunction. While it physiologically downregulates IL-1β-dependent signalling, thereby limiting adhesion molecule expression, leukocyte adhesion and transmigration in vitro and in vivo, its inactivation results in inflammatory over-activation of the endothelium. Interaction of SHP-2 and MyD88 at tyrosine 257 in the SH-2 binding motif of MyD88 controls inflammatory downstream signalling and cellular responses. Of particular importance, repression of SHP-2 activity via ROS in sepsis therefore accelerates the endothelial inflammatory response. Physiological integrity of SHP-2 activity is therefore essential to prevent an inflammatory state of the endothelium, as in sepsis, and maintaining SHP-2 activity may be a therapeutic target to prevent endothelial dysfunction and capillary leakage.
Funding sources
The funding sources were not involved in the design of the study; in the collection, analysis and interpretation of data; in the writing of the report; and in the decision to submit the paper for publication. The corresponding author had access to all data in the study and had final responsibility for the decision to submit for publication.
Declaration of interest
No conflict of interest exists.
Author contributions
YH planned and performed most of the experiments, analyzed the data and took part in writing the manuscript. MB and GH performed the majority of the western blots and GH also assisted in some of the animal experiments. PB and TC performed the IL-1β in vivo experiments. SW analyzed animal sepsis data. RC and LU planned and performed the in vivo experiments with the sepsis model. JP, FK, KP and UP revised the manuscript for intellectual content. AP produced the lentiviral particles and revised the manuscript. MW and AR performed the qRT-PCRs. BK, MH and SK organized and evaluated studies with patient material. HM designed the study, analyzed the data, performed several of the experiments and wrote the manuscript.
Acknowledgements
The authors would like to thank Dr. Pothoulakis and Dr. Rhee at the University of California, Los Angeles for kindly supplying the MyD88 WT and YF plasmids. This work was supported by German Research Foundation (DFG) within the Research Unit 917, the LMU Mentoring excellence Programme, the LMU FöFoLe Programme and the “Verein zur Förderung von Wissenschaft und Forschung“at the medical faculty, LMU. Sebastian Weis is supported by the Integrated Research and Treatment Center—Center for Sepsis Control and Care (CSCC) at the Jena University Hospital. The CSCC is funded by the German Ministry of Education and Research (BMBF No. 01EO1502). Luisa Ungelenk was funded by the DFG-Research Training Group 1715 “Molecular Signatures of Adaptive Stress Responses”.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ebiom.2019.03.034.
Appendix A. Supplementary data
Supplementary material
Supplementary material
References
- 1.Aird W.C. The role of the endothelium in severe sepsis and multiple organ dysfunction syndrome. Blood. 2003;101(10):3765–3777. doi: 10.1182/blood-2002-06-1887. [DOI] [PubMed] [Google Scholar]
- 2.Bauer M., Coldewey S.M., Leitner M., Loffler B., Weis S., Wetzker R. Deterioration of organ function as a Hallmark in Sepsis: the cellular perspective. Front Immunol. 2018;9:1460. doi: 10.3389/fimmu.2018.01460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hawiger J., Veach R.A., Zienkiewicz J. New paradigms in sepsis: from prevention to protection of failing microcirculation. J Thromb Haemost. 2015;13(10):1743–1756. doi: 10.1111/jth.13061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhao Y.J., Yi W.J., Wan X.J., Wang J., Tao T.Z., Li J.B. Blockade of ICAM-1 improves the outcome of polymicrobial sepsis via modulating neutrophil migration and reversing immunosuppression. Mediators Inflamm. 