

Abstract
Lipin‐1 is a Mg2+‐dependent phosphatidic acid phosphohydrolase involved in the generation of diacylglycerol during synthesis of phospholipids and triglycerides. Ethanol‐mediated inhibitory effects on adipose‐specific lipin‐1 expression were associated with experimental steatohepatitis in rodents. In the present study, using an adipose‐specific lipin‐1 overexpression transgenic (Lpin1‐Tg) mouse model, we tested a hypothesis that adipose‐specific lipin‐1 overexpression in mice might dampen ethanol‐induced liver damage. Experimental alcoholic steatohepatitis was induced by pair‐feeding ethanol to Lpin1‐Tg and wild‐type (WT) mice using the chronic‐plus‐binge ethanol feeding protocol. Unexpectedly, following the chronic‐plus‐binge ethanol challenge, Lpin1‐Tg mice exhibited much more pronounced steatosis, exacerbated inflammation, augmented elevation of serum liver enzymes, hepatobiliary damage, and fibrogenic responses compared with the WT mice. Mechanistically, overexpression of adipose lipin‐1 in mice facilitated the onset of hepatic ferroptosis, which is an iron‐dependent form of cell death, and subsequently induced ferroptotic liver damage in mice under ethanol exposure. Concurrently, adipose lipin‐1 overexpression induced defective adiponectin signaling pathways in ethanol‐fed mice. Conclusion: We identified ferroptosis as a mechanism in mediating the detrimental effects of adipose‐specific lipin‐1 overexpression in mice under chronic‐plus‐binge ethanol exposure. Our present study sheds light on potential therapeutic approaches for the prevention and treatment of human alcoholic steatohepatitis.
Abbreviations
- ALD
alcoholic liver disease
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- ALP
alkaline phosphatase
- CK19
cytokeratin 19
- E
ethanol
- Fe
iron
- Fe2+
ferrous, reduced form
- FFA
free fatty acid
- FGF
fibroblast growth factor
- GAPDH
glyceraldehyde‐3‐phosphate dehydrogenase
- GSH
glutathione
- GPX4
glutathione peroxidase‐4
- H&E
hematoxylin and eosin
- HMW
high molecular weight
- β‐OHB
β‐hydroxybutyrate
- ICAM
intercellular adhesion molecule
- LCN2
lipocalin‐2
- LPIN1
lipin‐1
- MCP
monocyte chemoattractant protein
- MDA
malondialdehyde
- MIP
macrophage inflammatory protein
- mRNA
messenger RNA
- NF‐κB
nuclear factor kappa B
- NADPH
nicotinamide adenine dinucleotide phosphate, reduced form
- SAA1
serum amyloid A‐1
- SIRT1
sirtuin 1
- TG
Lpin1‐Tg mice
- Tg
transgenic
- TIMP‐1
metalloproteinase 1
- WT
wild type
Alcoholic liver disease (ALD) comprises a clinical spectrum of detrimental conditions that include steatosis/steatohepatitis, hepatitis, fibrosis/cirrhosis, and liver cancer.1 Alcoholic steatohepatitis, an early stage of ALD, is a well‐known risk factor for the development and progression of ALD.1, 2 Despite intense investigations, pathogenic mechanisms of alcoholic steatohepatitis remain enigmatic, and there has been no effective therapy clinically.
Lipin‐1, a Mg2+‐dependent phosphatidic acid phosphohydrolase (PAP), is a bifunctional molecule that regulates lipid metabolism in various tissues including liver and adipose.3, 4, 5 Lipin‐1‐mediated PAP activity catalyzes the conversion of phosphatide to diacylglycerol, the immediate precursor of triacylglycerol. Nuclear lipin‐1 protein is capable of interacting and regulating multiple pivotal transcription factors and cofactors.3, 4, 5
Lipin‐1 plays a critical functional role in adipocyte development and maturation.6, 7, 8 Lipin‐1 is required during the initial stages of adipogenesis for the induction of key adipogenic factors, such as peroxisome proliferator‐activated receptor α.7, 8 In mice, lipin‐1 deficiency impairs adipocyte differentiation and causes lipodystrophy, whereas the overexpression of lipin‐1 in adipose tissue increases adipocyte triglyceride storage, enhances adipose mass, and promotes obesity.7, 8, 9, 10, 11
Several lines of evidence demonstrate that adipose lipin‐1 and adiponectin, an adipocyte‐derived hormone, exhibit cross‐regulation.12, 13, 14, 15 For instance, lipin‐1 messenger RNA (mRNA) expression levels were positively correlated with adiponectin mRNA in both visceral and subcutaneous adipose tissues in human.12 Decreased lipin‐1 expression in visceral adipose tissue was associated with insulin resistance in polycystic ovary syndrome patients.13 More importantly, adipose lipin‐1 mRNA levels were decreased in adiponectin‐deficient mice.15
Disrupted lipin‐1 signaling contributes to the development and progression of alcoholic steatohepatitis in cell‐specific or tissue‐specific manners.14, 16, 17, 18, 19, 20, 21, 22 Although hepatocyte‐specific lipin‐1 exerted protective effects against ethanol‐induced liver damage, myeloid‐specific lipin‐1 contributed to the ethanol‐mediated inflammatory process in mice.14, 19 Using experimental ALD rodent models, we and others demonstrated that ethanol exposure specifically inhibited lipin‐1 gene and protein expression in adipose tissues.17, 22 The ethanol‐mediated inhibitory effects on lipin‐1 were associated with the development of alcoholic fatty liver accompanied by reduced adiponectin production in the adipose tissues of mice.17, 22 Interestingly, lipopolysaccharide, one of the putative inducers of ALD, inhibited both lipin‐1 and adiponectin expression in the adipose of mice.15
Ferroptosis is an iron‐dependent form of oxidative programmed cell death featuring glutathione (GSH) depletion, detrimental glutathione peroxidase‐4 (GPX4) redox defense, disrupted glutamate antiporter xCT/SCL7A11 (system Xc‐), and increased levels of lipid hydroperoxides.23, 24 In recent years, ferroptosis has emerged as a central mediator of cell death, implicated in multiple pathological processes including ischemia/reperfusion‐induced kidney or liver damage, neurotoxicity, and neurodegenerative diseases.23, 24, 25, 26, 27, 28
Ethanol‐induced hepatocellular injury is associated with excessive iron accumulation, changes in the level of GSH, generation of reactive oxygen species and local inflammation, all of which can cause structural and functional damage of the liver.1, 2, 29 Iron overload, GSH depletion, and the consequent lipid peroxidation and inflammation are considered the hallmarks of ferroptosis.23, 24, 25 Nonetheless, whether and how ferroptosis is involved in the pathogenic mechanisms of alcoholic steatohepatitis is presently unknown.
In the present study, using adipose tissue–specific lipin‐1 overexpression transgenic (Lpin1‐Tg) mice in a chronic‐plus‐binge ethanol feeding protocol,30, 31 we tested a hypothesis that adipose‐specific overexpression of lipin‐1 might dampen ethanol‐induced liver injury in mice. To our surprise, adipose lipin‐1 overexpression exacerbated steatohepatitis in mice in response to chronic‐plus‐binge ethanol challenge. We also presented evidence demonstrating that the detrimental effects of overexpressing adipose‐specific lipin‐1 in ethanol‐challenged mice were mediated, at least in part, through promoting hepatic ferroptosis.
Materials and Methods
Animal Studies
Lpin1‐Tg mice on the C57BL/6 background were kindly provided by Dr. Zhanxiang Zhou (University of North Carolina at Greensboro, NC).30 Generation of Lpin1‐Tg mice was described in detail in the published study.30 Male C57BL/6J mice were purchased from Jackson Laboratory.
