

Summary
Aims
Glycogen synthase kinase 3β (GSK‐3β) is activated following hypoxic‐ischemic (HI) brain injury. TDZD‐8 is a specific GSK‐3β inhibitor. Currently, the impact of inhibiting GSK‐3β in neonatal HI injury is unknown. We aimed to investigate the effect of TDZD‐8 following neonatal HI brain injury.
Methods
Unilateral common carotid artery ligation followed by hypoxia was used to induce HI injury in postnatal day 7 mouse pups pretreated with TDZD‐8 or vehicle. The infarct volume, whole‐brain imaging, Nissl staining, and behavioral tests were used to evaluate the protective effect of TDZD‐8 on the neonatal brain and assess functional recovery after injury. Western blot was used to evaluate protein levels of phosphorylated protein kinase B (Akt), GSK‐3β, and cleaved caspase‐3. Protein levels of cleaved caspase‐3, neuronal marker, and glial fibrillary acidic protein were detected through immunohistochemistry.
Results
Pretreatment with TDZD‐8 significantly reduced brain damage and improved neurobehavioral outcomes following HI injury. TDZD‐8 reversed the reduction of phosphorylated Akt and GSK‐3β, and the activation of caspase‐3 induced by hypoxia‐ischemia. In addition, TDZD‐8 suppressed apoptotic cell death and reduced reactive astrogliosis.
Conclusion
TDZD‐8 has the therapeutic potential for hypoxic‐ischemic brain injury in neonates. The neuroprotective effect of TDZD‐8 appears to be mediated through its antiapoptotic activity and by reducing astrogliosis.
Keywords: GSK‐3β; neonatal hypoxia‐ischemia, neuroprotection; TDZD‐8
1. Introduction
Glycogen synthase kinase 3β (GSK‐3β) is a constitutively active serine/threonine kinase that is expressed in all tissues. In the central nervous system, GSK‐3β is highly expressed in brain regions including the cerebral cortex, hippocampus, and cerebellum.1, 2, 3, 4 Under physiological conditions, GSK‐3β plays important roles in neuronal polarization5, 6, neurogenesis7, 8 and axonal growth.9, 10 Under pathological conditions, overactivation of GSK‐3β is involved in pro‐apoptosis of neurons11 and dysregulation of this kinase has a devastating effect on neurodevelopment.12 GSK‐3β has been reported to be involved in stroke pathology. In transient middle cerebral artery occlusion models, GSK‐3β expression levels increased following 90 minutes of ischemia and 4 hours of reperfusion.13 Selective inhibition of the GSK‐3β isoform using pharmacological tools (ie, TDZD‐8, Chir025) resulted in reduction of cerebral infarction, oxidative stress, and apoptosis, suggesting the GSK‐3β isoform as a drug target for neuroprotection.14, 15 Inhibition of GSK‐3β is thought to mediate neuroprotection against stroke through multiple mechanisms, including regulation by PI3K/Akt prosurvival signaling, interaction with MAPK, and nuclear factor‐kappa B signaling pathways.14, 15, 16, 17, 18
Neonatal hypoxic‐ischemic (HI) brain injury and its related disease hypoxic‐ischemic encephalopathy (HIE) are global brain injuries that impact three to five per 1000 live births. Neonatal HI results in a mortality rate of 15%‐20%.19 In survivors, approximately 25% suffer from lifelong neurological deficits including mental retardation and epilepsy, making neonatal HI a leading cause of neurological impairment in children.19 Hypothermia has now emerged as an effective therapy of neonatal HI.20, 21 Given the incomplete protection from HI afforded by hypothermia, other neuroprotective strategies are currently in clinical trials including prophylactic barbiturates,22 erythropoietin,23 and allopurinol.24 Recent studies showed that inhibition of GSK‐3β by SB216763 and granulocyte‐colony stimulating factor elicited neuroprotection in neonatal rodents.18, 25 We have recently shown that HI‐induced brain injury in the neonatal mouse models26, 27, 28 is mediated via caspase‐3‐dependent cell death pathway. Inhibiting caspase‐3 resulted in neuroprotection in several neonatal hypoxic‐ischemic injury models.29, 30, 31 The Akt/GSK‐3β pathway is a major signaling that is known to tightly regulate caspase‐3 activation.11, 26, 27, 28 Thus, we hypothesized that inhibition of GSK‐3β would ameliorate neonatal hypoxic‐ischemic brain injury.
A newly identified non‐ATP competitive GSK‐3β inhibitor, 2‐Methyl‐4‐(phenylmethyl)‐1,2,4‐thiadiazolidine‐3,5‐dione (TDZD‐8), has been shown to have neuroprotective, antiinflammatory, and antioxidative effects.32, 33, 34 Prophylactic administration of TDZD‐8 in adult rats increased phosphorylation of GSK‐3β (Ser9) site and reduced brain damage following cerebral ischemia/reperfusion injury via inhibition of the apoptotic pathway and reactive oxygen species (ROS) production.15 GSK‐3β is involved in neuronal development processes, but it is unknown whether GSK‐3β inhibitor TDZD‐8 may play a protective role in hypoxic stress in immature animals. In this study, we report that TDZD‐8 prevented the reduction of p‐GSK‐3β and brain injury in the HI model, suggesting that phosphorylation of GSK‐3β at the Ser9 site plays a crucial role in neuroprotection in the neonatal stage.
2. Material and Methods
2.1. Animals
All protocols were carried out in compliance with the Canadian Council on Animal Care (CCAC guidelines), and all animal experimental procedures were approved by the local Animal Care and Use Program Committee (Office of Research Ethics at the University of Toronto). Timed‐pregnant CD1 mice were purchased from Charles River Laboratories (Sherbrooke, QC, Canada) and housed at an ambient temperature of 23±1°C and a 12‐hour light/dark cycle with food and water fed ad libitum. Pups were timed as postnatal day 0, P0, on their day of birth, and P7 pups of either sex were used for experiments. A total of 104 pups were used for this study. Body weight was used as a general indicator of health, and any pups below 4 grams at P7 were not used.
2.2. Reagents and drugs
Cresyl violet, 2,3,5‐triphenyl‐2H‐tetrazolium chloride (T8877, TTC) and Dimethyl sulfoxide (D2650, DMSO) were purchased from Sigma‐Aldrich (St. Louis, MO, USA). TDZD‐8 was supplied by Centro de Investigaciones Biologicas‐CSIC (CIB‐CSIC; Madrid, Spain).35
2.3. Drug administration
Pups were randomly selected to receive TDZD‐8 or vehicle treatment. TDZD‐8 (5 mg/kg) or vehicle control containing 5% DMSO and 5% Tween‐80 (P‐8074) in 0.9% saline36, 37 was administered to the pups 20 minutes prior to ischemia induction. These compounds were administered intraperitoneally (i.p.) with a volume to body weight injection ratio of 20 μL/g. Sham‐operated animals were injected with vehicle.
2.4. Hypoxic‐ischemic brain injury model
Hypoxic‐ischemic brain injury was induced in P7 pups as previously described.26, 27, 28 Briefly, anesthesia was induced and maintained with 3% and 1.5% inhaled isoflurane in oxygen, respectively. The right common carotid artery was exposed and ligated using a bipolar electrocoagulation device (Summit Hill Laboratories, Tinton Falls, NJ, USA). Body temperature was maintained during the surgery with a heat blanket. Following the surgery, pups were placed in a recovery cage under a heat lamp for 5 minutes until they regained consciousness and were returned to their home cage for 90 minutes. After the recovery period, pups were placed in an airtight hypoxic chamber (Biospherix, NY, USA) fed with humidified 7.5% oxygen in mixture with 92.5% nitrogen gas for 60 minutes. The chamber temperature was maintained at 37°C using a homoeothermic blanket control unit (MA, USA). After hypoxia, the pups were recovered under a heat lamp for 5 minutes and were returned to their home cage. Sham animals were anesthetized, and their common carotid artery was exposed and separated but no ligation or hypoxia took place.