2014;2014 doi: 10.1155/2014/195290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang D., Elner S.G., Bian Z.M., Till G.O., Petty H.R., Elner V.M. Pro-inflammatory cytokines increase reactive oxygen species through mitochondria and NADPH oxidase in cultured RPE cells. Exp Eye Res. 2007;85(4):462–472. doi: 10.1016/j.exer.2007.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhu W., London N.R., Gibson C.C., Davis C.T., Tong Z., Sorensen L.K. Interleukin receptor activates a MYD88-ARNO-ARF6 cascade to disrupt vascular stability. Nature. 2012;492(7428):252–255. doi: 10.1038/nature11603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ince C., Mayeux P.R., Nguyen T., Gomez H., Kellum J.A., Ospina-Tascon G.A. The endothelium in sepsis. Shock (Augusta GA) 2016;45(3):259–270. doi: 10.1097/SHK.0000000000000473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peters K., Unger R.E., Brunner J., Kirkpatrick C.J. Molecular basis of endothelial dysfunction in sepsis. Cardiovasc Res. 2003;60(1):49–57. doi: 10.1016/s0008-6363(03)00397-3. [DOI] [PubMed] [Google Scholar]
- 9.Deguine J., Barton G.M. MyD88: a central player in innate immune signaling. F1000prime reports. 2014;6:97. doi: 10.12703/P6-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Auron P.E. The interleukin 1 receptor: ligand interactions and signal transduction. Cytokine Growth Factor Rev. 1998;9(3–4):221–237. doi: 10.1016/s1359-6101(98)00018-5. [DOI] [PubMed] [Google Scholar]
- 11.Verstrepen L., Bekaert T., Chau T.L., Tavernier J., Chariot A., Beyaert R. TLR-4, IL-1R and TNF-R signaling to NF-kappaB: variations on a common theme. Cell Mol Life Sci. 2008;65(19):2964–2978. doi: 10.1007/s00018-008-8064-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rhee S.H., Kim H., Moyer M.P., Pothoulakis C. Role of MyD88 in phosphatidylinositol 3-kinase activation by flagellin/toll-like receptor 5 engagement in colonic epithelial cells. J Biol Chem. 2006;281(27):18560–18568. doi: 10.1074/jbc.M513861200. [DOI] [PubMed] [Google Scholar]
- 13.Ekman S., Kallin A., Engstrom U., Heldin C.H., Ronnstrand L. SHP-2 is involved in heterodimer specific loss of phosphorylation of Tyr771 in the PDGF beta-receptor. Oncogene. 2002;21(12):1870–1875. doi: 10.1038/sj.onc.1205210. [DOI] [PubMed] [Google Scholar]
- 14.Zhang S.Q., Tsiaras W.G., Araki T., Wen G., Minichiello L., Klein R. Receptor-specific regulation of phosphatidylinositol 3'-kinase activation by the protein tyrosine phosphatase Shp2. Mol Cell Biol. 2002;22(12):4062–4072. doi: 10.1128/MCB.22.12.4062-4072.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Holgado-Madruga M., Wong A.J. Role of the Grb2-associated binder 1/SHP-2 interaction in cell growth and transformation. Cancer Res. 2004;64(6):2007–2015. doi: 10.1158/0008-5472.can-03-2886. [DOI] [PubMed] [Google Scholar]
- 16.Kaneshiro S., Ebina K., Shi K., Higuchi C., Hirao M., Okamoto M. IL-6 negatively regulates osteoblast differentiation through the SHP2/MEK2 and SHP2/Akt2 pathways in vitro. J Bone Miner Metab. 2014;32(4):378–392. doi: 10.1007/s00774-013-0514-1. [DOI] [PubMed] [Google Scholar]
- 17.Lerner-Marmarosh N., Yoshizumi M., Che W., Surapisitchat J., Kawakatsu H., Akaike M. Inhibition of tumor necrosis factor-[alpha]-induced SHP-2 phosphatase activity by shear stress: a mechanism to reduce endothelial inflammation. Arterioscler Thromb Vasc Biol. 2003;23(10):1775–1781. doi: 10.1161/01.ATV.0000094432.98445.36. [DOI] [PubMed] [Google Scholar]