A mouse model of chronic‐plus‐binge alcohol feeding (also referred as the Gao‐binge model) was used as previously described.14, 31 The 7‐8‐month‐old male Lpin1‐Tg mice and their age‐matched littermate wild‐type (WT) controls were divided into four dietary groups: (1) WT control, (2) WT plus ethanol (identical to the control diet but with 5% weight per volume ethanol added), (3) Lpin1‐Tg control, and (4) Lpin1‐Tg plus ethanol. All mice were fed a Lieber‐DeCarli liquid diet (Bio‐Serv, Frenchtown, NJ) for 5 days. Ethanol groups were then fed Lieber‐DeCarli liquid diets containing 5% weight per volume ethanol for 10 days while control mice were pair‐fed to their ethanol‐fed counterparts for 10 days. At day 11, mice in the ethanol groups were gavaged a single dose of ethanol (5 g/kg body weight, 31.25% ethanol), whereas mice in control groups were gavaged an isocaloric dose of dextrin maltose. The mice were euthanized, and blood and tissue samples were collected 9 hours after gavage. For mice on the ethanol‐containing diet, animal cages were placed on heating pads to maintain body temperature, to prevent ethanol‐induced hypothermia. Liquid diets were freshly prepared from powder daily. During the feeding period, Lpin1‐Tg mice and their WT counterparts were observed to consume similar volumes of ethanol‐containing diets. All animal experiments were approved by the Institutional Animal Care and Use Committee at Northeast Ohio Medical University.
Enzyme‐Linked Immunosorbent Assays of Mouse Serum
Serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels were measured using ALT and AST assay kits (Pointe Scientific, Austin, TX). Serum alkaline phosphatase (ALP) levels were determined using an ALP quantification kit (Biovision, Milpitas, CA). Plasma triglyceride and cholesterol were determined using the Sigma Diagnostics Triglyceride and Infinity Cholesterol Reagent (Thermo Fisher Scientific, Waltham, MA). Plasma levels of total and high‐molecular‐weight (HMW) forms of adiponectin were determined with a Mouse Adiponectin Assay Kit (ALPCO, Salem, NH). Serum fibroblast growth factor 15 (FGF15) levels were analyzed with an enzyme‐linked immunosorbent assay (ELISA) kit (LifeSpan Bio, Seattle, WA). Serum lipocalin‐2 (LCN2) and serum amyloid A‐1 (SAA1) concentrations were measured with ELISA kits from Abcam (Cambridge, MA). Serum β‐hydroxybutyrate (β‐OHB) was measured using a β‐OHB colorimetric assay kit (Biovision, Milpitas, CA).
Histology
Liver tissue was fixed in 10% formalin and embedded in paraffin as described previously.14, 18 Hematoxylin and eosin (H&E) staining, sirius red staining, and Perls’ Prussian blue staining were performed with a commercial staining solution (Sigma‐Aldrich, St. Louis, MO). Immunohistochemistry staining of cytokeratin 19 (CK19) was performed with a commercial primary antibody (Biocare Medical, LLC, Concord, CA, and Abcam), followed by a horseradish peroxidase–based detection kit (Vector Laboratories, Inc., Burlingame, CA).
Measurement of Hepatic Lipid
Liver cholesterol and triglyceride levels were measured using Infinity Cholesterol and Triglyceride Reagents (Thermo Fisher Scientific) as described previously.14, 18
Lipid Peroxidation Assay
The hepatic lipid peroxidation assays were performed as described previously.19 The hepatic lipid peroxidation was expressed as the extent of malondialdehyde (MDA) production using a thiobarbituric reactive substances analysis kit from Cayman Chemical (Ann Arbor, MI).
GPX Enzyme Activity Assay
Liver GPX enzyme activity was analyzed with a commercialized colorimetric assay kit (Biovision, Milpitas, CA).
Bile Acid Analysis
Liver, gallbladder, and intestine bile acid contents were analyzed with a Total Bile Acid Assay kit (Biovision, Milpitas, CA) as described previously.32
Iron Assay
Hepatic iron contents were determined with a commercial iron assay kit from Abcam, according to the manufacturer’s instructions.32
GSH and Reduced Form of Nicotinamide Adenine Dinucleotide Phosphate Assays
The hepatic GSH level was assessed using a GSH luminescent assay kit purchased from Promega (Madison, WI) according to the manufacturer’s instructions.25 The hepatic reduced form of nicotinamide adenine dinucleotide phosphate (NADPH) level was assessed using a NADPH colorimetric assay kit purchased from Abcam.
Protein Analysis
Western blot analyses were performed on liver tissue extracts as described previously.14, 18 The sirtuin 1 (SIRT1) antibody was obtained from Millipore (07‐131, Cleveland, OH). The LCN2 and SAA1 antibodies were purchased from Abcam (Ab63929 and Ab171030). The GPX4 and glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) antibodies were purchased from Proteintech Group (14432‐1‐AP and 60004‐1‐Ig; Rosemont, IL).
RNA Analysis
Total RNA was purified from tissues using TRIZOL (Invitrogen, Waltham, MA).14, 18 Quantitative real‐time polymerase chain reaction assays were performed with the PowerUp SYBR Green qPCR Master Mix (Fisher Scientific, Chicago, IL) and BIO‐RAD thermocycler systems (Bio‐Rad Laboratories, Hercules, CA). Primer sets were either purchased or designed (Table 1).
Table 1.
Primer Sequence for Real‐Time PCR
| Gene Abbreviations | Direction | Sequence |
|---|---|---|
| Collagen 1a | Forward | 5’‐ACCTCAAGATGTGCCACTC‐3’ |
| Reverse | 5’‐TGCCTCCTCCAAACCAGAC‐3’ | |
| FGF15 | Forward | 5’‐GCAGTACCTGTACTCCGCTG‐3’ |
| Reverse | 5’‐GTTTTGGTCCTCCTCGCAGT‐3’ | |
| E‐selectin | Forward | 5’‐AGCAGAGTTTCACGTTGCAGG‐3’ |
| Reverse | 5’‐TGGCGCAGATAAGGCTTCA‐3’ | |
| GAPDH | Forward | 5’‐TGACCTCAACTACATGGTCTACA‐3’ |
| Reverse | 5’‐CTTCCCATTCTCGGCCTTG‐3’ | |
| IL‐1β | Forward | 5’‐TCGCTCAGGGTCACAAGAAA‐3’ |
| Reverse | 5’‐CATCAGAGGCAAGGAGGAAAAC‐3’ | |
| ICAM‐1 | Forward | 5’‐CAATTTCTCATGCCGCACAG‐3’ |
| Reverse | 5’‐AGCTGGAAGATCGAAAGTCCG‐3’ | |
| Lpin‐1α | Forward | 5’‐GGTCCCCCAGCCCCAGTCCTT‐3’ |
| Reverse | 5’‐GCAGCCTGTGGCAATTCA‐3’ | |
| Lpin‐1β | Forward | 5’‐CAGCCTGGTAGATTGCCAGA‐3’ |
| Reverse | 5’‐GCAGCCTGTGGCAATTCA‐3’ | |
| Lcn2 | Forward | 5’‐TGGCCCTGAGTGTCATGTG‐3’ |
| Reverse | 5’‐CTCTTGTAGCTCATAGATGGTGC‐3’ | |
| MCP‐1 | Forward | 5’‐TTAAAAACCTGGATCGGAACCAA‐3’ |
| Reverse | 5’‐GCATTAGCTTCAGATTTACGGGT‐3’ | |
| MIP‐1α | Forward | 5’‐TGAGAGTCTTGGAGGCAGCGA‐3’ |
| Reverse | 5’‐TGTGGCTACTTGGCAGCAAACA‐3’ | |
| MIP‐1β | Forward | 5’‐AACACCATGAAGCTCTGCGT‐3’ |
| Reverse | 5’‐AGAAACAGCAGGAAGTGGGA‐3’ | |
| TIMP‐1 | Forward | 5’‐TCTTGGTTCCCTGGCGTACTCT‐3’ |
| Reverse | 5’‐GTGAGTGTCACTCTCCAGTTTGC‐3’ | |
| Cyp7a1 | Forward | 5’‐CACCATTCCTGCAACCTTCTGG‐3’ |
| Reverse | 5’‐ATGGCATTCCCTCCAGAGCTGA‐3’ | |
| SAA1 | Forward | 5’‐TTAGCTCAGTAGGTTGTGCTGCTGG‐3’ |
| Reverse | 5’‐ACAATGTTTCCCCAGAGAGCA‐3’ | |
| AdipoR1 | Forward | 5’‐TCCCAGGAACACTCCTGCTC‐3’ |
| Reverse | 5’‐CTTCTACTGCTCCCCACAGC‐3’ | |
| AdipoR2 | Forward | 5’‐AGCCTCTATATCACCGGAGCTG‐3’ |
| Reverse | 5’‐GCTGATGAGAGTGAAACCAGATGT‐3’ | |
| TNF‐α | Forward | 5’‐AGGCTGCCCCGACTACGT‐3’ |
| Reverse | 5’‐GACTTTCTCCTGGTATGAGATAGCAAA‐3’ | |
| VCAM‐1 | Forward | 5’‐TGAACCCAAACAGAGGCAGAGT‐3’ |
| Reverse | 5’‐GGTATCCCATCACTTGAGCAGG‐3’ | |
| FGFR4 | Forward | 5’‐TCCGACAAGGATTTGGCAGACC‐3’ |
| Reverse | 5’‐TGGCGGCACATTCCACAATCAC‐3’ | |
| Akr1c6 | Forward | 5’‐CTCACCATTATATTGCTGCCTGT‐3’ |
| Reverse | 5’‐TCTCTTTGCCATAGCGTTTTTCT‐3’ | |
| Gls2 | Forward | 5’‐GGTCTGCGCTATAACAAACTCT‐3’ |
| Reverse | 5’‐CATGACACTGCCTGACTCACA‐3’ | |
| Slc1a5 | Forward | 5’‐CATCAACGACTCTGTTGTAGACC‐3’ |
| Reverse | 5’‐CGCTGGATACAGGATTGCGG‐3’ | |
| Adiponectin | Forward | 5’‐TGTTCCTCTTAATCCTGCCAA‐3’ |
| Reverse | 5’‐CAAACCTGCACAAGTTCCCTT‐3’ |
Statistical Analysis
Data were analyzed using GraphPad Prism 7.0 software (San Diego, CA) using two‐tailed Student t tests or one‐way analysis of variance test followed by a Tukey post hoc test. All data were expressed as the mean ± SEM. Significance was defined as P < 0.05.