2.5. Whole‐brain imaging, histological assessments, and infarct volume measurement
TTC staining/Infarct Volume Measurement26, 27, 28: Twenty‐four hours after HI injury, whole brains were removed from the pups and sectioned coronally into ~1‐mm slices used for TTC staining. Slices were stained with 1% TTC and placed in a dark incubator maintained at 37°C for 20 minutes.
Whole‐Brain Imaging/Nissl Staining: Seven days after HI injury, whole brains were removed, fixed, imaged, and sectioned coronally into ~100‐μm slices used for Nissl staining (1% cresyl violet).
The corrected infarct volumes for TTC and whole‐brain imaging were calculated as follows: Corrected infarct volume (%)=(contralateral hemisphere volume−ipsilateral hemisphere+infarct volume)/contralateral hemisphere volume×100%.28, 38, 39 The hemispheric and infarct volumes for the whole brains were traced and then quantified using the ImageJ software (National institute of Health, Bethesda, MD, USA). For TTC quantification, the hemispheric/infarct areas of each brain slice was traced and quantified using the same software.
2.6. Neurobehavioral assessments
2.6.1. Geotaxis reflex
This reflex assessed the animal's balance and proprioception.26, 27, 28, 40 Neonatal mice were positioned on a 45° inclined wooden board such that the animal's head was pointed downward, and the time latency for them to rotate 90° (either left or right) was determined. The maximum time allocated for each trial of behavioral observation was 20 seconds, and any trial past the maximum time was scored as 20 seconds.40
2.6.2. Cliff avoidance reflex
This test was used to assess the animal's capacities in both locomotion and external sensation.26, 27, 28, 40 Mouse pups were first placed on the wooden board such that their front paws were in an overhanging position relative to the board. The amount of time taken for the mouse to successfully evade the cliff by turning the body 90° (either left or right) was determined. A maximum time of 20 seconds was used in a similar manner as compared to the geotaxis reflex test.40
2.7. Western blot
Western blot procedures were performed as previously described.26, 27, 28 Twenty‐four hours after HI, the ipsilateral hemispheres were removed and frozen in dry ice. To study the developmental expression of GSK‐3β, protein was extracted from cortex. The brain samples were homogenized in RIPA buffer with proteinase and phosphatase inhibitors. Samples of the mouse brain (30 μg) were separated on a 10% SDS‐PAGE gel that was transferred to a nitrocellulose membrane (350 mA, 90 minutes). Blots were blocked with 5% nonfat dry milk in Tris‐buffered saline, and incubated with primary and secondary antibodies at 4°C overnight and room temperature, respectively (anticleaved caspase‐3, #9664S, 1:1000; antiphospho‐Akt (#9271S, Ser473, 1:1000); anti‐Akt (#9272S, 1:1000); antiphospho‐GSK‐3β (#9323S, Ser9, 1:1000); anti‐GSK‐3β (#9832S, 1:1000); anti‐GAPDH (#2118S, 1:10,000). All the primary antibodies were from Cell Signaling Technology (Danvers, MA, USA). Protein signals of interest were visualized using enhanced chemiluminescent reagents (PerkinElmer, Boston, MA, USA) and analyzed by exposure to film (HyBlot CL, Metuchen, NJ, USA).
2.8. Immunohistochemistry and confocal imaging
Brain samples were collected 7 days after HI at P14 and fixed in 4% paraformaldehyde/30% sucrose solution at 4°C overnight. Brains were coronally sectioned into ~50‐μm slices using a vibratome (Tissue Sectioning System Microtome Vibratome, HuiYou, China) and immunohistochemically stained as previously described.28 In brief, samples were probed with mouse antineuronal nuclei (NeuN) antibody (MAB377, 1:500; Chemicon, Temecula, CA, USA), antiglial fibrillary acidic protein (GFAP) (ab7260, 1:1000; Abcam, Cambridge, MA, USA), anticaspase‐3 (#9661, 1:200; Cell Signaling Technology) antibodies overnight at 4°C, and labeled with DAPI (1 μg/mL; Cell Signaling Technology). Next, the sections were incubated with secondary antibodies Alexa 488 and 568 (#835724, #632115, 1:200; Cell Signaling Technology) for 1 hour at room temperature and mounted on glass coverslips with ProLong Gold antifade reagent (P36930; Thermo Fisher Scientific, Burlington, Canada). Confocal laser scanning microscope (LSM700 Zeiss; Oberkochen, Germany) was used to image the immunolabeled brain slices. Three brains per treatment group were collected, and three to five coronal slices per brain were imaged. All of the treatment groups were imaged at the same laser settings with 40× lens. Cortical areas directly adjacent to the injury site were imaged, and the number of cells per field was quantified using Cell Counter plugin for ImageJ software (National institute of Health, Bethesda, MD, USA).
2.9. In situ hybridization and 3D imaging
In situ hybridization (ISH) data were collected from Allen Brain Atlas—Developing Mouse Brain website (http://developingmouse.brain-map.org/). Data were taken from C57BL/6J mice at developmental time points: E18.5, P4, P14, and P28; 3D imaging was performed using the Allen Developing Mouse Brain Atlas—Brain Explorer 2 software.41 Gene expression data measured as intensity level were plotted as a series of grid points for each brain slices superimposed on each other.
2.10. Data analysis
All statistical analyses were performed using statistical software SigmaPlot (Systat Software, San Jose, CA, USA). Data are presented as mean±SEM. Student's t test was used to determine the statistical difference between two groups. Multiple‐group statistical analysis was performed using one‐way ANOVA test followed by Fisher LSD Method for multiple pairwise comparisons. A P‐value of <.05 (P<.05) was considered statistically significant. All experiments were conducted in a blinded manner; experimenters did not know the treatment conditions. All experiments were repeated at least three times.
3. Results
3.1. TDZD‐8 pretreatment attenuates infarct volume following hypoxic‐ischemic brain injury
First, we investigated whether TDZD‐8 can reduce brain damage using a hypoxic‐ischemic brain injury model in neonatal mice. Subsequent experimental procedures and timeline of experiments are shown in Figure 1A. Twenty‐four hours after HI, brains from the pups were coronally sectioned and stained with TTC and the damaged areas were shown in white (Figure 1B). The infarct volume for the vehicle‐treated HI group was 44.7±3.11% (n=9). Infarct volume was significantly reduced in the TDZD‐8‐treated HI group (33.7±2.90%, n=10, *P<.05) (Figure 1C). These results indicate that TDZD‐8 pretreatment attenuates brain damage induced by HI in neonatal mice.
Figure 1.