- 18.MacGillivray M., Herrera-Abreu M.T., Chow C.W., Shek C., Wang Q., Vachon E. The protein tyrosine phosphatase SHP-2 regulates interleukin-1-induced ERK activation in fibroblasts. J Biol Chem. 2003;278(29):27190–27198. doi: 10.1074/jbc.M213083200. [DOI] [PubMed] [Google Scholar]
- 19.Ren Y., Meng S., Mei L., Zhao Z.J., Jove R., Wu J. Roles of Gab1 and SHP2 in Paxillin tyrosine Dephosphorylation and Src activation in response to epidermal growth factor. J Biol Chem. 2004;279(9):8497–8505. doi: 10.1074/jbc.M312575200. [DOI] [PubMed] [Google Scholar]
- 20.Wu C.J., O'Rourke D.M., Feng G.S., Johnson G.R., Wang Q., Greene M.I. The tyrosine phosphatase SHP-2 is required for mediating phosphatidylinositol 3-kinase/Akt activation by growth factors. Oncogene. 2001;20(42):6018–6025. doi: 10.1038/sj.onc.1204699. [DOI] [PubMed] [Google Scholar]
- 21.Mannell H., Hellwig N., Gloe T., Plank C., Sohn H.Y., Groesser L. Inhibition of the tyrosine phosphatase SHP-2 suppresses angiogenesis in vitro and in vivo. J Vasc Res. 2008;45(2):153–163. doi: 10.1159/000110081. [DOI] [PubMed] [Google Scholar]
- 22.Zhu J.-X., Cao G., Williams J.T., DeLisser H.M. SHP-2 phosphatase activity is required for PECAM-1-dependent cell motility. Am J Physiol Cell Physiol. 2010;299(4):C854–C865. doi: 10.1152/ajpcell.00436.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boedtkjer E., Aalkjaer C. Insulin inhibits Na+/H+ exchange in vascular smooth muscle and endothelial cells in situ: involvement of H2O2 and tyrosine phosphatase SHP-2. Am J Physiol Heart Circ Physiol. 2009;296(2):H247–H255. doi: 10.1152/ajpheart.00725.2008. [DOI] [PubMed] [Google Scholar]
- 24.Ukropec J.A., Hollinger M.K., Woolkalis M.J. Regulation of VE-cadherin linkage to the cytoskeleton in endothelial cells exposed to fluid shear stress. Exp Cell Res. 2002;273(2):240–247. doi: 10.1006/excr.2001.5453. [DOI] [PubMed] [Google Scholar]
- 25.Dixit M., Loot A.E., Mohamed A., Fisslthaler B., Boulanger C.M., Ceacareanu B. Gab1, SHP2, and protein kinase a are crucial for the activation of the endothelial NO synthase by fluid shear stress. Circ Res. 2005;97(12):1236–1244. doi: 10.1161/01.RES.0000195611.59811.ab. [DOI] [PubMed] [Google Scholar]
- 26.Heun Y., Pogoda K., Anton M., Pircher J., Pfeifer A., Woernle M. HIF-1alpha dependent wound healing angiogenesis in vivo can be controlled by site-specific Lentiviral magnetic targeting of SHP-2. Mol. Ther. 2017;25(7):1616–1627. doi: 10.1016/j.ymthe.2017.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mannell H.K., Pircher J., Chaudhry D.I., Alig S.K., Koch E.G., Mettler R. ARNO regulates VEGF-dependent tissue responses by stabilizing endothelial VEGFR-2 surface expression. Cardiovasc Res. 2012;93(1):111–119. doi: 10.1093/cvr/cvr265. [DOI] [PubMed] [Google Scholar]
- 28.Kontaridis M.I., Liu X., Zhang L., Bennett A.M. Role of SHP-2 in fibroblast growth factor receptor-mediated suppression of myogenesis in C2C12 myoblasts. Mol Cell Biol. 2002;22(11):3875–3891. doi: 10.1128/MCB.22.11.3875-3891.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hofmann A., Wenzel D., Becher U.M., Freitag D.F., Klein A.M., Eberbeck D. Combined targeting of lentiviral vectors and positioning of transduced cells by magnetic nanoparticles. Proc Natl Acad Sci U S A. 2009;106(1):44–49. doi: 10.1073/pnas.0803746106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Trueck C., Zimmermann K., Mykhaylyk O., Anton M., Vosen S., Wenzel D. Optimization of magnetic nanoparticle-assisted lentiviral gene transfer. Pharm Res. 2012;29(5):1255–1269. doi: 10.1007/s11095-011-0660-x. [DOI] [PubMed] [Google Scholar]