Results
Adipose‐Specific Lipin‐1 Overexpression Exacerbated Steatohepatitis in Mice After Chronic‐Plus‐Binge Ethanol Challenge
The involvement of adipose‐specific lipin‐1 overexpression in experimental alcoholic steatohepatitis was examined using WT and Lpin1‐Tg mice with a chronic‐plus‐binge ethanol feeding protocol.14, 31, 32 Lpin1‐Tg mice fed with or without ethanol exhibited a specific and significant approximate 1.8‐fold increase in lipin‐1 mRNA in adipose tissue compared with the WT controls (Supporting Fig. S1A). Consist with our previous findings,18 ethanol feeding to WT mice significantly increased hepatic lipin‐1 mRNA expression levels compared with WT controls (Supporting Fig. S1B.). Hepatic lipin‐1 mRNA levels in Lpin1‐Tg mice fed with or without ethanol were not altered compared with WT controls (Supporting Fig. S1B). Ethanol feeding caused a significant increase in the liver/body weight ratio in both WT and Lpin1‐Tg mice compared with their respective controls (Table 2). The percentage of white adipose tissue weight in body weight was not altered by the overexpression of lipin‐1 in adipose or by the ethanol feeding (Table 2). Histology analysis revealed that adipocyte size were similar among the four groups (Supporting Fig. S1C).
Table 2.
Selected Parameters in WT and Lpin1‐Tg Mice Following Chronic‐Plus‐Binge Ethanol Feeding
| Parameters | WT | WT + E | Lpin1‐Tg | Lpin1‐Tg + E |
|---|---|---|---|---|
| Starting body weight (g) | 33.84 ± 0.63* | 32.92 ± 1.41* | 33.56 ± 0.82* | 33.43 ± 0.91* |
| Final body weight (g) | 31.67 ± 0.88* | 30.40 ± 1.35*† | 31.36 ± 0.89*† | 29.01 ± 0.70† |
| Liver weight (g) | 0.9633 ± 0.0335† | 1.2980 ± 0.1158* | 0.9013 ± 0.0637† | 1.2100 ± 0.0678* |
| Liver weight/body weight (%) | 3.052 ± 0.100† | 4.263 ± 0.295* | 2.840 ± 0.194† | 4.152 ± 0.156* |
| WAT weight (g) | 1.128 ± 0.087* | 1.076 ± 0.056* | 1.1250 ± 0.159* | 0.9716 ± 0.1124* |
| WAT weight/body weight (%) | 3.534 ± 0.198* | 3.540 ± 0.099* | 3.496 ± 0.422* | 3.373 ± 0.435* |
| Liver cholesterol (µg/mg liver) | 2.860 ± 0.170* | 2.853 ± 0.207* | 3.550 ± 0.339* | 3.174 ± 0.177* |
| Serums | ||||
| Triglycerides (mg/dL) | 29.98 ± 2.32‡ | 98.25 ± 12.67* | 26.48 ± 2.71‡ | 70.24 ± 5.76† |
| Cholesterol (mg/dL) | 84.30 ± 6.42* | 84.07 ± 1.22* | 65.64 ± 12.48* | 72.90 ± 4.04* |
| β‐OHB (mM) | 0.454 ± 0.052† | 1.867 ± 0.522* | 0.692 ± 0.113† | 1.478 ± 0.193* |
| FFA (µmol/µL) | 0.344 ± 0.060‡ | 1.262 ± 0.096* | 0.454 ± 0.093‡ | 0.819 ± 0.115† |
Ten‐week to 12‐week‐old male WT and adipose‐specific Lpin1‐Tg mice were divided into four groups as follows: (1) pair‐fed WT control; (2) ethanol‐fed WT; (3) pair‐fed Lpin1‐Tg control; and (4) ethanol‐fed Lpin1‐Tg. Results are shown as means ± SEM (n = 5‐12 mice). Means without a common symbol (e.g., *, …) differ, P < 0.05.
Abbreviations: WAT, white adipose tissue.
Ethanol administration significantly increased the serum levels of free fatty acid (FFA), triacylglycerol, and β‐OHB (Table 2). Adipose overexpression of lipin‐1 partially but significantly attenuated elevations of serum FFA and triglyceride in accompany with a slight lower level of β‐OHB in the ethanol‐fed mice in comparison with ethanol‐fed WT mice (Table 2).
Liver triglyceride contents, serum markers of liver damage, AST, and ALT were significantly higher in the ethanol‐fed WT mice compared with WT control mice (Fig. 1A‐E). Surprisingly, in comparison with ethanol‐fed WT mice, ethanol‐fed Lpin1‐Tg mice showed the exacerbated onset and progression of steatosis and liver damage, as revealed by marked accumulation of lipid droplets, greater hepatic triglyceride contents, robustly augmented elevation of plasma ALT, and AST (Fig. 1A‐E). Hepatic cholesterol contents were unchanged in all groups (Table 2).
Figure 1.
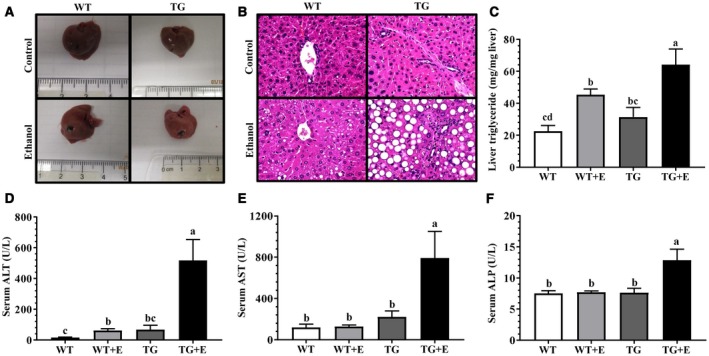
Adipose‐specific lipin‐1 overexpression exacerbated the ethanol‐induced liver injury. Male Lpin1‐Tg and WT mice were fed an ethanol‐containing diet for 10 days followed by a single gavage of ethanol. Lpin1‐Tg and WT control mice were pair‐fed with control diets without ethanol for 10 days followed by a single gavage of maltose. (A) Pictures of livers. (B) H&E (original magnification × 200) of liver sections. (C) Hepatic triglyceride levels. (D) Serum ALT. (E) Serum AST. (F) Serum ALP. Results are shown as means ± SEM (n = 5‐9 mice). Means without a common letter differ, P < 0.05.