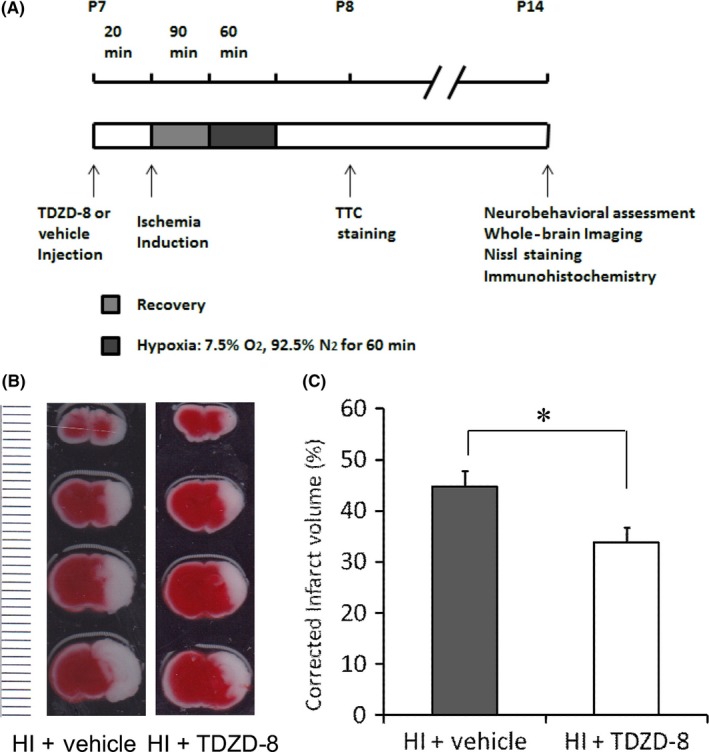
TDZD‐8 pretreatment reduced infarct volume 24 hours following neonatal hypoxic‐ischemic brain injury. (A) Timeline of neonatal hypoxic‐ischemic injury and experimental procedures. Seven‐day‐old pups (P7) were randomly selected to be injected with either TDZD‐8 or vehicle solution 20 minutes prior to ischemia induction. This was followed by 90 minutes of recovery and 60 minutes of hypoxia with 7.5% O2 and 92.5% N2. TTC staining was performed 24 hours after HI (P8). Neurobehavioral assessment, whole‐brain imaging, Nissl staining, and immunohistochemistry were performed 7 days after the HI (P14). (B) Effect of TDZD‐8 pretreatment on infarct volume. Representative TTC images of coronal brain slices 24 hours after HI injury show brain damage areas in white color (the dotted line on the left of the image represented “mm” in size for each line). (C) Summary of brain infarction volumes of TTC images. Pretreatment with TDZD‐8 significantly reduced the infarct volume compared to the vehicle‐treated group (*P<.05, Student's t test); HI+vehicle group, n=9; HI+TDZD‐8 group, n=20
Whole‐brain imaging showed that the vehicle‐treated HI brains had a considerable portion of the brain missing indicating severe brain injury from HI, while the sham group had fully intact brains. Brains from the TDZD‐8‐treated HI group were less morphologically damaged compared to that of the vehicle‐treated brains (Figure 2A). Subsequent staining with cresyl violet showed that the TDZD‐8‐treated group sustained less brain damage compared to the vehicle‐treated HI group (Figure 2A). Analysis of the liquefaction corrected infarction volumes of the whole brains revealed that hypoxic‐ischemic injury induced brain damage in the vehicle‐treated group (53.2±5.10%, n=18) and that injury was significantly attenuated in the TDZD‐8‐treated group (36.1±3.84%, n=20; *P<.01) (Figure 2B). These results demonstrated that TDZD‐8 pretreatment reduces brain damage induced by HI 7 days after injury in neonatal mice.
Figure 2.
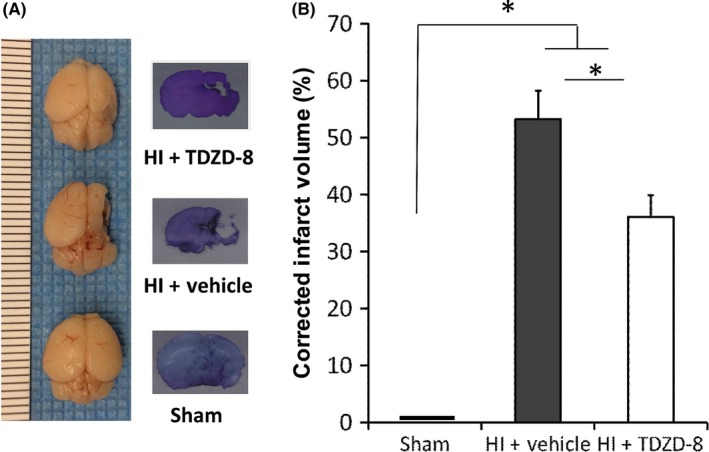
TDZD‐8 pretreatment reduced infarct volume 7 days following neonatal hypoxic‐ischemic brain injury. (A) Effect of TDZD‐8 on brain damage. Representative whole‐brain images and Nissl staining taken 7 days after HI injury are shown (the dotted line on the left of the whole‐brain image represented “mm” in size for each line). (B) Analysis of brain damage for whole‐brain images. Hypoxic‐ischemic injury induced significant brain damage in the vehicle‐treated group compared to the sham group. Pretreatment with TDZD‐8 significantly reduced the infarct volume and improved whole‐brain morphology compared to the vehicle‐treated group 7 days after the HI injury (*P<.01 vs sham group or vehicle group; One‐way ANOVA followed by Fisher LSD Method); sham group, n=12; HI+vehicle group, n=18; HI+TDZD‐8 group, n=20
3.2. TDZD‐8 improves neurobehavioral outcome following hypoxic‐ischemic brain injury
We next addressed whether the reduction in brain damage observed in the TDZD‐8‐treated HI group led to improved sensorimotor function in this model. Seven days after HI, we assessed the neurobehavioral function of mice using the cliff avoidance test and geotaxis reflex.
3.2.1. Cliff avoidance test
The vehicle control group (7 days: 6.14±1.63 seconds) displayed a significantly longer latency to complete the test in comparison with the sham group (7 days: 2.03±0.15 seconds). TDZD‐8 pretreatment (7 days: 3.02±0.45 seconds) significantly reduced the latency to complete the test in comparison with the vehicle group (Figure 3A, *P<.05). These results indicated that TDZD‐8 improves the locomotion and reduces maladaptive behavior response of the mice following HI.
Figure 3.
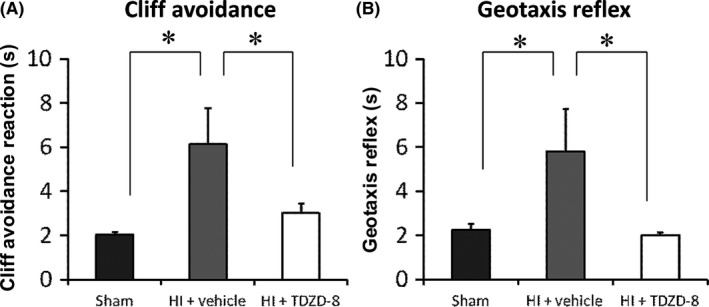
TDZD‐8 pretreatment improved neurobehavioral outcome assessed at 7 days following neonatal hypoxic‐ischemic brain injury. (A) Cliff avoidance test. (B) Geotaxis Reflex test. TDZD‐8 pretreatment significantly improved cliff avoidance and geotaxis test scores in comparison with the vehicle control group but had no difference compared to the sham group (*P<.05 vs sham group or vehicle group; One‐way ANOVA followed by Fisher LSD Method); sham group, n=18; HI+vehicle group, n=12; HI+TDZD‐8 group, n=11
3.2.2. Geotaxis reflex test
Mice in the vehicle‐treated HI group (7 days: 5.80±1.96 seconds) took significantly longer to complete the geotaxis test in comparison with the sham group (7 days: 2.26±0.26 seconds). Mice that were pretreated with TDZD‐8 (7 days: 2.02±0.14 seconds) had significantly improved geotaxis test scores compared to the vehicle‐treated group (Figure 3B, *P<.05). This showed that TDZD‐8 improves balance and proprioception in these mice following HI.