- 31.Krötz F., Engelbrecht B., Buerkle M.A., Bassermann F., Bridell H., Gloe T. The tyrosine phosphatase, SHP-1, is a negative regulator of endothelial superoxide formation. J Am Coll Cardiol. 2005;45(10):1700–1706. doi: 10.1016/j.jacc.2005.02.039. [DOI] [PubMed] [Google Scholar]
- 32.Weckbach L.T., Groesser L., Borgolte J., Pagel J.I., Pogoda F., Schymeinsky J. Midkine acts as proangiogenic cytokine in hypoxia-induced angiogenesis. Am J Physiol Heart Circ Physiol. 2012;303(4):H429–H438. doi: 10.1152/ajpheart.00934.2011. [DOI] [PubMed] [Google Scholar]
- 33.Baez S. An open cremaster muscle preparation for the study of blood vessels by in vivo microscopy. Microvasc Res. 1973;5(3):384–394. doi: 10.1016/0026-2862(73)90054-x. [DOI] [PubMed] [Google Scholar]
- 34.Gonnert F.A., Recknagel P., Seidel M., Jbeily N., Dahlke K., Bockmeyer C.L. Characteristics of clinical sepsis reflected in a reliable and reproducible rodent sepsis model. J Surg Res. 2011;170(1):e123–e134. doi: 10.1016/j.jss.2011.05.019. [DOI] [PubMed] [Google Scholar]
- 35.Weis S., Carlos A.R., Moita M.R., Singh S., Blankenhaus B., Cardoso S. Metabolic adaptation establishes disease tolerance to Sepsis. Cell. 2017;169(7):1263–1275. doi: 10.1016/j.cell.2017.05.031. [e14] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mannell H., Hammitzsch A., Mettler R., Pohl U., Krötz F. Suppression of DNA-PKcs enhances FGF-2 dependent human endothelial cell proliferation via negative regulation of Akt. Cell Signal. 2010;22(1):88–96. doi: 10.1016/j.cellsig.2009.09.015. [DOI] [PubMed] [Google Scholar]
- 37.Alig S.K., Stampnik Y., Pircher J., Rotter R., Gaitzsch E., Ribeiro A. The tyrosine phosphatase SHP-1 regulates hypoxia inducible factor-1alpha (HIF-1alpha) protein levels in endothelial cells under hypoxia. PloS One. 2015;10(3) doi: 10.1371/journal.pone.0121113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Blom C., Deller B.L., Fraser D.D., Patterson E.K., Martin C.M., Young B. Human severe sepsis cytokine mixture increases beta2-integrin-dependent polymorphonuclear leukocyte adhesion to cerebral microvascular endothelial cells in vitro. Crit Care. 2015;19:149. doi: 10.1186/s13054-015-0883-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meng T.C., Fukada T., Tonks N.K. Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Mol Cell. 2002;9(2):387–399. doi: 10.1016/s1097-2765(02)00445-8. [DOI] [PubMed] [Google Scholar]
- 40.Biswal S., Remick D.G. Sepsis: redox mechanisms and therapeutic opportunities. Antioxid Redox Signal. 2007;9(11):1959–1961. doi: 10.1089/ars.2007.1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fida N.M., Al-Mughales J., Farouq M. Interleukin-1alpha, interleukin-6 and tumor necrosis factor-alpha levels in children with sepsis and meningitis. Pediatrics Int. 2006;48(2):118–124. doi: 10.1111/j.1442-200X.2006.02152.x. [DOI] [PubMed] [Google Scholar]
- 42.Tiganis T., Bennett Anton M. Protein tyrosine phosphatase function: the substrate perspective. Biochem J. 2007;402(Pt 1):1–15. doi: 10.1042/BJ20061548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xu S., Liu X., Bao Y., Zhu X., Han C., Zhang P. Constitutive MHC class I molecules negatively regulate TLR-triggered inflammatory responses via the Fps-SHP-2 pathway. Nat Immunol. 2012;13(6):551–559. doi: 10.1038/ni.2283. [DOI] [PubMed] [Google Scholar]
- 44.An H., Zhao W., Hou J., Zhang Y., Xie Y., Zheng Y. SHP-2 phosphatase negatively regulates the TRIF adaptor protein-dependent type I interferon and proinflammatory cytokine production. Immunity. 2006;25(6):919–928. doi: 10.1016/j.immuni.2006.10.014. [DOI] [PubMed] [Google Scholar]