Ethanol‐fed Lpin1‐Tg mice also exhibited hepatobiliary damage, as demonstrated by substantially elevated levels of serum ALP, an indicator of bile duct damage (Fig. 1F). Accordingly, immunohistochemical staining for CK19, a marker for cholangiocytes, showed more bile ducts in the center of fibrotic areas in the livers of ethanol‐fed Lpin1‐Tg mice than in mice of the other three groups (Fig. 2A; Supporting Fig. S2A).
Figure 2.
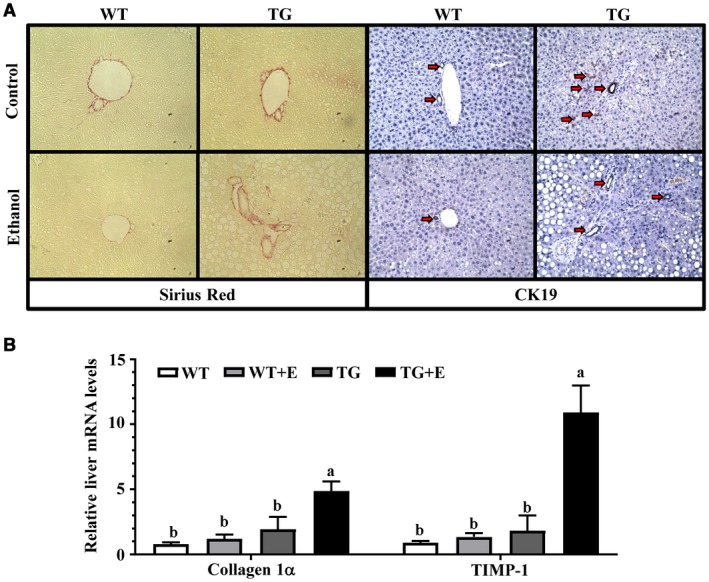
Adipose‐specific lipin‐1 overexpression induced liver fibrosis in ethanol‐fed mice. Mice were fed as described in Fig. 1. (A) Sirius red and CK19 staining (original magnification × 100). (B) Relative liver mRNA levels of collagen 1α and TIMP‐1. Results are shown as means ± SEM (n = 5‐9 mice). Means without a common letter differ, P < 0.05.
Immunohistochemical staining for sirius red displayed apparent fibrotic changes in the portal areas of the livers of ethanol‐fed Lpin1‐Tg mice in comparison with livers of ethanol‐fed WT mice (Fig. 2A; Supporting Fig. S2B). Accordingly, the mRNA expression levels of two markers of liver fibrosis, collagen 1α and tissue inhibitor of metalloproteinase 1 (Timp‐1), were significantly enhanced in the ethanol‐fed Lpin1‐Tg mice in comparison with ethanol‐fed WT mice (Fig. 2B).
Collectively, these data demonstrated that adipose‐specific overexpression of lipin‐1 exacerbated steatohepatitis, aggravated hepatobiliary damage, and induced mild fibrotic injury in response to chronic‐plus‐binge ethanol challenge in mice.
Lipin‐1 Overexpression in Adipose Exacerbated Inflammatory Responses in Ethanol‐Administered Mice
Adipose‐specific lipin‐1 overexpression augmented the ethanol‐mediated increases in mRNA levels of multiple inflammation mediators, including tumor necrosis factor alpha (TNF‐α), interleukin‐1β (IL‐1β), monocyte chemoattractant protein 1 (MCP‐1), macrophage inflammatory protein 1α (MIP‐1α), MIP‐1β, E‐selectin, intercellular adhesion molecule‐1 (Icam‐1), and vascular cell adhesion molecule‐1 (Vcam‐1) in the ethanol‐fed mice (Fig. 3A).
Figure 3.
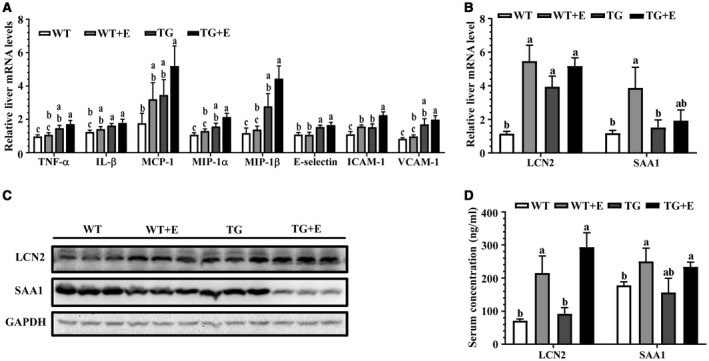
Adipose‐specific lipin‐1 overexpression exacerbated the ethanol‐induced inflammatory responses in mice. Mice were fed as described in Fig. 1. (A) Relative liver mRNA levels of TNF‐α, IL‐1β, MCP‐1, MIP‐1α, MIP‐1β, E‐selectin, ICAM‐1, and VCAM‐1. (B) Relative liver mRNA levels of LCN2 and SAA1. (C) Representative western blot analysis of hepatic expression levels of LCN2 and SAA1. (D) Serum concentrations of LCN2 and SAA1. Results are shown as means ± SEM (n = 5‐9 mice). Means without a common letter differ, P < 0.05.
We further evaluated two major proinflammatory molecules, LCN2 and SAA1, which play detrimental roles in alcoholic steatohepatitis in rodents and human.14, 16
LCN2 mRNAs were significantly elevated in the ethanol‐fed WT and Lpin1‐Tg mice fed with or without ethanol (Fig. 3B). The ethanol‐mediated increases in hepatic protein expression and circulating LCN2 were slightly augmented in the ethanol‐fed Lpin1‐Tg mice, but their levels did not reach statistical significance (Fig. 3C,D; Supporting Fig. S2C).
Although SAA1 mRNA was elevated in the ethanol‐fed WT mice compared with WT controls, SAA1 mRNA was normalized in the ethanol‐fed Lpin1‐Tg mice (Fig. 3B). Surprisingly, hepatic gene and protein expression levels of SAA1 were drastically reduced in the ethanol‐fed Lpin1‐Tg mice compared with WT controls (Fig. 3B,C; Supporting Fig. S2C). Interestingly, circulating SAA1 levels were induced slightly by ethanol administration to WT and Lpin1‐Tg mice to the same extent as WT control (Fig. 3D). Taken together, these observations demonstrated that Lpin1‐Tg mice fed with ethanol were prone to developing hepatic inflammation.
Adipose‐Specific Lipin‐1 Overexpression Exacerbated Aberrant Iron Homeostasis in Ethanol‐Fed Mice
We investigated whether iron dysregulation exacerbated the ethanol‐induced liver damage in mice with adipose‐specific lipin‐1 overexpression.29 Ethanol feeding did not alter the total concentrations of iron and ferrous (Fe2+, reduced form) in the livers of WT mice compared with WT controls (Fig. 4A). Remarkably, hepatic iron (Fe) or Fe2+ concentrations were markedly augmented about 3‐fold in the ethanol‐fed Lpin1‐Tg mice compared with WT controls and with ethanol‐fed WT mice (Fig. 4A). Perls’ Prussian blue staining revealed that ethanol feeding caused formations of iron‐positive hepatocytes around the central veins in the livers of WT mice, which were substantially augmented by adipose‐specific lipin‐1 overexpression (Fig. 4B). Prominent iron staining was spotted in the bile duct areas around the portal veins in the livers of ethanol‐fed Lpin1‐Tg mice (Fig. 4B).
Figure 4.