3.3. Developmental expression of GSK‐3β and Akt
To understand the role of GSK‐3β in neuronal death following hypoxic‐ischemic injury in neonatal mice, we studied the physiological gene expression of the kinase during development. ISH data obtained from Allen Brain Atlas: Developing Mouse Brain41 revealed that GSK‐3β gene expression is localized in the mouse cortex and hippocampus during early and late stages of brain development measured at E18.5, P4, P14, and P28 (Figure 4A). Importantly, the cortex and hippocampus are brain areas most vulnerable to hypoxic‐ischemic injury in rodents.42 While gene expression of GSK‐3β was similar in E18.5 and P14 but lower at P28 times points, highest intensity levels were seen at P4 (Figure 4A).
Figure 4.
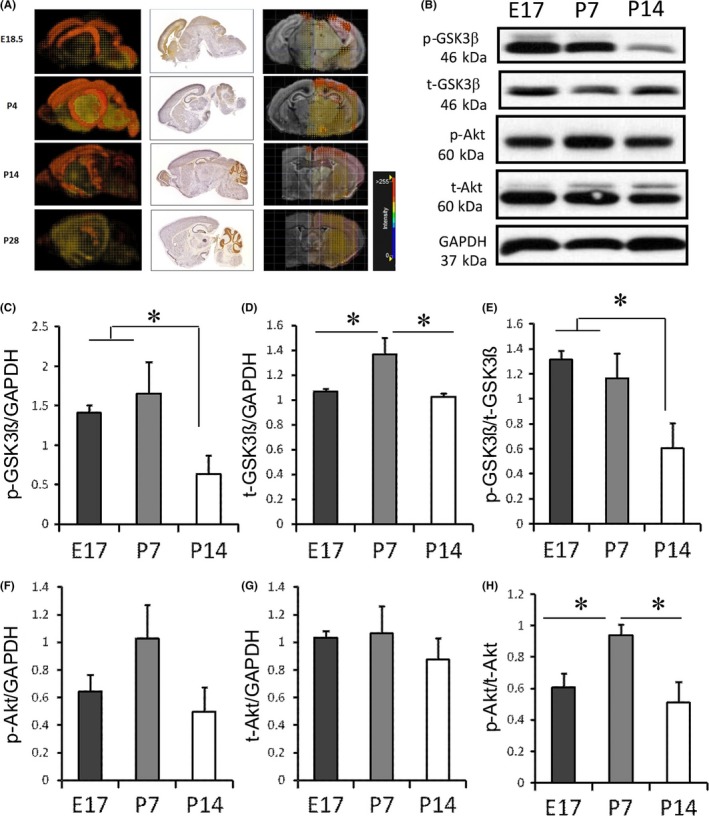
Developmental changes in GSK‐3β and Akt levels. (A) 3D gene expression patterns of GSK‐3β in coronal and sagittal slice in C57BL mice were determined at E18.5, P4, P14, and P28, using ISH. Images were obtained from Website: © 2015 Allen Institute for Brain Science. Allen Developing Mouse Brain Atlas [Internet]. Available from: http://developingmouse.brain-map.org. (B) Western blots showing protein levels of GSK‐3β and Akt at E17, P7, and P14. Analysis of band intensity showing the ratio of (C) p‐GSK3β to GAPDH, (D) t‐GSK3β to GAPDH, and (E) p‐GSK3β to t‐GSK3β. Analysis of band intensity showing the ratio of (F) p‐Akt to GAPDH, (G) t‐Akt to GAPDH, and (H) p‐Akt to t‐Akt. (*P<.05 vs sham group or TDZD‐8 treatment group; One‐way ANOVA followed by Fisher LSD Method). E17 group, n=5; P7 group, n=4; P14 group, n=5
Next, we studied the protein expression levels of total GSK‐3β at E17, P7, and P14 time points (Figure 4B‐E). The level of phosphorylated GSK‐3β (p‐GSK‐3β), the inactive form of the protein, was labeled with an antibody binding to the Serine 9. As shown in Figure 4C, the p‐GSK‐3β level was significantly higher at E17 (1.41±0.10) and P7 (1.65±0.40) than at P14 (0.64±0.23). Also, we found that total GSK‐3β protein levels was highest at P7 (1.36±0.14) compared to E17 (1.07±0.02) and P14 (1.02±0.03) time points (Figure 4D); there was no significant difference between E17 and P14. The p‐GSK‐3β to t‐GSK‐3β ratio was compatible between E17 (1.32±0.07) and P7 (1.16±0.20) animals, but decreased significantly in P14 (0.61±0.20) (Figure 4E), indicating that GSK‐3β activity was low from prenatal to postnatal day 7 but increased 14 days following birth. We also measured the changes in Akt expression levels at these time points (Figure 4B, F‐H). The p‐Akt to t‐Akt ratio was significantly higher at age P7 (0.94±0.07) compared to E17 (0.61±0.09) and P14 (0.51±0.13) (P<.05; n=5). The p‐Akt to t‐Akt ratios between E17 and P14 were compatible (Figure 4H).
3.4. TDZD‐8 pretreatment increases Serine‐9‐phosphorylated GSK‐3β (p‐GSK‐3β) and phosphorylated Akt (p‐Akt) levels after hypoxic‐ischemic brain injury
Next, we asked whether TDZD‐8 treatment would affect the level of p‐GSK‐3β after HI injury. We found that the ratio of p‐GSK‐3β/GSK‐3β was significantly increased in the TDZD‐8‐treated HI group (0.69±0.02) compared to the vehicle‐treated HI group (0.36±0.13, P<.05, Figure 5A, B). Our results showed that p‐Akt/t‐Akt expression ratio was significantly increased after pretreatment with TDZD‐8 (0.85±0.11) compared to the vehicle‐treated group (0.47±0.12, P<.05, Figure 5C). This finding supports the hypothesis that TDZD‐8 promotes prosurvival signaling by directly or indirectly activating p‐Akt following the HI injury.
Figure 5.