- 45.Coulombe G., Leblanc C., Cagnol S., Maloum F., Lemieux E., Perreault N. Epithelial tyrosine phosphatase SHP-2 protects against intestinal inflammation in mice. Mol Cell Biol. 2013;33(11):2275–2284. doi: 10.1128/MCB.00043-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dinarello C.A., Simon A., van der Meer J.W. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat Rev Drug Discov. 2012;11(8):633–652. doi: 10.1038/nrd3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Alfaidi M., Wilson H., Daigneault M., Burnett A., Ridger V., Chamberlain J. Neutrophil elastase promotes interleukin-1beta secretion from human coronary endothelium. J Biol Chem. 2015;290(40):24067–24078. doi: 10.1074/jbc.M115.659029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.De Martin R., Hoeth M., Hofer-Warbinek R., Schmid J.A. The transcription factor NF-kappa B and the regulation of vascular cell function. Arterioscler Thromb Vasc Biol. 2000;20(11):E83–E88. doi: 10.1161/01.atv.20.11.e83. [DOI] [PubMed] [Google Scholar]
- 49.Neel B.G. Structure and function of SH2-domain containing tyrosine phosphatases. Semin Cell Biol. 1993;4(6):419–432. doi: 10.1006/scel.1993.1050. [DOI] [PubMed] [Google Scholar]
- 50.Neel B.G., Tonks N.K. Protein tyrosine phosphatases in signal transduction. Curr Opin Cell Biol. 1997;9(2):193–204. doi: 10.1016/s0955-0674(97)80063-4. [DOI] [PubMed] [Google Scholar]
- 51.Adachi O., Kawai T., Takeda K., Matsumoto M., Tsutsui H., Sakagami M. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity. 1998;9(1):143–150. doi: 10.1016/s1074-7613(00)80596-8. [DOI] [PubMed] [Google Scholar]
- 52.Kaisho T., Akira S. Toll-like receptor function and signaling. J Allergy Clin Immunol. 2006;117(5):979–987. doi: 10.1016/j.jaci.2006.02.023. [quiz 88] [DOI] [PubMed] [Google Scholar]
- 53.Neel B.G., Gu H., Pao L. The 'Shp'ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem Sci. 2003;28(6):284–293. doi: 10.1016/S0968-0004(03)00091-4. [DOI] [PubMed] [Google Scholar]
- 54.Xi C.X., Xiong F., Zhou Z., Mei L., Xiong W.C. PYK2 interacts with MyD88 and regulates MyD88-mediated NF-kappaB activation in macrophages. J Leukoc Biol. 2010;87(3):415–423. doi: 10.1189/jlb.0309125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ojaniemi M., Glumoff V., Harju K., Liljeroos M., Vuori K., Hallman M. Phosphatidylinositol 3-kinase is involved in Toll-like receptor 4-mediated cytokine expression in mouse macrophages. Eur J Immunol. 2003;33(3):597–605. doi: 10.1002/eji.200323376. [DOI] [PubMed] [Google Scholar]
- 56.Gelman A.E., LaRosa D.F., Zhang J., Walsh P.T., Choi Y., Sunyer J.O. The adaptor molecule MyD88 activates PI-3 kinase signaling in CD4+ T cells and enables CpG oligodeoxynucleotide-mediated costimulation. Immunity. 2006;25(5):783–793. doi: 10.1016/j.immuni.2006.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Laird M.H., Rhee S.H., Perkins D.J., Medvedev A.E., Piao W., Fenton M.J. TLR4/MyD88/PI3K interactions regulate TLR4 signaling. J Leukoc Biol. 2009;85(6):966–977. doi: 10.1189/jlb.1208763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lubrano V., Balzan S. Enzymatic antioxidant system in vascular inflammation and coronary artery disease. World J Exp Med. 2015;5(4):218–224. doi: 10.5493/wjem.v5.i4.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Salmeen A., Barford D. Functions and mechanisms of redox regulation of cysteine-based phosphatases. Antioxid Redox Signal. 2005;7(5–6):560–577. doi: 10.1089/ars.2005.7.560. [DOI] [PubMed] [Google Scholar]
- 60.Nardozza A.P., D'Orazio M., Trapannone R., Corallino S., Filomeni G., Tartaglia M. Reactive oxygen species and epidermal growth factor are antagonistic cues controlling SHP-2 dimerization. Mol Cell Biol. 2012;32(10):1998–2009. doi: 10.1128/MCB.06674-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material
Supplementary material