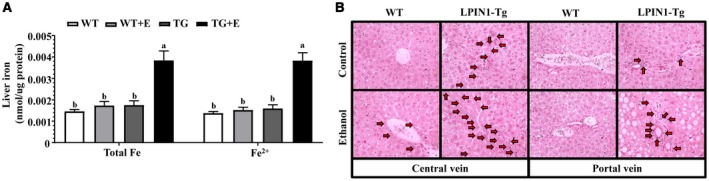
Adipose‐specific lipin‐1 overexpression exacerbated aberrant iron homeostasis in ethanol‐fed mice. Mice were fed as described in Fig. 1. (A) Liver total Fe and Fe2+ concentrations. (B) Perls’ Prussian blue staining of liver sections (original magnification × 200). Results are shown as means ± SEM (n = 5‐9 mice). Means without a common letter differ, P < 0.05.
Interestingly, the mRNA expression of intestinal ferroportin, an iron exporter, were markedly elevated in the ethanol‐fed Lpin1‐Tg mice compared with all other groups, suggesting that ferroportin may be involved in exacerbated aberrant iron homeostasis in ethanol‐fed Lpin1‐Tg mice (Supporting Fig. S2D).
Taken together, our results showed that adipose‐specific lipin‐1 overexpression caused iron overload in liver and altered hepatic iron distribution in the ethanol‐fed mice. The exacerbated aberrant iron homeostasis might contribute to liver injury and bile duct damage in Lpin1‐Tg mice in response to chronic‐plus‐binge ethanol challenge.
Lipin‐1 Overexpression in Adipose Induced Hepatic Ferroptosis in Mice After Ethanol Feeding
We hypothesized that greater accumulation of iron and altered iron distribution in the livers of ethanol‐fed Lpin1‐Tg mice might be involved in inducing ferroptosis, a form of iron‐dependent cell death. Hepatic ferroptosis was assessed by measuring three markers of ferroptosis: lipid peroxidation, GSH, and NADPH.25
Ethanol feeding to WT mice mildly but significantly generated oxidative stress, as demonstrated by the slight increase in hepatic MDA, a lipid peroxidation end product, compared with WT controls (Fig. 5A). Ethanol feeding to Lpin1‐Tg mice significantly augmented hepatic MDA levels compared with all other groups (Fig. 5A).
Figure 5.
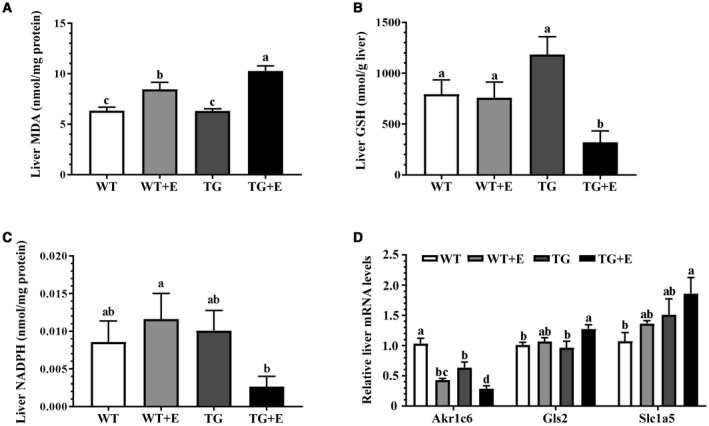
Lipin‐1 overexpression in adipose tissue–induced hepatic ferroptosis in the ethanol‐fed mice. Mice were fed as described in Fig. 1. (A) Liver MDA contents. (B) Liver GSH contents. (C) Liver NADPH levels. (D) Relative liver mRNA levels of Akr1c6, Gls2, and Slc1a5. Results are shown as means ± SEM (n = 5‐9 mice). Means without a common letter differ, P < 0.05.
In comparison with the WT controls, liver GSH contents were not significantly altered in ethanol‐fed WT mice and in Lpin1‐Tg control mice (Fig. 5B). Strikingly, hepatic GSH levels were the lowest in the ethanol‐fed Lpin1‐Tg mice compared with the other three groups (Fig. 5B). Hepatic NADPH was not altered in WT mice fed with or without ethanol and in the Lpin1‐Tg control mice (Fig. 5C). However, ethanol administration to the Lpin1‐Tg mice caused significant decreases in hepatic NADPH levels compared with ethanol‐fed WT mice (Fig. 5C).
We further analyzed the mRNA expression of several genes regulating ferroptosis. The mRNA expression of aldo‐keto reductase family 1 member C6, a protein to eliminate the peroxidized lipid and prevent ferroptosis, was decreased by ethanol administration to WT mice and further significantly reduced in ethanol‐fed Lpin1‐Tg mice compared with all other groups (Fig. 5D). Ethanol administration to Lpin1‐Tg mice increased mRNA abundances of glutaminase 2 (Gls2) and solute carrier family 1 (neutral amino acid transporter) member 5 (Slc1a5), two molecules facilitating the ferroptosis process, compared with WT controls (Fig. 5D). Note that liver GPX4 gene and protein expression and enzymatic activity were not significantly altered in these groups, implying involvement of GPX4‐independent mechanisms (Supporting Fig. S3). Collectively, these data demonstrated that ferroptosis occurred in the livers of ethanol‐fed Lpin1‐Tg mice.
Adipose‐Specific Overexpression of Lipin‐1 Exacerbated the Ethanol‐Mediated Impairment of Adipose Adiponectin and Hepatic Adiponectin Receptors in Mice
Plasma circulating levels of total or HMW adiponectin were not altered in the ethanol‐fed WT or control Lpin1‐Tg mice compared with WT control mice (Fig. 6A). Strikingly, Lpin1‐Tg mice showed substantially higher serum concentrations of total and HMW adiponectin in response to ethanol challenge compared with other groups (Fig. 6A).
Figure 6.
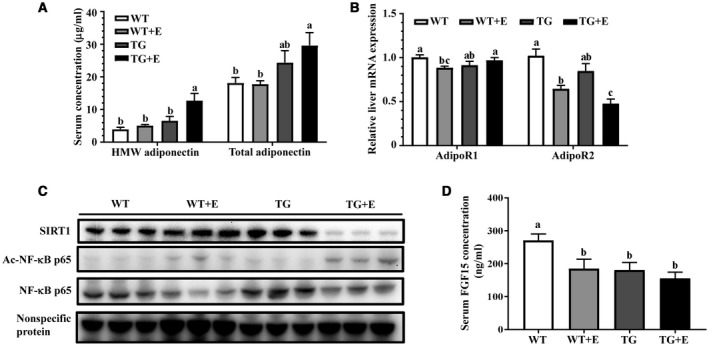
Adipose‐specific overexpression lipin‐1 in mice exacerbated the ethanol‐mediated defective hepatic adiponectin signaling. Mice were fed as described in Fig. 1. (A) Serum concentrations of HMW and total adiponectin. (B) Relative liver mRNA levels of AdipoR1 and AdipoR2. (C) Representative western blot analysis of hepatic expression levels of SIRT1, acetylated NF‐κB, and NF‐κB. (D) Serum FGF15 levels. Results are shown as means ± SEM (n = 5‐9 mice). Means without a common letter differ, P < 0.05. Abbreviations: Ac, acetylated.
The mRNA expression levels of adiponectin was not altered by ethanol feeding to the WT and Lpin1‐Tg mice compared with WT control mice, suggesting posttranscription regulation of adiponectin in the ethanol‐fed Lpin1‐Tg mice (Supporting Fig. S4A).
Lipin‐1α is one of the variants generated through splicing lipin‐1β with exon 7 skipping.4, 5, 19 Ethanol feeding to WT mice mildly but significantly increased adipose lipin‐1α mRNA abundance compared with WT control mice (Supporting Fig. S4B). Strikingly, the ethanol‐mediated increases in lipin‐1α mRNA levels were robustly augmented in the Lpin1‐Tg mice, suggesting that lipin‐1α might be involved in the adiponectin generation from the adipose of ethanol‐fed mice (Supporting Fig. S4B).