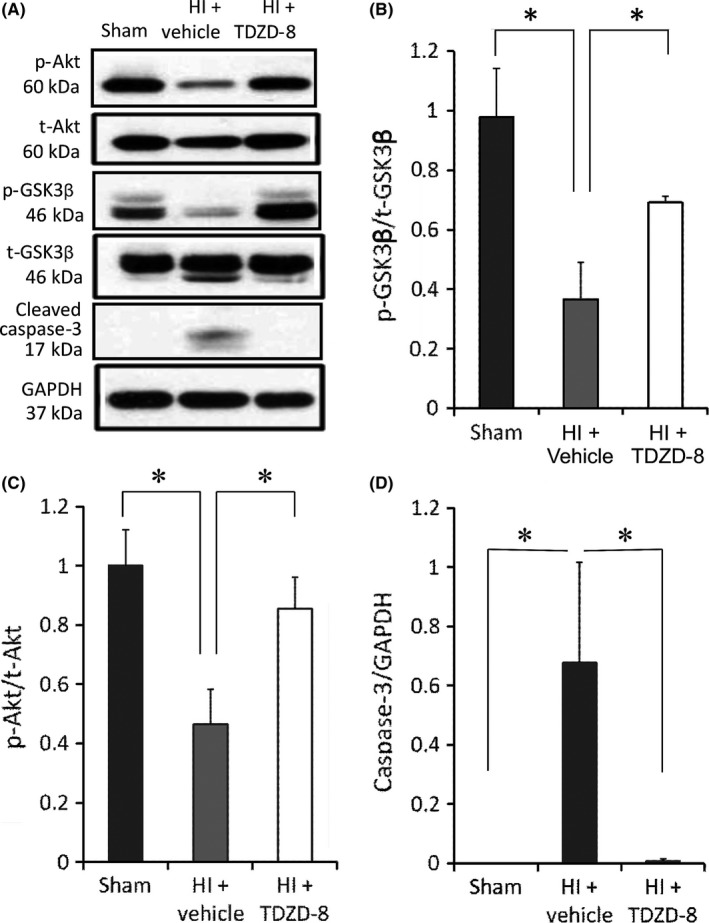
The effects of TDZD‐8 on the expression levels of cell survival and apoptotic markers following HI. Samples were collected 24 hours after HI from the ipsilateral brain hemispheres. (A) Representative Western blot gel showing protein expression levels from the sham, vehicle‐treated HI, and TDZD‐8‐treated HI groups. Analysis of band intensity showing the ratio of (B) p‐GSK‐3β to t‐GSK‐3β, (C) p‐Akt to t‐Akt, and (D) cleaved caspase‐3 to GAPDH. (*P<.05 vs sham group or TDZD‐8 treatment group; One‐way ANOVA followed by Fisher LSD Method). Sham group, n=5; HI+vehicle group, n=5; HI+TDZD‐8 group, n=5
3.5. TDZD‐8 pretreatment suppresses apoptotic signaling after hypoxic‐ischemic brain injury
We have previously shown that mitochondrial‐mediated apoptosis is one of the main mechanisms for cell death following neonatal hypoxic‐ischemic brain injury.26, 27, 28 Here, we addressed whether TDZD‐8 rescues neuronal cells from apoptosis. Our results showed that the expression levels of cleaved caspase‐3 were significantly reduced in the TDZD‐8 treatment group (0.01±0.01) compared to the vehicle‐treated group (0.68±0.34, *P<.05, Figure 5D). Immunohistochemistry results also reveal a significant decrease in DAPI‐positive cells containing cleaved caspase‐3 marker in the ipsilateral hemispheres of the TDZD‐8 treatment group (39.4±3.93) compared to the ipsilateral hemispheres of the vehicle‐treated group(68.0±7.13); the sham group had the lowest levels of cleaved caspase‐3 (Figure 6A, B).
Figure 6.
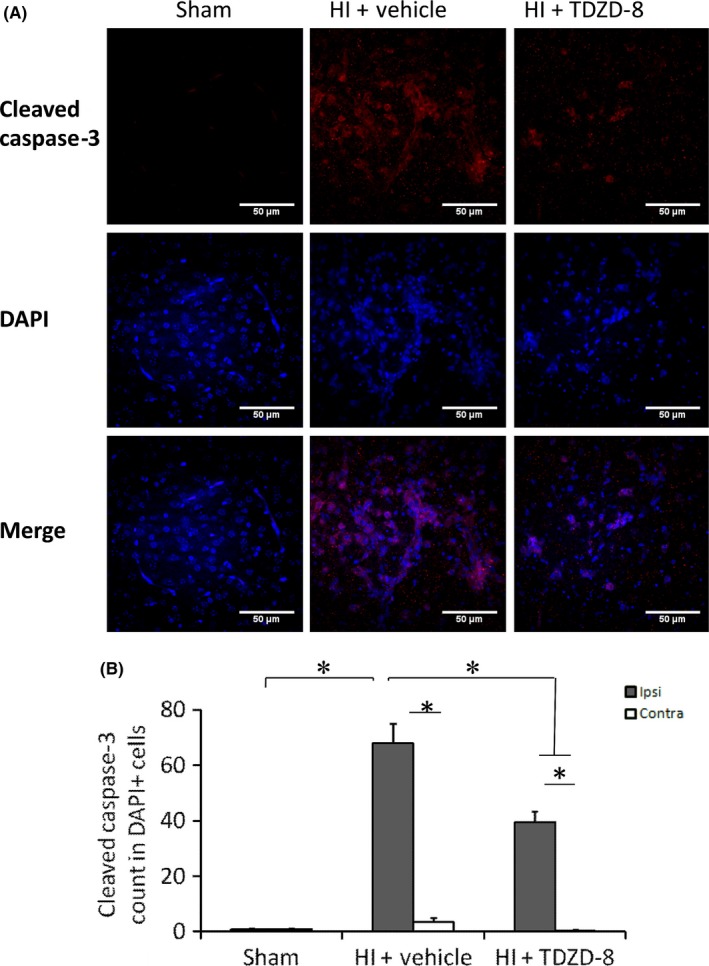
Immunohistochemistry staining showing the effects of TDZD‐8 on cleaved caspase‐3 staining following hypoxic‐ischemic brain injury. Brain samples were collected 7 days after HI and sliced coronally with a thickness of ~50 μm. (A) Representative images for cleaved caspase‐3 and DAPI staining in sham, vehicle, and TDZD‐8 groups following HI. (B) Analysis of cleaved caspase‐3 count in DAPI‐positive cells. (P<.05 for all comparisons; One‐way ANOVA followed by Fisher LSD Method). For ipsilateral brain, sham group, n=5; HI+vehicle group, n=9; HI+TDZD‐8 group, n=7. For contralateral brain, sham group, n=5; HI+vehicle group, n=8; HI+TDZD‐8 group, n=5
3.6. TDZD‐8 reduces GFAP‐positive cells and maintains NeuN‐positive cells following hypoxic‐ischemic brain injury
To investigate the nonneuronal cell involvement in hypoxia‐ischemia, we performed immunohistochemical analysis to detect GFAP‐positive cells in brains 7 days following HI. Our IHC results showed that there was a significant reduction in GFAP‐positive cells in the ipsilateral hemisphere of the TDZD‐8 treatment group (15.8±3.34%) in comparison with the ipsilateral hemisphere of the vehicle‐treated group (39.8±4.00%); the contralateral hemisphere and sham brains had the lowest number of GFAP‐positive cells (Figure 7A, C). Interestingly, there were significantly more NeuN‐positive cells in the TDZD‐8 treatment group (58.6±6.12%) compared to the vehicle‐treated group (31.3±4.25%, Figure 7A, B). This finding indicated that fewer neurons were lost to injury in the TDZD‐8 treated group as compared to that of the vehicle‐treated group.
Figure 7.
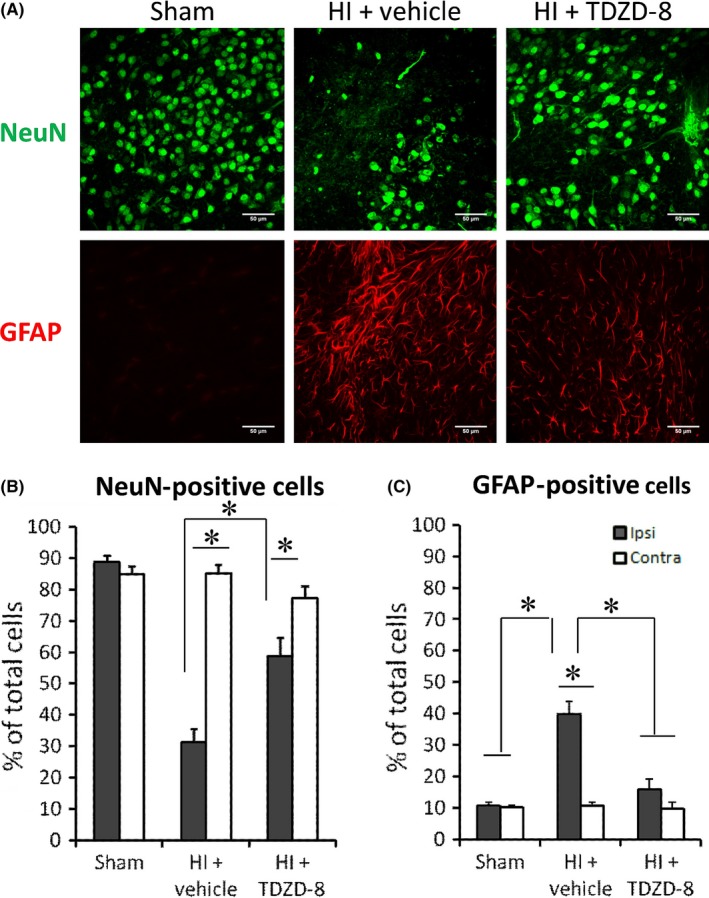
Immunohistochemistry staining showing the effects of TDZD‐8 on NeuN‐positive cells and GFAP‐positive cells following hypoxic‐ischemic brain injury. Brain samples were collected 7 days after HI and sliced into coronal sections with a thickness of ~50 μm. (A) Representative images for NeuN and GFAP staining. (B) Analysis of NeuN‐positive cells as a percentage of DAPI‐positive cells in sham, vehicle, and treatment groups. (C) Analysis of GFAP‐positive cells as a percentage of DAPI‐positive cells. (P<.05 for all comparisons; One‐way ANOVA followed by Fisher LSD Method). For ipsilateral brain, sham group, n=9; HI+vehicle group, n=9; HI+TDZD‐8 group, n=10. For contralateral brain, sham group, n=9; HI+vehicle group, n=9; HI+TDZD‐8 group, n=8
4. Discussion
In this study, we showed that the levels of GSK‐3β mRNA and total protein were significantly higher in P7 pups than at E17 and 14 days and the ratio of p‐GSK‐3β/t‐GSK‐3β was reduced significantly at P14 compared to E17 and P7. We also demonstrated that treatment with TDZD‐8 (i) reduced cerebral infarct volume following the HI injury; (ii) improved neurobehavioral recovery outcomes after the HI injury and increased the phosphorylation of GSK‐3β; (iii) reduced cleaved caspase‐3 protein levels and increased the expression of prosurvival molecule p‐Akt; and (iv) reduced HI‐induced upregulation of GFAP‐positive cells and improved neuronal survival. These findings suggest that TDZD‐8 elicits neuroprotection and neurobehavioral recovery by restoring p‐GSK‐3β and p‐Akt expression levels. In addition, our results indicate that TDZD‐8 may reduce HI‐activated astrocytes, a marker for neurological injury.43
It has been show that TDZD‐8 reduced brain injury following cerebral ischemia in adult mice15; however, its effect in neonatal mice remains unclear. We found that pretreatment with TDZD‐8 reduced the brain damage and improved neurobehavioural test scores of the mice following HI. During development, the p‐Akt/t‐Akt and p‐GSK‐3β/t‐GSK‐3β protein ratios are high at the age of P7, which corresponds to a high Akt and low GSK‐3β activity. Following HI, the p‐Akt/t‐Akt and p‐GSK‐3β/t‐GSK‐3β levels decreased significantly in our vehicle control group, which indicates that these molecules play an important role in the pathology of HI. The reduction of p‐Akt and p‐GSK‐3β levels was found to cause an increase in cleaved caspase‐3 levels indicating the involvement of apoptotic signaling.44, 45 Our results verified that TDZD‐8 reduced brain injury following HI injury in neonatal mice and support that pretreatment with TDZD‐8 elicited neuroprotection through restoring p‐Akt/t‐Akt and p‐GSK‐3β/t‐GSK‐3β levels which led to suppression of cleaved caspase‐3‐mediated apoptotic signaling.
Caspase‐3 has been a prominent marker of apoptosis and was shown to be upregulated following neonatal HI, while inhibition of caspase‐3 resulted in neuroprotection against hypoxic‐ischemia.30 Previous studies have shown that phosphorylation of Akt (p‐Akt) inhibited GSK‐3β leading to the suppression of caspase‐3‐mediated apoptotic signaling; this suggests a possible link between GSK‐3β regulation and cell death in our model.11, 46, 47 Specifically, upstream activation of GSK‐3β after hypoxic‐ischemic injury leads to decreased Bcl‐2 expression, which in turn activates caspase‐3.48 In this study, we found that cleaved caspase‐3 protein expression levels were very low in the treatment group compared with the number of cleaved caspase‐3 containing cells in our immunohistochemistry data (Figure 6A, B). The differences seen in cleaved caspase‐3 levels could be attributed to the differences in the age of the brain samples, timing of caspase‐3 expression, and the pharmacological half‐life of TDZD‐8. Specifically, the brain samples used for Western blot analysis were collected 24 hours following HI whereas the brain samples used for immunohistochemistry were collected 7 days following HI. While caspase‐3 levels are present during both the early and late phases of ischemia, caspase‐3 levels are predominately expressed from 72 hours to 7 days after ischemia injury.49 Moreover, TDZD‐8 has a half‐life of 120 minutes50; thus, it is likely that TDZD‐8 exerted a greater effect early after the onset of injury (24 hours) compared to a later stage (7 days). This supports the idea that during early stages of ischemia, TDZD‐8‐mediated phosphorylation of GSK‐3β leads to decrease of caspase‐3 expression seen in our Western blot. However, during later stages of ischemia, it is possible that p‐GSK‐3β is dephosphorylated as the effect of TDZD‐8 becomes diminished resulting in a relative increase in caspase‐3 levels seen in the immunohistochemistry data. Additionally, sex differences exist in caspase‐3 activation in both adult and neonatal mice after stroke. Female mice usually display a stronger activation of caspase‐3,51 while, in the present study, we used both male and female pups. The differences between male and female animals in their response to TDZD‐8 are still needed to be illustrated in the future.
Hypoxic‐ischemic injury can compromise the normal function of astrocytes leading to detrimental secondary effects on neuronal cells. Specifically, pathologically activated astrocytes (reactive astrocytes) have been shown to mediate the inflammatory response52, 53, 54 and express caspase‐3 apoptotic markers55 leading to secondary cerebral damage. In this study, we used GFAP, an indicator for reactive astrogliosis, as a marker for neurological damage following neonatal HI injury. GFAP is a sensitive marker for reactive astrocytes.56, 57 In Figure 7A, it can be seen that the vehicle group had severe reactive astrogliosis with overlapping and hypertrophic astrocytes. In the TDZD‐8‐treated group, reactive astrocytes were fewer in number and shared fewer overlapping regions. Further studies need to address the link between GSK‐3β and reactive astrogliosis following HI injury.
5. Conclusion
We showed that pretreatment with specific GSK‐3β inhibitor TDZD‐8 was neuroprotective against neonatal hypoxic‐ischemic brain injury. The primary mechanism of action is suggested to involve the reduction of HI‐induced apoptosis and reactive astrogliosis and upregulation of prosurvival signaling. TDZD‐8 pretreatment reduced brain damage and improved behavioral recovery, thus making it a potential preventative drug in treatment of neonatal hypoxic‐ischemic brain injury. It is important to note that our present work mainly focused on the preventive effect of TDZD‐8 against hypoxic‐ischemic insult. While it is clinically relevant in specific situations, future studies are needed to investigate the therapeutic effects of TDZD‐8 after the hypoxic‐ischemic injury, and the potential therapeutic window for administering TDZD‐8 after an injury. Given the widespread use of therapeutic hypothermia in the clinical care of neonates with HI, the impact of TDZD‐8 on brain injury when administered with hypothermia also needs to be determined.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgments
Supported by Discovery Grants from National Sciences and Engineering Research Council of Canada to HSS (RGPIN‐2016‐04574) and to ZPF (RGPIN‐2014‐06471), from Heart and Stroke Foundation of Canada (G‐13‐0003069) and CIHR (FRN#132571) to HSS.