The serum levels of leptin, another adipocyte‐derived hormone, were significantly increased in the ethanol‐fed WT and Lpin1‐Tg mice, to an extent similar to their respective controls (Supporting Fig. S4C).
Although ethanol feeding significantly reduced hepatic mRNA expression levels of AdipoR2 compared with WT controls,37 the decreases in hepatic AdipoR2 were more pronounced in ethanol‐fed Lpin1‐Tg mice (Fig. 6B). Taken together, our results demonstrated that adipose‐specific lipin‐1 overexpression markedly enhanced the circulating total and HMW adiponectin levels and reduced the mRNA abundance of hepatic AdipoR2 in the ethanol‐fed mice.
Adipose‐Specific Lipin‐1 Overexpression Exacerbated the Impairment of Hepatic SIRT1 in Response to Ethanol Challenge in Mice
Disruption of SIRT1, an NAD+‐dependent class III protein deacetylase, is a central mechanism underlying ALD.33 Thus, we investigated the involvement of SIRT1 signaling in the exacerbated alcoholic steatohepatitis by adipose‐specific lipin‐1 overexpression in mice.
Protein expression levels of SIRT1 were nearly depleted by ethanol administration to Lpin1‐Tg mice compared with all other groups (Fig. 6C; Supporting Fig. S5A). Given that adiponectin up‐regulated SIRT1 through both AdipoR1/R2 in cultured hepatic cells,17, 33 the drastically decreased hepatic SIRT1 protein levels in the ethanol‐fed mice was likely due to an impaired hepatic adiponectin signaling.
The ethanol‐mediated increases in acetylated nuclear factor kappa B (NF‐κB), which is a downstream target of SIRT1, were significantly augmented in the livers of ethanol‐fed Lpin1‐Tg mice compared with the other three groups (Fig. 6C; Supporting Fig. S5B). Thus, hepatic SIRT1 enzymatic activity after ethanol administration was inhibited by adipose‐specific lipin‐1 overexpression in mice. Collectively, these results demonstrated that adipose specific overexpression of lipin‐1 severely inhibited hepatic SIRT1 protein expression and disrupted SIRT1 signaling in mice after ethanol administration.
Adipose Specific Lipin‐1 Overexpression Caused Defective Adiponectin‐FGF15 Axis in Ethanol‐Fed Mice
FGF15, an ileum‐derived hormone, and adiponectin cross‐regulated each other, and the adiponectin‐FGF15 axis was coordinately regulated by ethanol exposure in mice.14, 32, 34 The substantially increased circulating adiponectin in the ethanol‐fed Lpin1‐Tg mice led us to investigate whether ileum FGF15 was concomitantly elevated.
In comparison to the WT control mice, the serum levels of FGF15 were significantly decreased in ethanol‐fed WT mice and Lpin1‐Tg mice fed with or without ethanol (Fig. 6D). Accordingly, ileum FGF15 mRNA levels were decreased in the ethanol‐fed WT mice and in Lpin1‐Tg mice fed with or without ethanol (Supporting Fig. S6A).
The mRNA expression of hepatic fibroblast growth factor receptor 4, a receptor for FGF15,38 was reduced significantly in the ethanol‐fed WT and ethanol‐fed Lpin1‐Tg mice to the same extent as WT controls (Supporting Fig. S6A).
FGF15 is a negative regulator of hepatic Cyp7a1, the rate‐limiting enzyme for bile acid synthesis in the liver.35 The expression of hepatic Cyp7a1 was decreased drastically in both WT and Lpin1‐Tg mice after ethanol administration compared with WT controls (Supporting Fig. S6C). Consistently, hepatic bile acid contents and bile acid pool size were not significantly altered in WT and Lpin1‐Tg mice with or without ethanol feeding (Supporting Fig. S7). The unchanged hepatic bile acid in our chronic‐plus‐binge mouse model differed from an earlier study.34 Hepatic bile acid levels were significantly elevated in mice after 8 weeks of chronic ethanol feeding.34 The discrepancy might be the results of different ethanol feeding protocols used in these studies. Taken together, these results demonstrated that adipose‐specific lipin‐1 overexpression caused defective adiponectin‐FGF15 axis in ethanol‐fed mice.
Discussion
Ethanol‐mediated inhibitory effects on adipose‐specific lipin‐1 expression were associated with alcoholic fatty liver in rodents, suggesting that adipose‐specific lipin‐1 overexpression might ameliorate ethanol‐induced liver damage.17, 22 In the present study, using a Lpin1‐Tg mouse model, we unveiled an unexpected role of adipose‐specific lipin‐1 in experimental alcoholic steatohepatitis in mice. We found that adipose‐specific lipin‐1 overexpression in mice enhanced steatosis, caused hepatobiliary damage, induced mild fibrosis, and exacerbated liver damage in response to chronic‐plus‐binge ethanol challenge. Mechanistically, the exacerbated steatohepatitis in the livers of ethanol‐fed Lpin1‐Tg mice was associated with excessive iron accumulation, altered iron distribution, reduced levels of GSH and NADPH, elevated MDA levels, and impaired ferroptosis‐related gene expression along with aggravated inflammatory response, all of which indicated the presence of hepatic ferroptosis. Concurrently, adipose lipin‐1 overexpression induced defective adiponectin signaling, including impaired adiponectin‐SIRT1 signaling and disrupted adiponectin‐FGF15 axis after ethanol administration in mice. Altogether, we found that adipose lipin‐1 overexpression aggravated experimental alcoholic steatohepatitis in mice, through inducing ferroptotic liver damage (Fig. 7).
Figure 7.
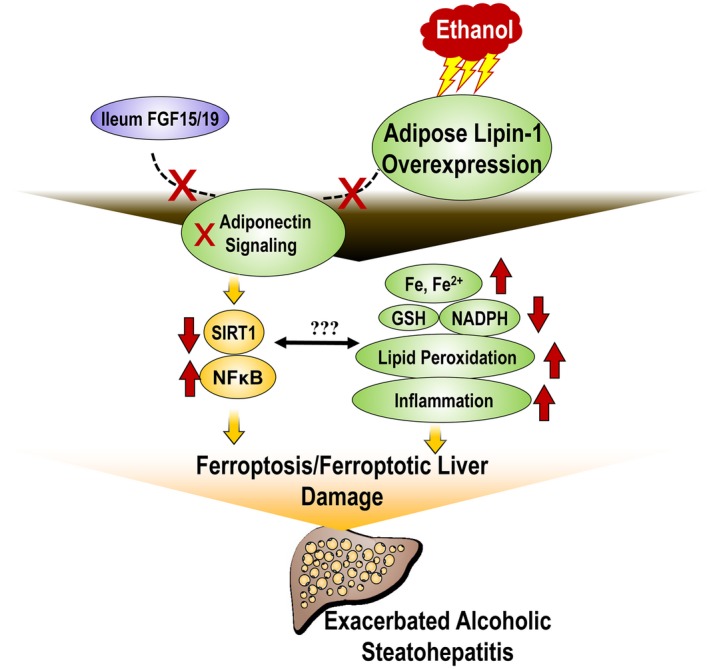
Proposed role of adipose‐specific lipin‐1 in mediating the detrimental effects of ethanol in liver
Although ethanol exposure rendered liver susceptible to the ferroptic process in Lpin1‐Tg mice, ethanol administration to WT mice was not sufficient to steer the hepatic ferroptosis signaling cascade of events and induce hepatic ferroptosis. Aside from aberrant iron metabolism and lipid peroxidation signaling, the coexistence of multiple factors is required to execute ferroptosis.23, 24, 25, 26, 27, 28 For instance, inactivation of GPX4 is essential for the execution of ferroptosis.28 It was reported that hepatic GPX4 expression was decreased in livers of adolescent rats exposed to alcohol intermittent binge drinking.36 Surprisingly, we found that the GPX4 mRNA gene and protein expression levels as well as GPX4 enzymatic activity were not significantly altered in the livers of ethanol‐fed WT or Lpin1‐Tg mice fed with or without ethanol. Depleting cysteine and repressing glutamate antiporter xCT/SCL7A11 are known to contribute to the ferroptosis process.23, 24 In this scenario, ethanol may sensitize liver to ferroptosis by depleting cysteine and repressing glutamate antiporter xCT/SCL7A11 in Lpin1‐Tg mice. The detailed mechanisms of ethanol in promoting hepatic cell death and inducing liver damage through ferroptosis are currently under investigation in our laboratory.