Huang S, Wang H, Turlova E, et al. GSK‐3β inhibitor TDZD‐8 reduces neonatal hypoxic‐ischemic brain injury in mice. CNS Neurosci Ther. 2017;23:405‐415. 10.1111/cns.12683
The first two authors contributed equally to this work.
Contributor Information
Hong‐Shuo Sun, Email: hss.sun@utoronto.ca.
Zhong‐Ping Feng, Email: zp.feng@utoronto.ca.
References
- 1. Woodgett JR. Molecular‐cloning and expression of glycogen‐synthase kinase‐3 factor‐A. EMBO J. 1990;9:2431‐2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yao HB, Shaw PC, Wong CC, et al. Expression of glycogen synthase kinase‐3 isoforms in mouse tissues and their transcription in the brain. J Chem Neuroanat. 2002;23:291‐297. [DOI] [PubMed] [Google Scholar]
- 3. Mukai F, Ishiguro K, Sano Y, et al. Alternative splicing isoform of tau protein kinase I/glycogen synthase kinase 3 beta. J Neurochem. 2002;81:1073‐1083. [DOI] [PubMed] [Google Scholar]
- 4. Hur EM, Zhou FQ. GSK3 signalling in neural development. Nat Rev Neurosci. 2010;11:539‐551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shi SH, Cheng T, Jan LY, et al. APC and GSK‐3 beta are involved in mPar3 targeting to the nascent axon and establishment of neuronal polarity. Curr Biol. 2004;14:2025‐2032. [DOI] [PubMed] [Google Scholar]
- 6. Yoshimura T, Kawano Y, Arimura N, et al. GSK‐3 beta regulates phosphorylation of CRMP‐2 and neuronal polarity. Cell. 2005;120:137‐149. [DOI] [PubMed] [Google Scholar]
- 7. Spittaels K, Van den Haute C, Van Dorpe J, et al. Neonatal neuronal overexpression of glycogen synthase kinase‐3 beta reduces brain size in transgenic mice. Neuroscience. 2002;113:797‐808. [DOI] [PubMed] [Google Scholar]
- 8. Kim WY, Wang X, Wu Y, et al. GSK‐3 is a master regulator of neural progenitor homeostasis. Nat Neurosci. 2009;12:1390‐1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhou FQ, Zhou J, Dedhar S, et al. NGF‐induced axon growth is mediated by localized inactivation of GSK‐30 and functions of the microtubule plus end binding protein APC. Neuron. 2004;42:897‐912. [DOI] [PubMed] [Google Scholar]
- 10. Trivedi N, Marsh P, Goold RG, et al. Glycogen synthase kinase‐3 beta phosphorylation of MAP1B at Ser1260 and Thr1265 is spatially restricted to growing axons. J Cell Sci. 2005;118:993‐1005. [DOI] [PubMed] [Google Scholar]
- 11. Pap M, Cooper GM. Role of glycogen synthase kinase‐3 in the phosphatidylinositol 3‐kinase/Akt cell survival pathway. J Biol Chem. 1998;273:19929‐19932. [DOI] [PubMed] [Google Scholar]
- 12. Doble BW, Woodgett JR. GSK‐3: tricks of the trade for a multi‐tasking kinase. J Cell Sci. 2003;116:1175‐1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sasaki C, Hayashi T, Zhang WR, et al. Different expression of glycogen synthase kinase‐3 beta between young and old rat brains after transient middle cerebral artery occlusion. Neurol Res. 2001;23:588‐592. [DOI] [PubMed] [Google Scholar]
- 14. Kelly S, Zhao H, Hua Sun G, et al. Glycogen synthase kinase 3 beta inhibitor Chir025 reduces neuronal death resulting from oxygen‐glucose deprivation, glutamate excitotoxicity, and cerebral ischemia. Exp Neurol. 2004;188:378‐386. [DOI] [PubMed] [Google Scholar]
- 15. Collino M, Thiemermann C, Mastrocola R, et al. Treatment with the glycogen synthase kinase‐3 beta inhibitor, TDZD‐8, affects transient cerebral ischemia/reperfusion injury in the rat hippocampus. Shock. 2008;30:299‐307. [DOI] [PubMed] [Google Scholar]
- 16. Li Q, Li H, Roughton K, et al. Lithium reduces apoptosis and autophagy after neonatal hypoxia‐ischemia. Cell Death Dis. 2010;1:e56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zheng Z, Kim JY, Ma H, et al. Anti‐inflammatory effects of the 70 kDa heat shock protein in experimental stroke. J Cereb Blood Flow Metab. 2008;28:53‐63. [DOI] [PubMed] [Google Scholar]
- 18. Li L, Klebe D, Doycheva D, et al. G‐CSF ameliorates neuronal apoptosis through GSK‐3 beta inhibition in neonatal hypoxia‐ischemia in rats. Exp Neurol. 2015;263:141‐149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dickey EJ, Long SN, Hunt RW. Hypoxic ischemic encephalopathy–what can we learn from humans? J Vet Intern Med. 2011;25:1231‐1240. [DOI] [PubMed] [Google Scholar]
- 20. Shankaran S, Laptook AR, Ehrenkranz RA, et al. Whole‐body hypothermia for neonates with hypoxic‐ischemic encephalopathy. N Engl J Med. 2005;353:1574‐1584. [DOI] [PubMed] [Google Scholar]
- 21. Zhou WH, Cheng GQ, Shao XM, et al. Selective head cooling with mild systemic hypothermia after neonatal hypoxic‐ischemic encephalopathy: a multicenter randomized controlled trial in China. J Pediatr. 2010;157:367‐372. [DOI] [PubMed] [Google Scholar]
- 22. Hall RT, Hall FK, Daily DK. High‐dose phenobarbital therapy in term newborn infants with severe perinatal asphyxia: a randomized, prospective study with three‐year follow‐up. J Pediatr. 1998;132:345‐348. [DOI] [PubMed] [Google Scholar]
- 23. Zhu C, Kang W, Xu F, et al. Erythropoietin improved neurologic outcomes in newborns with hypoxic‐ischemic encephalopathy. Pediatrics. 2009;124:E218‐E226. [DOI] [PubMed] [Google Scholar]
- 24. Van Bel F, Shadid M, Moison RM, et al. Effect of allopurinol on postasphyxial free radical formation, cerebral hemodynamics, and electrical brain activity. Pediatrics. 1998;101:185‐193. [DOI] [PubMed] [Google Scholar]
- 25. D'Angelo B, Ek CJ, Sun Y, et al. GSK3beta inhibition protects the immature brain from hypoxic‐ischaemic insult via reduced STAT3 signalling. Neuropharmacology. 2016;101:13‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Xiao AJ, Chen W, Xu B, et al. Marine compound xyloketal B reduces neonatal hypoxic‐ischemic brain injury. Mar Drugs. 2015;13:29‐47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chen W, Xu B, Xiao A, et al. TRPM7 inhibitor carvacrol protects brain from neonatal hypoxic‐ischemic injury. Mol Brain. 2015;8:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sun HS, Xu B, Chen W, et al. Neuronal K‐ATP channels mediate hypoxic preconditioning and reduce subsequent neonatal hypoxic‐ischemic brain injury. Exp Neurol. 2015;263:161‐171. [DOI] [PubMed] [Google Scholar]
- 29. Bredesen DE. Neural apoptosis. Ann Neurol. 1995;38:839‐851. [DOI] [PubMed] [Google Scholar]
- 30. Cheng Y, Deshmukh M, D'Costa A, et al. Caspase inhibitor affords neuroprotection with delayed administration in a rat model of neonatal hypoxic‐ischemic brain injury. J Clin Invest. 1998;101:1992‐1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Han W, Sun YY, Wang XY, et al. Long‐term administration of the caspase inhibitor Q‐VD‐OPh reduced brain injury induced by neonatal hypoxia‐ischemia. Dev Neurosci. 2014;36:64‐72. [DOI] [PubMed] [Google Scholar]
- 32. Rojo AI, de Sagarra MR, Cuadrado A, et al. GSK‐3 beta down‐regulates the transcription factor Nrf2 after oxidant damage: relevance to exposure of neuronal cells to oxidative stress. J Neurochem. 2008;105:192‐202. [DOI] [PubMed] [Google Scholar]
- 33. Salazar M, Rojo AI, Velasco D, et al. Glycogen synthase kinase‐3 beta inhibits the xenobiotic and antioxidant cell response by direct phosphorylation and nuclear exclusion of the transcription factor Nrf2. J Biol Chem. 2006;281:14841‐14851. [DOI] [PubMed] [Google Scholar]
- 34. Chen G, Bower KA, Ma CL, et al. Glycogen synthase kinase 3 beta (GSK3 beta) mediates 6‐hydroxydopamine‐induced neuronal death. FASEB J. 2004;18:1162‐1164. [DOI] [PubMed] [Google Scholar]
- 35. Martinez A, Alonso M, Castro A, et al. First non‐ATP competitive glycogen synthase kinase 3 beta (GSK‐3 beta) inhibitors: Thiadiazolidinones (TDZD) as potential drugs for the treatment of Alzheimer's disease. J Med Chem. 2002;45:1292‐1299. [DOI] [PubMed] [Google Scholar]
- 36. Serenó L, Coma M, Rodríguez M, et al. A novel GSK‐3 beta inhibitor reduces Alzheimer's pathology and rescues neuronal loss in vivo. Neurobiol Dis. 2009;35:359‐367. [DOI] [PubMed] [Google Scholar]
- 37. Franklin AV, King MK, Palomo V, et al. Glycogen synthase kinase‐3 inhibitors reverse deficits in long‐term potentiation and cognition in fragile X mice. Biol Psychiatry. 2014;75:198‐206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Uehara H, Yoshioka H, Kawase S, et al. A new model of white matter injury in neonatal rats with bilateral carotid artery occlusion. Brain Res. 1999;837:213‐220. [DOI] [PubMed] [Google Scholar]
- 39. Steinberg GK, George CP, DeLaPaz R, et al. Dextromethorphan protects against cerebral injury following transient focal ischemia in rabbits. Stroke. 1988;19:1112‐1118. [DOI] [PubMed] [Google Scholar]
- 40. Ten VS, Bradley‐Moore M, Gingrich JA, et al. Brain injury and neurofunctional deficit in neonatal mice with hypoxic‐ischemic encephalopathy. Behav Brain Res. 2003;145:209‐219. [DOI] [PubMed] [Google Scholar]
- 41. Anonymous . Website: © 2015 Allen Institute for Brain Science. Allen Developing Mouse Brain Atlas [Internet]. 2015. http://developingmouse.brain-map.org. Accessed October 6, 2015.
- 42. Tsuji M, Wilson MA, Lange MS, et al. Minocycline worsens hypoxic‐ischemic brain injury in a neonatal mouse model. Exp Neurol. 2004;189:58‐65. [DOI] [PubMed] [Google Scholar]
- 43. Cespedes AE, Arango C, Cardona GP. Injury markers in two models of cerebral ischemia. Biomedica. 2013;33:292‐305. [PubMed] [Google Scholar]
- 44. Pitsikas N, Brambilla A, Besozzi C, et al. Effects of cerestat and NBQX on functional and morphological outcomes in rat focal cerebral ischemia. Pharmacol Biochem Behav. 2001;68:443‐447. [DOI] [PubMed] [Google Scholar]
- 45. Boast CA, Gerhardt SC, Pastor G, et al. The N‐methyl‐d‐aspartate antagonists Cgs‐19755 and Cpp reduce ischemic brain‐damage in gerbils. Brain Res. 1988;442:345‐348. [DOI] [PubMed] [Google Scholar]
- 46. Xiong T, Tang J, Zhao J, et al. Involvement of the Akt/Gsk‐3 Beta/Crmp‐2 pathway in axonal injury after hypoxic‐ischemic brain damage in neonatal rat. Neuroscience. 2012;216:123‐132. [DOI] [PubMed] [Google Scholar]
- 47. Wu Y, Shang Y, Sun S, et al. Erythropoietin prevents PC12 cells from 1‐methyl‐4‐phenylpyridinium ion‐induced apoptosis via the Akt/GSK‐3 beta/caspase‐3 mediated signaling pathway. Apoptosis. 2007;12:1365‐1375. [DOI] [PubMed] [Google Scholar]
- 48. Yun SI, Yoon HY, Chung YS. Glycogen synthase kinase‐3 beta regulates etoposide‐induced apoptosis via Bcl‐2 mediated caspase‐3 activation in C3H10T1/2 cells. Apoptosis. 2009;14:771‐777. [DOI] [PubMed] [Google Scholar]
- 49. Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999;22:391‐397. [DOI] [PubMed] [Google Scholar]
- 50. Jordan CT. The leukemic stem cell. Best Pract Res Clin Haematol. 2007;20:13‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhu C, Xu F, Wang X, et al. Different apoptotic mechanisms are activated in male and female brains after neonatal hypoxia‐ischaemia. J Neurochem. 2006;96:1016‐1027. [DOI] [PubMed] [Google Scholar]
- 52. Bona E, Andersson AL, Blomgren K, et al. Chemokine and inflammatory cell response to hypoxia‐ischemia in immature rats. Pediatr Res. 1999;45:500‐509. [DOI] [PubMed] [Google Scholar]
- 53. Glabinski AR, Tani M, Strieter RM, et al. Synchronous synthesis of alpha‐ and beta‐chemokines by cells of diverse lineage in the central nervous system of mice with relapses of chronic experimental autoimmune encephalomyelitis. Am J Pathol. 1997;150:617‐630. [PMC free article] [PubMed] [Google Scholar]
- 54. Nedergaard M, Dirnagl U. Role of glial cells in cerebral ischemia. Glia. 2005;50:281‐286. [DOI] [PubMed] [Google Scholar]
- 55. Benjelloun N, Joly LM, Palmier B, et al. Apoptotic mitochondrial pathway in neurones and astrocytes after neonatal hypoxia‐ischaemia in the rat brain. Neuropathol Appl Neurobiol. 2003;29:350‐360. [DOI] [PubMed] [Google Scholar]
- 56. Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol. 2010;119:7‐35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Herrmann JE, Imura T, Song B, et al. STAT3 is a critical regulator of astrogliosis and scar formation after spinal cord injury. J Neurosci. 2008;28:7231‐7243. [DOI] [PMC free article] [PubMed] [Google Scholar]