The hepatic ferroptotic signaling in the liver damage induced by adipose lipin‐1 overexpression in ethanol‐fed mice remains to be elucidated. We found that, despite circulating adiponectin being markedly elevated, adiponectin signaling was severely impaired in ethanol‐fed Lpin1‐Tg mice. We speculate that adipose‐specific lipin‐1‐adiponectin axis may serve as a crucial mediator in the cross‐communications among adipose, ileum, and liver in the ethanol‐fed Lpin1‐Tg mice. Enhanced adipose lipin‐1‐adiponectin axis or defective adiponectin‐SIRT1 or FGF15 signaling in ethanol‐fed Lpin1‐Tg mice may directly or indirectly promote hepatic iron accumulation, drive hepatic ferroptosis, and cause ferroptotic liver damage.
We previously found that liver‐specific deletion of SIRT1 dramatically augmented the production of hepatic MDA, a lipid peroxidation end product, and promoted hepatic inflammatory responses in ethanol‐administrated mice.20 Given that lipid peroxidation is the hallmark of ferroptosis, we speculate that drastic reduction of SIRT1 protein expression and impairment of SIRT1 signaling may enhance ferroptosis in the livers of ethanol‐fed Lpin1‐Tg mice.
Coordinated elevations of adiponectin and FGF15 protected against ethanol‐induced liver damage in mice.14, 32 However, ileum FGF15 mRNA and its circulating levels were not elevated despite substantially increased levels of circulating adiponectin in the ethanol‐fed Lpin1‐Tg mice. It is likely that the disrupted adiponectin‐FGF15 axis exacerbated liver damage in the ethanol‐fed Lpin1‐Tg mice. The hepatic ferroptotic process in ethanol‐fed Lpin1‐Tg mice may also be triggered by the aberrant adiponectin‐FGF15 axis. The ethanol‐mediated interplay between adipose lipin‐1 and adiponectin‐FGF15 axis warrants future investigation.
It remains unresolved how overexpression of adipose‐specific lipin‐1 induces adiponectin production in the adipose of ethanol‐fed mice. Lipin‐1α and lipin‐1β are two major variants generated by lipin‐1 pre‐mRNA splicing.4, 5, 19 In concordance with drastic elevation of circulating adiponectin, adipose mRNA expression of lipin‐1α, but not lipin‐1β, was robustly increased by ethanol feeding to Lpin1‐Tg mice. Accordingly, secretion of adiponectin protein into 3T3‐L1 adipocytes culture medium was markedly enhanced by overexpression of lipin‐1α, but not of lipin‐1β (M. You, unpublished observation). Therefore, we speculate that ethanol exposure may enhance the generation of lipin‐1α through inducing lipin‐1 mRNA splicing and subsequently cause adiponectin release from adipose of ethanol‐fed Lpin1‐Tg mice.
It is intriguing that hepatic SAA1 protein expression was markedly reduced in the ethanol‐fed Lpin1‐Tg mice. Although hepatocytes are the major source of SAA1, SAA1 is also capable of mediating a crosstalk among hepatocytes, macrophages, and hepatic stellate cells (HSCs) in the injured liver.39, 40 Several lines of evidence suggest that SAA1 exerts an antifibrogenic effect. In a mouse model of carbon tetrachloride–induced hepatic fibrogenesis/cirrhosis, reducing serum SAA levels increased the severity of hepatic fibrosis.39 SAA can also modulate fibrogenic responses in the liver by promoting HSC death under certain conditions.40 It is possible that the reduced hepatic SAA1 protein expression may contribute to the fibrotic damage in Lpin1‐Tg mice in response to ethanol challenge. Additional studies are required to further dissect the mechanisms of SAA1 in mediating the fibrotic effects of adipose‐specific lipin‐1 and ethanol in liver.
The exacerbated liver damage in our chronic‐plus‐binge mouse model differed from an earlier study.30 Zhou et al. demonstrated that adipose lipin‐1 overexpression protected mice against 8‐week chronic ethanol‐induced liver injury.30 Drinking patterns significantly influence the pathogenesis of ALD in humans.1, 2 The discrepancy could be the results of different ethanol feeding protocols used in these two studies (chronic plus binge versus chronic 8 weeks).
Another possible factor contributing to the discrepancy between our observations and those of Zhou et al. could be the difference in adiponectin production in ethanol‐fed Lpin1‐Tg mice. Notably, circulating adiponectin was markedly elevated in our chronic‐plus‐binge ethanol‐fed Lpin1‐Tg mice, whereas adiponectin production was not altered in Lpin1‐Tg mice after 8 weeks of chronic ethanol feeding.30 Furthermore, in contrast to the findings of Zhou et al.,30 the size of adipocyte was not changed in WT or Lpin1‐Tg mice after chronic‐plus‐binge ethanol feeding. We speculate that the differential alterations of adipocyte size and adiponectin production in these two studies could also lead to distinct responses of livers in Lpin1‐Tg mice to the ethanol challenge.
In summary, this study demonstrated that adipose‐specific lipin‐1 overexpression accelerated iron accumulation, caused lipid peroxidation, reduced GSH and GAPDH, and promoted ferroptotic liver damage in mice after ethanol administration. Our findings shed light on mechanisms underlying the alcohol‐induced liver damage. Ferroptotic signaling routes are amenable to pharmacological intervention with iron chelators (e.g., deferoxamine) and lipophilic antioxidants (e.g., ferrostatin‐1 and liproxstatin‐1).23, 24 Potential therapeutic strategies for human alcoholic steatohepatitis may be developed from either modifying adipose‐specific lipin‐1 or using ferroptosis antagonists.
Supporting information
Supported by the National Institute on Alcohol Abuse and Alcoholism (Grant/Award Numbers P50AA024333, R01AA013623, and R01AA015951).
Potential conflict of interest: Nothing to report.
References
Author names in bold designate shared co‐first authorship.
- 1. Gao B, Bataller R. Alcoholic liver disease: pathogenesis and new therapeutic targets. Gastroenterology 2011;141:1572‐1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Greuter T, Malhi H, Gores GJ, Shah VH. Therapeutic opportunities for alcoholic steatohepatitis and nonalcoholic steatohepatitis: exploiting similarities and differences in pathogenesis. JCI Insight 2017;2:e95354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Péterfy M, Phan J, Xu P, Reue K. Lipodystrophy in the fld mouse results from mutation of a new gene encoding a nuclear protein, lipin. Nat Genet 2001;27:121‐124. [DOI] [PubMed] [Google Scholar]
- 4. Wang H, Airola MV, Reue K. How lipid droplets “TAG” along: glycerolipid synthetic enzymes and lipid storage. Biochim Biophys Acta 2017;1862:1131‐1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Csaki LS, Dwyer JR, Fong LG, Tontonoz P, Young SG, Reue K. Lipins, lipinopathies, and the modulation of cellular lipid storage and signaling. Prog Lipid Res 2013;52:305‐316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Csaki LS, Dwyer JR, Li X, Nguyen MH, Dewald J, Brindley DN, et al. Lipin‐1 and lipin‐3 together determine adiposity in vivo. Mol Metab 2013;3:145‐154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhang P, Takeuchi K, Csaki LS, Reue K. Lipin‐1 phosphatidic phosphatase activity modulates phosphatidate levels to promote peroxisome proliferator‐activated receptor γ (PPARγ) gene expression during adipogenesis. J Biol Chem 2012;287:3485‐3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Phan J, Péterfy M, Reue K. Lipin expression preceding peroxisome proliferator‐activated receptor‐gamma is critical for adipogenesis in vivo and in vitro. J Biol Chem 2004;279:29558‐29564. [DOI] [PubMed] [Google Scholar]
- 9. Mitra MS, Chen Z, Ren H, Harris TE, Chambers KT, Hall AM, et al. Mice with an adipocyte‐specific lipin 1 separation‐of‐function allele reveal unexpected roles for phosphatidic acid in metabolic regulation. Proc Natl Acad Sci U S A 2013;110:642‐647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Xu J, Lee WN, Phan J, Saad MF, Reue K, Kurland IJ. Lipin deficiency impairs diurnal metabolic fuel switching. Diabetes 2006;55:3429‐3438. [DOI] [PubMed] [Google Scholar]
- 11. Phan J, Reue K. Lipin, a lipodystrophy and obesity gene. Cell Metab 2005;1:73‐83. [DOI] [PubMed] [Google Scholar]
- 12. Chang YC, Chang LY, Chang TJ, Jiang YD, Lee KC, Kuo SS, et al. The associations of LPIN1 gene expression in adipose tissue with metabolic phenotypes in the Chinese population. Obesity (Silver Spring) 2010;18:7‐12. [DOI] [PubMed] [Google Scholar]
- 13. Mlinar B, Pfeifer M, Vrtacnik‐Bokal E, Jensterle M, Marc J. Decreased lipin 1 beta expression in visceral adipose tissue is associated with insulin resistance in polycystic ovary syndrome. Eur J Endocrinol 2008;159:833‐839. [DOI] [PubMed] [Google Scholar]
- 14. Wang J, Kim C, Jogasuria A, Han Y, Hu X, Wu J, et al. Myeloid cell‐specific lipin‐1 deficiency stimulates endocrine adiponectin‐FGF15 axis and ameliorates ethanol‐induced liver injury in mice. Sci Rep 2016;6:34117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. González CR, Novelle MG, Caminos JE, Vázquez MJ, Luque RM, López M, et al. Regulation of lipin1 by nutritional status, adiponectin, sex and pituitary function in rat white adipose tissue. Physiol Behav 2012;105:777‐783. [DOI] [PubMed] [Google Scholar]
- 16. Jiang Z, Zhou J, Zhou D, Zhu Z, Sun L, Nanji AA. The adiponectin‐SIRT1‐AMPK pathway in alcoholic fatty liver disease in the rat. Alcohol Clin Exp Res 2015;39:424‐433. [DOI] [PubMed] [Google Scholar]
- 17. Shen Z, Liang X, Rogers CQ, Rideout D, You M. Involvement of adiponectin‐SIRT1‐AMPK signaling in the protective action of rosiglitazone against alcoholic fatty liver in mice. Am J Physiol Gastrointest Liver Physiol 2010;298:G364‐G374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hu M, Wang F, Li X, Rogers CQ, Liang X, Finck BN, et al. Regulation of hepatic lipin‐1 by ethanol: role of AMP‐activated protein kinase/sterol regulatory element‐binding protein 1 signaling in mice. Hepatology 2012;55:437‐446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hu M, Yin H, Mitra MS, Liang X, Ajmo JM, Nadra K, et al. Hepatic‐specific lipin‐1 deficiency exacerbates experimental alcohol‐induced steatohepatitis in mice. Hepatology 2013;58:1953‐1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yin H, Hu M, Liang X, Ajmo JM, Li X, Bataller R, et al. Deletion of SIRT1 from hepatocytes in mice disrupts lipin‐1 signaling and aggravates alcoholic fatty liver. Gastroenterology 2014;146:801‐811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chen H, Shen F, Sherban A, Nocon A, Li Y, Wang H, et al. DEP domain‐containing mTOR‐interacting protein suppresses lipogenesis and ameliorates hepatic steatosis and acute‐on‐chronic liver injury in alcoholic liver disease. Hepatology 2018;68:496‐514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang W, Zhong W, Sun X, Sun Q, Tan X, Li Q, et al. Visceral white adipose tissue is susceptible to alcoholinduced lipodystrophy in rats: role of acetaldehyde. Alcohol Clin Exp Res 2015;39:416‐423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell 2017;171:273‐285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cao JY, Dixon SJ. Mechanisms of ferroptosis. Cell Mol Life Sci 2016;73:2195‐2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang H, An P, Xie E, Wu Q, Fang X, Gao H, et al. Characterization of ferroptosis in murine models of hemochromatosis. Hepatology 2017;66:449‐465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sui M, Jiang X, Chen J, Yang H, Zhu Y. Magnesium isoglycyrrhizinate ameliorates liver fibrosis and hepatic stellate cell activation by regulating ferroptosis signaling pathway. Biomed Pharmacother 2018;106:125‐133. [DOI] [PubMed] [Google Scholar]
- 27. Tuo QZ, Lei P, Jackman KA, Li XL, Xiong H, Li XL, et al. Tau‐mediated iron export prevents ferroptotic damage after ischemic stroke. Mol Psychiatry 2017;22:1520‐1530. [DOI] [PubMed] [Google Scholar]
- 28. Wenzel SE, Tyurina YY, Zhao J, St Croix CM, Dar HH, Mao G, et al. PEBP1 wardens ferroptosis by enabling lipoxygenase generation of lipid death signals. Cell 2017;171(628–641):e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kohgo Y, Ohtake T, Ikuta K, Suzuki Y, Torimoto Y, Kato J. Dysregulation of systemic iron metabolism in alcoholic liver diseases. J Gastroenterol Hepatol 2008;23(Suppl 1):S78‐S81. [DOI] [PubMed] [Google Scholar]
- 30. Zhang W, Zhong W, Sun Q, Sun X, Zhou Z. Adipose specific lipin1 overexpression in mice protects against alcohol‐induced liver injury. Sci Rep 2018;8:408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bertola A, Mathews S, Ki SH, Wang H, Gao B. Mouse model of chronic and binge ethanol feeding (the NIAAA model). Nat Protoc 2013;8:627‐637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hu X, Jogasuria A, Wang J, Kim C, Han Y, Shen H, et al. MitoNEET deficiency alleviates experimental alcoholic steatohepatitis in mice by stimulating endocrine adiponectin‐Fgf15 axis. J Biol Chem 2016;291:22482‐22495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. You M, Jogasuria A, Taylor C, Wu J. Sirtuin 1 signaling and alcoholic fatty liver disease. Hepatobiliary Surg Nutr 2015;4:88‐100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hartmann P, Hochrath K, Horvath A, Chen P, Seebauer CT, Llorente C, et al. Modulation of the intestinal bile acid/farnesoid X receptor/fibroblast growth factor 15 axis improves alcoholic liver disease in mice. Hepatology 2018;67:2150‐2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Inagaki T, Choi M, Moschetta A, Peng L, Cummins CL, McDonald JG, et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab 2005;2:217‐225. [DOI] [PubMed] [Google Scholar]
- 36. Ojeda ML, Carreras O, Sobrino P, Murillo ML, Nogales F. Biological implications of selenium in adolescent rats exposed to binge drinking: oxidative, immunologic and apoptotic balance. Toxicol Appl Pharmacol 2017;329:165‐172. [DOI] [PubMed] [Google Scholar]
- 37. Liang X, Hu M, Rogers CQ, Shen Z, You M. Role of SIRT1‐FoxO1 signaling in dietary saturated fat‐dependent upregulation of liver adiponectin receptor 2 in ethanol‐administered mice. Antioxid Redox Signal 2011;15:425‐435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ge H, Zhang J, Gong Y, Gupte J, Ye J, Weiszmann J, et al. Fibroblast growth factor receptor 4 (FGFR4) deficiency improves insulin resistance and glucose metabolism under diet‐induced obesity conditions. J Biol Chem 2014;289:30470‐30480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang Y, Huang H, Sun R, Chen B, Han F, Li Q, et al. Serum amyloid a induces M2b‐like macrophage polarization during liver inflammation. Oncotarget 2017;8:109238‐109246. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 40. Siegmund SV, Schlosser M, Schildberg FA, Seki E, De Minicis S, Uchinami H, et al. Serum amyloid a induces inflammation, proliferation and cell death in activated hepatic stellate cells. PLoS ONE 2016;11:e0150893. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials