

Summary
Aims
To date, no reliable methods have proven effective for treating spinal cord injury (SCI). Even systemic administration of methylprednisolone (MP) remains controversial. We previously reported that intrathecal (i.t.) administration of granulocyte colony‐stimulating factor (G‐CSF) improves outcome after experimental spinal cord ischemic insults in rats. The present study aimed to examine the neuroprotective efficacy of i.t. G‐CSF or MP in rats with SCI.
Methods
Female rats were subjected to spinal cord contusion injury at T10 using NYU impactor. We i.t. administered G‐CSF (10 μg) or MP (one bolus of 100 μg, followed by 18 μg/h infusion for 23 h) immediately after SCI.
Results
Both G‐CSF and MP significantly improved the rats’ motor function after SCI. Immunofluorescence staining revealed suppressed expression of transforming growth factor‐beta 1 (TGF‐β1), chondroitin sulfate proteoglycans (neurocan and phosphacan), OX‐42 and tumor necrosis factor alpha after i.t. G‐CSF, but not MP, in rats with SCI. In addition, G‐CSF significantly decreased the expression of astrocytic TGF‐β1 and glial fibrillary acidic protein around the injury site. Furthermore, rats with G‐CSF treatment showed increased neurofilament expression beyond the glial scars.
Conclusion
Direct i.t. administration of G‐CSF provides a promising therapeutic option for SCI or related spinal diseases.
Keywords: G‐CSF, Intrathecal, Methylprednisolone, Spinal cord injury, Transforming growth factor
Introduction
An estimated 3 million people worldwide have spinal cord injury (SCI). Every year, more than 20,000 new cases of SCI are reported (International Campaign for Cures of Spinal Cord Injury Paralysis). The clinical trials, National Acute SCI Study (NASCIS) 2 and 3, suggested that high dose of methylprednisolone (MP) provided neurological improvement in acute SCI 1, 2. Therefore, the MP treatment became the standard of care in acute SCI 3. However, improvements in the functional outcome of humans with SCI using MP are still controversial, and high systemic doses of MP are accompanied by adverse side effects 4, 5. Moreover, clinical trials have clearly indicated that MP does not significantly attenuate SCI‐induced neurological dysfunction 6, 7. Thus, discovering other pharmacological agents that can promote functional recovery is desirable.
Granulocyte colony‐stimulating factor (G‐CSF) is a granulocyte regulator that was originally used to treat neutropenia. Our previous reports and several other recent studies have indicated that in addition to its hematopoietic effects, G‐CSF possesses nonhematopoietic functions and can be used as a potential drug for neurodegenerative diseases, including stroke and spinal cord ischemic, mechanical, and transection injuries 8, 9, 10, 11, 12, 13. A phase I/IIa clinical trial of SCI has reported neurological improvement after G‐CSF administration 14, 15. Based on primary safety endpoints of clinical trial IIa, Schabitz et al. 16 concluded that the number of leukocytes substantially increases after systemic G‐CSF administration in humans and reduced rapidly after the end of treatment, and suggest the G‐CSF is safe in patients. To date, G‐CSF is one of the few growth factors approved for clinical use.
Intrathecal (i.t.) administration may be a more promising option compared to systemic administration. Because i.t. administration requires a smaller dose (approximately 1–2% of the systemic dose) and has a direct effect on the injured region, this allows a reduction in the adverse side effects and prevents the degradation of the drug's effect compared to systemic injection. Furthermore, compared to systemic injection, the direct effect on the injured region by i.t. administration allows the direct evaluation of the neuroprotective mechanisms of G‐CSF on the central nervous system (CNS) in SCI. Investigating the cellular mechanisms that may contribute to the neuroprotective or adverse effects of G‐CSF is important for its use in clinical practice.
It is accepted that glial cells (astrocytes and microglia) in the CNS likely participate in neuroinflammation and glial scar formation. These are critical events in the detrimental responses after CNS insults. Our previous study indicated that i.t. G‐CSF can modulate spinal cord ischemia‐induced glial cell (astrocyte and microglia) activation and upregulate growth factors expression in glial cells 11. Kawabe et al. 17 also found that G‐CSF has the capacity to activate glial cells and enhance the expression of vascular endothelial growth factor (VEGF) after SCI. Thus, in SCI, we hypothesized that the direct neuroprotective effects of G‐CSF were due to its modulation of glial cell functions. In the present study, we used Wistar rats with experimental SCI induced by the New York University (NYU) impactor device. Our aims were to assess the effects of direct spinal administration of G‐CSF on the injury‐induced glial scar, as well as to examine the effects of G‐CSF on neurodegeneration and glial activation.
Materials and Methods
Implantation of i.t. Catheters
Female Wistar rats (weight, 300–320 g) were used in the experiments. As described in a previous study 11, i.t. catheters were inserted via the atlantooccipital membrane into the i.t. space at the level of T12 in the spinal cord and externalized and fixed to the cranial aspect of the head. Rats with i.t. catheters were housed individually and exposed to a daily 12‐h light/dark cycle with access to food and water ad libitum. The rats that showed abnormal locomotor behavior or the presence of fresh blood in the cerebrospinal fluid were excluded from the study. The use of animals conformed to the Guiding Principles in the Care and Use of Animals of the American Physiology Society and was approved by the National Sun Yat‐Sen University Animal Care and Use Committee.
Spinal Cord Contusion Injury in Rats
A 4‐day recovery period was allowed after implantation of the i.t. catheter, and spinal cord contusion injury was subsequently performed. Spinal cord contusion was performed under 2.5% isoflurane anesthesia and prophylactic administration of cephalosporin. While anesthetized, rats received a spinal cord contusion injury utilizing the NYU impactor device (W.M. Keck Center for Collaborative Neuroscience, Rutgers University, NJ, USA). Laminectomy of the caudal portion of T9 and all of T10 was performed to expose the spinal cord. The intact T10 spinal cord was contused with the NYU weight‐drop device (from a height of 50 mm). Bladder evacuation was applied twice daily for at least 7 days.
Drug Treatment
The dose of i.t. MP was calculated and modified according to Xu et al. 18 and clinical treatment 14. The systemic dosage of MP in humans was converted to a corresponding i.t. dosage in rats as follows: 30 mg/kg (human systemic) = 0.3 mg/kg (i.t. for human) = 0.1 mg/rat (i.t.) administered as a bolus. During the 23 h following SCI, 5.4 mg/(kg h) (human systemic) = 0.054 mg/(kg h) (i.t. for humans) = 0.018 mg/(rat h) (i.t.). As the average weight of animals in this study was 300 g, we divided the dose for humans by 3 to determine the dose for rats (0.3 mg/kg [human]/3 = 0.1 mg/300 g for rat). The i.t. dosage of G‐CSF was 10 μg, in accordance with our previous studies 9, 10, 11. The rats received drug treatment immediately after using the NYU impactor to generate the SCI. The rats were divided into four groups, as follows:
Sham group: T9–T10 vertebrae were exposed without injury, and the rats received i.t. injection of artificial cerebrospinal fluid (aCSF, 26.5 μL) and aCSF (1 μL/h) infused by osmotic pump (Alzet, Cupertino, CA, USA) for 23 h after the injury.
SCI control group: rats with SCI received an i.t. injection of a bolus of aCSF (26.5 μL) and then had aCSF (1 μL/h) infused by osmotic pump for 23 h after the injury.
SCI + MP group: rats with SCI received an i.t. injection of a bolus of MP (0.1 mg; Solu Medrol®; Pharmacia & Upjohn Company, Kalamazoo, MI, USA) and then had MP infused (18 μg/[μL h]) by an osmotic pump for 23 h after the injury.
SCI + G‐CSF group: rats with SCI received a bolus of G‐CSF via i.t. injection (10 μg/23 μL; Chugai Pharmaceutical Co., Kitaku, Tokyo, Japan), followed by a flush with 3.5 μL of aCSF, and then had aCSF infused by osmotic pump (1 μL/h) for 23 h after the injury.
Behavioral Analysis
The 21‐point Basso–Beattie–Bresnahan (BBB) locomotion scale 19 was used to evaluate the improvement in hind limb function in the treated animals with SCI (n = 8 per group). The BBB scores were recorded every 3 days following SCI by independent examiners who were unaware of the experimental groups.
Immunohistochemistry and Quantification
The immunohistochemical procedures and the quantification of immunochemistry were performed as previous described 9, 10, 11, 20, 21. Briefly, under isoflurane anesthesia, the rats (n = 4 per each group per each time point) were intracardially perfused and the thoracic vertebrae (T8–T12) were harvested. The spinal cord tissues harvested at the different time points from different treatment groups were then mounted on the same block in optimal cutting temperature compound to decrease the variation. Next, 20‐μm‐thick sections incubated for 1 h at room temperature with 4% normal horse serum in 0.01% Triton X‐100 and PBS follow by incubated overnight at 4°C with primary antibodies (Table 1) in PBS. The sections were then incubated with Cy™3‐labeled donkey anti‐rabbit antibody (Jackson ImmunoResearch Laboratories Inc., West Grove, PA, USA) and/or Alexa Fluor 488‐labeled chicken anti‐mouse antibody (Molecular Probes Inc., Eugene, OR, USA) for 1 h at room temperature. The sections were examined using a Leica TCS SP5II fluorescence microscope (Leica, Wetzlar, Germany), and images were captured using a SPOT Xplorer Digital camera (Diagnostic Instruments Inc., Sterling Heights, MI, USA). For quantification of immunofluorescence staining in each group, every fourth section at approximately 400 μm rostral to the lesion epicenter was selected from a series of consecutive thoracic spinal cord sections, and four successive sections were measured. Immunofluorescence was measured, and images were acquired from whole portions of the spinal cord sections under a 2.5× objective. Confocal images (Figures 2C and 5C) were captured by Leica TCS SP5II equipped with Leica HyD (Hybrid Detector). The exposure time was the same for all spinal cord sections in the same microscopic slide.
Table 1.
Antibodies used in this study
| Primary antibody | Supplier | Catalog # | Host |
|---|---|---|---|
| Cd11b (OX‐42) | Serotec (Raleigh, NC, USA) | MCA275 | Mouse |
| GFAP | Chemicon (Temecula, CA, USA) | MAB3402 | Mouse |
| GFAP | GeneTex (Irvine, CA, USA) | GTX61295 | Rabbit |
| Neurocan | Chemicon (Temecula, CA, USA) | MAB5212 | Mouse |
| Neurofilament | Chemicon (Temecula, CA, USA) | MAB5262 | Mouse |
| PCNA | Cell signaling (Danvers, MA, USA) | 2586 | Mouse |
| Phosphacan | Chemicon (Temecula, CA, USA) | MAB5210 | Mouse |
| TGF‐β1 | Abcam (Cambridge, MA, USA) | ab92486 | Rabbit |
| TNF‐α | Invitrogen (Carlsbad, CA, USA) | ARC3012 | Rabbit |
| Vimentin | Invitrogen (Carlsbad, CA, USA) | 18‐0052 | Mouse |
GFAP, glial fibrillary acidic protein; PCNA, proliferating cell nuclear antigen; TGF‐β1, transforming growth factor‐beta 1; TNF‐α, tumor necrosis factor alpha.
Figure 2.

Immunohistochemistry and IR analyses of the effects of i.t. G‐CSF on spinal TGF‐β1 expression in rats with SCI. (A) Spinal cord sections from the sham, SCI control, SCI + MP, and SCI + G‐CSF groups were harvested at 6 h, 3 days, and 7 days after SCI and incubated with anti‐TGF‐β1 antibody. These results show SCI‐induced upregulation of spinal TGF‐β1 IR at 6 h, 3 days, and 7 days after injury. The i.t. G‐CSF clearly inhibited the SCI‐induced TGF‐β1 upregulation. (B) Quantification of the IR of TGF‐β1 was expressed as the fold change compared to control animals (at each time point), which were considered to be 1. Administration of i.t. G‐CSF significantly attenuated the SCI‐induced upregulation of TGF‐β1 at 6 h, 3 days, and 7 days after injury. (C) Double immunofluorescence of spinal TGF‐β1 (red) plus the astrocyte‐specific marker GFAP (green) in the SCI and G‐CSF groups at 3 days after SCI. Confocal images showed that astrocytes were colocalized with TGF‐β1 in the SCI control group. G‐CSF attenuated the SCI‐induced TGF‐β1 expression in astrocytes. + P < 0.05, compared with the sham group; *P < 0.05, compared with the SCI control group; # P < 0.05, compared with the SCI + MP group. Scale bar: 50 μm.
Statistical Analysis
All data are presented as the mean ± the standard error of the mean (SEM). Neurological outcomes after SCI were analyzed by two‐way repeated‐measures analyses of variance (ANOVA) with group and time point as independent variables fol‐lowed by post hoc pairwise multiple comparison using the Student–Newman–Keuls method. For immunohistochemical analyses, significant differences between the treatment groups were assessed and were analyzed using one‐way ANOVA followed by Student–Newman–Keuls post hoc tests for multiple comparisons. A P value of <0.05 was considered statistically significant.
Results
Administration of i.t. G‐CSF Improved Neurological Outcomes After SCI
The motor function of the rats was monitored in accordance with the guidelines of the BBB locomotor scores. The i.t. injection of G‐CSF significantly inhibited the SCI‐induced neurological dysfunction from day 6 to day 30 after injury (Figure 1). The i.t. continuous infusion of MP significantly improved the functional outcome in SCI rats at days 21 to 30 after injury. The rats from the sham group and those treated with i.t. G‐CSF, MP, or aCSF without SCI exhibited a normal neurological outcome from day 1 to day 30 after injection (data not shown). For analyzing the time series results, the area under the curve (AUC) value of the calculated BBB scores was determined for the various experimental times 22. There were statistical differences between the SCI control and SCI + G‐CSF groups, SCI control and SCI + MP groups, and between the SCI + G‐CSF and SCI + MP groups. These results demonstrate significant improvement of the G‐CSF‐ and MP‐treated groups following SCI. Moreover, the attenuation of SCI‐induced motor dysfunctions in the G‐CSF‐treated group was better than that in the SCI + MP group.
Figure 1.
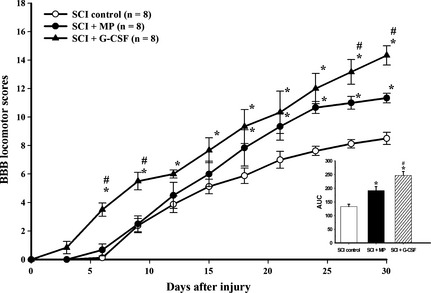
Administration of i.t. G‐CSF or MP improves locomotor functions after SCI. A significant improvement in hind limb motor function was observed in the G‐CSF‐treated group compared with the SCI control or SCI + MP groups after SCI. The AUC (bar chart) presents the evolution of the BBB score over time for each group. Both treatments showed significantly improved recovery chronically after SCI. *P < 0.05, compared with the SCI control group; # P < 0.05, compared with the SCI + MP group.
Effects of G‐CSF on Spinal Transforming Growth Factor‐beta 1 (TGF‐β1) Expression in SCI
As shown in Figure 2A, TGF‐β1 immunoreactivity (IR) around the lesion site was clearly increased at 6 h, 3 days, and 7 days after SCI. Compared to the SCI control group, the group that received i.t. G‐CSF showed obviously decreased spinal TGF‐β1 IR, while MP administration attenuated the SCI‐induced upregulation of TGF‐β1 expression at day 3 after SCI. Quantification of the TGF‐β1 IR further confirmed that the TGF‐β1 levels were significantly upregulated 6 h, 3 days, and 7 days after SCI, and that i.t. G‐CSF significantly decreased spinal TGF‐β1 expression in rats with SCI (Figure 2B). MP administration significantly inhibited the SCI‐induced TGF‐β1 expression at days 3 and 7 after SCI. Using double immunofluorescence staining, we found that the TGF‐β1 IR signal was colocalized with the astrocyte‐specific marker glial fibrillary acidic protein (GFAP) at day 3 after SCI. Furthermore, i.t. G‐CSF inhibited the SCI‐induced TGF‐β1 expression in astrocytes at day 3 after SCI (Figure 2C).
Effects of G‐CSF on Neurocan and Phosphacan Expression in SCI
Expression of the chondroitin sulfate proteoglycans (CSPGs) neurocan and phosphacan around the lesion site was examined by immunohistochemistry at 3, 7, and 30 days after SCI. As shown in Figure 3, compared to the sham group, both neurocan and phosphacan IR around the lesion site were obviously increased from 3 to 30 days in the SCI control, SCI + MP, and SCI + G‐CSF groups after injury. Compared with the SCI control group, the group that received i.t. G‐CSF demonstrated inhibited neurocan and phosphacan expression at days 7 and 30 after injury. The quantification results further confirmed that neurocan and phosphacan IR significantly increased 3 to 30 days after SCI. The levels of neurocan and phosphacan IR in the SCI + G‐CSF group were significantly lower than the levels in the SCI control group at 7 and 30 days after SCI. MP administration significantly inhibited only the SCI‐induced neurocan upregulation from 3 to 30 days after SCI.
Figure 3.

The effect of i.t. G‐CSF on the expression of spinal CSPGs in SCI rats. The spinal cord tissues were immunofluorescence stained for neurocan (A) and phosphacan (B) at 3 days, 7 days, and 30 days after SCI. The results show SCI‐induced upregulation of spinal neurocan and phosphacan IR from 3 to 30 days after injury. The i.t. G‐CSF inhibited both neurocan and phosphacan upregulation at 7 days and 30 days after SCI. Quantification of the IR of neurocan and phosphacan is expressed as the fold change compared to control animals (at each time point), which was considered to be 1. Administration of i.t. G‐CSF significantly attenuated the SCI‐induced upregulation of neurocan and phosphacan at 6 and 30 days after injury. ▵ indicates the lesion area. + P < 0.05, compared with the sham group; *P < 0.05, compared with the SCI control group; # P < 0.05, compared with the SCI + MP group. Scale bar: 200 μm.
G‐CSF Increases Axon Distribution Within the Lesion Site
As shown in Figure 4, the spinal sections are colabeled with antibodies against an axon marker (heavy‐chain neurofilament, NF) and astrocyte marker (GFAP), thus illustrating the distribution of axons within the epicenter of the lesion. There are NF axons within the GFAP IR border of the lesion in the above groups. GFAP IR accumulated at the edge of the contusion injury region. The NF IR profiles in both the SCI control and SCI + MP groups were less than that in the SCI + G‐CSF group. The quantification results showed that there were significant differences in NF IR (axon density) between the SCI control and SCI + MP groups. Specifically, there was a significant increase in the axonal density of the SCI + G‐CSF group compared with that of the SCI control and SCI + MP groups.
Figure 4.

The effect of i.t. G‐CSF on neurofilaments (NFs) in the lesion core at 30 days after SCI. Double immunostaining showing the relationship between the astrocyte‐specific marker GFAP (green) and the axon marker NF (red) expression at the lesion epicenter in the SCI control, SCI + MP, and SCI + G‐CSF groups. Quantification of the same area of IR of NFs is expressed as the fold change compared to SCI animals, which was considered to be 100%. Administration of i.t. G‐CSF, but not MP, significantly attenuated the SCI‐induced downregulation of NF IR. *P < 0.05, compared with the SCI control group; # P < 0.05, compared with the SCI + MP group. Scale bar: 500 μm.
Effects of G‐CSF on SCI‐induced Astrocyte and Microglial Cell Activation
Upregulation of GFAP and OX‐42 IR at the lesion site revealed astrocytic and microglial activation. The GFAP IR (Figure 5A) and OX‐42 IR (Figure 5B) were obviously increased from days 3 to 14 after SCI. Moreover, compared with the sham group, the SCI control, SCI + MP, and SCI + G‐CSF groups had markedly higher GFAP and OX‐42 IR. Administration of G‐CSF enhanced the SCI‐induced upregulation of GFAP IR at 7 and 14 days (Figure 5A) and clearly decreased OX‐42 IR from 3 to 30 days after SCI. Compared with the SCI control group, the SCI + MP group showed upregulated GFAP IR and downregulated OX‐42 IR at 14 days after SCI. Quantification showed that both the GFAP and OX‐42 IR were significantly increased in the SCI control, SCI + MP, and SCI + G‐CSF groups from 3 to 30 days after injury. G‐CSF administration significantly increased the SCI‐induced GFAP IR upregulation at 7 and 14 days after injury. MP administration significantly enhanced GFAP IR at 14 days after injury. At Day 30 after SCI, morphological analysis of astrocytes showed extensive gliosis, hypertrophic soma with overlapping processes, and formation of dense plexus in the SCI control group. Compared to the SCI control group, the SCI + MP group showed lesser overlapping processes. The SCI + G‐CSF group showed lesser gliosis than the vehicle‐treated and SCI + MP groups. To determine the effect of G‐CSF on gliosis, we performed immunohistochemical staining for two gliosis markers, vimentin and proliferating cell nuclear antigen (PCNA), on 7 days after SCI (Figure 5C). We observed intense PCNA IR at the lesion border in the SCI control and SCI + MP groups, and most of the PCNA IR signal colocalized with the GFAP IR signal. PCNA IR was lesser in the SCI + G‐CSF group compared to the SCI control and SCI + MP groups. The vimentin IR signal intensity increased in the SCI control, SCI + MP, and SCI + G‐CSF groups after SCI. G‐CSF treatment attenuated the SCI‐induced upregulation of vimentin at 7 days after SCI. The SCI + G‐CSF group had a significantly decreased level of OX‐42 IR expression in the epicenter of the injury site from 3 to 14 days after SCI compared with the SCI control and SCI + MP groups. MP administration significantly inhibited the SCI‐induced OX‐42 IR upregulation only at 14 days after SCI (Figure 5B). There were no significant differences between the SCI control, SCI + MP, and SCI + G‐CSF groups at 30 days after SCI (data not shown).
Figure 5.

Immunohistochemistry and IR analyses of the effect of i.t. G‐CSF on astrocyte and microglial cell activation after SCI. Spinal cord sections from the sham, SCI control, SCI + MP, and SCI + G‐CSF groups were harvested at 3, 7, and 14 days after SCI and incubated with antibodies for the astrocyte‐specific marker GFAP (A) or the microglial cell‐specific maker OX‐42 (B). Immunohistochemistry for proliferating cell nuclear antigen (PCNA) and vimentin was performed to evaluate gliosis at 7 days after SCI, and the images were captured by confocal microscopy. The quantification was performed from nonconfocal immunofluorescence images (C). The results show SCI‐induced upregulation of spinal GFAP and OX‐42 IR from 3 to 14 days after injury. The i.t. G‐CSF clearly enhanced GFAP and inhibited OX‐42 expression after SCI. Quantification of the IR of GFAP and OX‐42 is expressed as the fold change compared to control animals (at each time point), which was considered to be 100%. G‐CSF significantly upregulated GFAP and downregulated OX‐42 expression after SCI. + P < 0.05, compared with the sham group; *P < 0.05, compared with the SCI control group; # P < 0.05, compared with the SCI + MP group. Scale bar: 200 μm; 50 μm in 30 days; 100 μm in C.
Effects of G‐CSF on SCI‐induced Upregulation of Spinal Tumor Necrosis Factor Alpha (TNF‐α) Expression
Near the epicenter of the injury site, TNF‐α IR at 3 to 14 days after injury was clearly increased in the SCI control, SCI + MP, and SCI + G‐CSF groups compared with the sham group (Figure 6). Administration of G‐CSF clearly inhibited the SCI‐induced upregulation of TNF‐α expression from 3 to 14 days after SCI. Conversely, MP administration did not attenuate TNF‐α upregulation after SCI in our experiment. Quantification of the TNF‐α IR further confirmed that the TNF‐α levels were significantly increased from 3 to 14 days after injury in the SCI control, SCI + MP, and SCI + G‐CSF groups. G‐CSF, but not MP, administration significantly decreased TNF‐α expression after SCI.
Figure 6.

The effect of G‐CSF on the upregulation of spinal TNF‐α expression after SCI. The spinal cord tissues were immunostained for TNF‐α at 3, 7, and 14 days after SCI. The expression of TNF‐α IR was increased from 3 to 14 days in the SCI and MP groups, as compared to the TNF‐α IR of the sham or SCI + G‐CSF groups. Quantification of the TNF‐α IR is expressed as the fold change compared to control animals (at each time point), which was considered to be 1. G‐CSF, but not MP, significantly attenuated the SCI‐induced upregulation of TNF‐α from 3 to 14 days after injury. + P < 0.05, compared with the sham group; *P < 0.05, compared with the SCI control group; # P < 0.05, compared with the SCI + MP group. Scale bar: 50 μm.
Discussion
In the present study, we clearly demonstrated that an i.t. injection of G‐CSF significantly improved the neurological outcomes and spinal tissue integrity in rats after SCI. The neuroprotective potential of local G‐CSF administration was better than that for MP administration. From the immunofluorescence analyses, we found that G‐CSF, but not MP, inhibited the SCI‐induced upregulation of TGF‐β1 and two CSPG (neurocan and phosphacan) proteins around the lesion epicenter. Moreover, i.t. G‐CSF inhibited microglial cell overactivation and proinflammatory TNF‐α upregulation after SCI. We suggest that the direct central administration of G‐CSF inhibited CSPGs and spinal neuroinflammation in SCI, which may result in increased axon redistribution within the lesion site. Collectively, the present findings support the hypothesis that direct spinal G‐CSF administration can inhibit TGF‐β1 and CSPG, which may play critical roles in the attenuation of neurological dysfunctions due to SCI.
The Advantages of i.t. Delivery for SCI
To date, few pharmacological agents have been used to treat SCI. Thus, reducing systemic exposure to MP by administering the drug directly into the spinal cerebrospinal fluid may confer important clinical benefits. Compared to systemic administration, i.t. MP administration resulted in a 30‐fold increase in MP concentration in the spinal cord and a greater than 300‐fold increase in the therapeutic index of MP 23. The present results clearly indicate that central administration of MP and G‐CSF improved the SCI‐induced neurological dysfunctions in rats. Potential therapeutic interventions include the use of drug cocktails with antiinflammatory, neurotrophic, and anti‐excitotoxic agents in the acute phase after SCI. Therefore, we strongly propose that direct delivery of neuroprotective plus antiinflammatory agents to the injured region of the spinal cord by i.t. injection may serve as a positive therapeutic strategy and may decrease the systemic adverse side effects for patients with SCI.
Effects of G‐CSF on CSPGs
The glial scar not only forms a physical barrier to axonal regeneration, but also expresses CSPGs, which limit neurite outgrowth across the lesion site 24, 25. CSPGs, including neurocan, versican, brevican, phosphacan, and NG2, which are present in massive quantities in glial scars, are secreted by reactivated astrocytes after CNS insults 26. The present study provides evidence that exogenous G‐CSF by i.t. administration can suppress the expression of two subtypes of CSPG, neurocan and phosphacan, at the spinal level from 3 to 30 days after SCI. Moreover, G‐CSF also inhibited the SCI‐induced intensive expression of GFAP around the cavity at 30 days after SCI (Figure 5). Our results clearly indicate that G‐CSF can attenuate glial scar formation and improve neurological recovery after SCI. Our findings are partially supported by Chung et al. 27, who suggested that systemic G‐CSF can suppress glial scar formation after SCI in rats, possibly by restricting the expression of GFAP and CSPG, which might facilitate functional recovery after SCI. Although the antiglial scar effects of G‐CSF have been described following SCI, the modulation of the glial scar and CSPG by G‐CSF and the mechanisms involved have not been elucidated.
Many studies have demonstrated that TGF‐β1 is a potent inducer of glial scars 28, 29, 30 and their associated neurite outgrowth inhibitor molecules, including CSPG formation 31, 32, 33. Similar to previous studies, our current data also show a rapid and dramatic increase in TGF‐β1 expression after SCI 34, 35. Previous studies have demonstrated that TGF‐β1 participates in the organization of the glial scar in response to a number of CNS pathological insults 36, 37, 38. They also strongly propose that inhibiting TGF‐β1 expression is a promising strategy for repairing CNS injuries. Moreover, CNS injury‐induced inflammatory processes can contribute to/enhance the upregulation of CSPG expression, leading directly to environments that are nonpermissive to neurite growth 39, 40. Mounting evidence suggests that G‐CSF exerts antiinflammatory effects, thereby inhibiting signaling pathways and reducing the overproduction of pro‐inflammatory cytokines 9, 41, 42. Here, we report for the first time that G‐CSF can suppress the expression of spinal astrocytic TGF‐β1 after SCI, possibly through antiinflammatory processes.
The Neuroprotective Effects of G‐CSF on SCI‐induced TGF‐β1 Upregulation
There are at least two possibilities for how the direct i.t. G‐CSF administration improved the axon distribution in the lesion site after SCI via modulating the TGF‐β1 expression. One is that TGF‐β1 signaling directly inhibits axon growth and promotes glial scar formation. It is known that the neurite growth inhibitor small GTPase Rho and its downstream effector serine/threonine kinase Rho kinase (Rho/ROCK) can be activated by TGF‐β1 43, 44, 45. Several studies have also indicated that inhibition of Rho/ROCK increases neurite outgrowth and accelerates neurological recovery after injury 46, 47. The present study found that G‐CSF attenuated the SCI‐induced spinal TGF‐β1 upregulation. Inhibition of TGF‐β1 may downregulate the Rho/ROCK pathway, resulting in axon growth. Neurite outgrowth is considered a critical process for neuroregeneration and neurological recovery. Our present findings support this notion, as our double immunohistochemistry results show that G‐CSF increased the axon distribution in the lesion site after SCI.
Another possible mechanism underlying the protective role of G‐CSF in the modulation of TGF‐β1 after SCI is the attenuation of glial scar formation. Kohta et al. 29 found that inhibiting TGF‐β1 promotes functional recovery and axon growth after SCI. In vitro and in vivo experiments suggest that inhibiting Rho/ROCK signaling attenuates CSPG expression 48, 49. Our present results clearly show that i.t. G‐CSF attenuates SCI‐induced spinal TGF‐β1 and CSPG upregulation, which is accompanied by inhibited axon growth. Thus, we suggest that G‐CSF downregulates TGF‐β1 expression, which decreases the downstream Rho/ROCK activity, resulting in an environment that is permissive to axon growth.
Effects of G‐CSF on SCI‐induced Glial Cell Activation
Studies reported that G‐CSF and G‐CSF receptor are expressed in glial cells and are upregulated in response to neuronal injury 50, 51. Administration of G‐CSF can enhance the expression of the spinal neurogenesis factors GDNF and VEGF‐A in astrocytes in naïve and spinal ischemic rats 11. It is well known that astrocytes participate in anisomorphic astrogliosis (reactive gliosis) and isomorphic astrogliosis (astrocyte activation) in neuropathological and neuroprotective reactions, respectively. Reactive gliosis can release proinflammatory mediators and contribute to secondary damage to the CNS 52. In contrast with anisomorphic astrogliosis, isomorphic astrogliosis is also accompanied by astrocyte hypertrophy and can rescue or protect injured neuronal cells by releasing neurotrophic factors 52, 53. Our present immunohistochemical observations showed that G‐CSF, but not MP, significantly enhanced the expression of GFAP at 7, 14 (Figure 5A), and 30 (data not shown) days after SCI. We propose that the i.t. G‐CSF activated astrocytes (isomorphic astrogliosis), thus reducing insults by releasing neurotrophic factors, which play important roles in the development of secondary injury after SCI. Immunohistochemical analysis on 7 days after SCI showed reduced levels of the anisomorphic astrogliosis markers PCNA and vimentin in the SCI + G‐CSF group (Figure 5C). Further, morphological analysis performed 30 days after SCI (Figure 5A) revealed that G‐CSF treatment had attenuated hypertrophy of astrocytes, and astrocytic process overlapping was lesser in the SCI + G‐CSF group than in the SCI control and SCI + MP groups. Therefore, we suggest that G‐CSF may directly produce a beneficial effect by activating astrocytes (isomorphic astrogliosis).
Microglial cells are involved in signaling cascades that are associated with proinflammatory mediators and their receptor systems 54. Previous studies have shown that implanting grafts with microglia or macrophages can promote axon regeneration after SCI 55, 56. Similar to the dual opposing functions of astrocytes, evidences indicate that microglia are beneficial because they are capable of supplying neurotrophic factors 57, 58. The above evidence suggests that microglial activation may have beneficial effects after neurodegenerative insults. However, the long‐term activation of microglia results in pathological forms of inflammation, resulting in the development/progression of neurodegenerative diseases 59, 60. Our present data clearly show upregulation of the SCI‐induced OX‐42 from 3 to 14 days after injury. The SCI‐induced spinal microglial activation was inhibited by i.t. G‐CSF and MP. However, there were no significant differences in OX‐42 between the SCI control, SCI + G‐CSF, and SCI + MP groups at 30 days after SCI (data not shown). Moreover, i.t. G‐CSF or MP attenuated SCI‐induced pro‐inflammatory TNF‐α expression in the spinal cord. G‐CSF may attenuate SCI‐induced neuroinflammatory responses after SCI. However, the effects of i.t. G‐CSF on spinal cord ischemic injury‐induced spinal glial activation in the present study were different from our results in previous studies. Our previous studies demonstrated that i.t. G‐CSF enhanced spinal ischemia‐induced microglial activation and inhibited spinal ischemia‐induced astrocyte activation at 48 h after injury. We speculate that G‐CSF has different effects on microglia and astrocytes depending on which neuronal insult model and analysis time points are used.
Summary
Systemic G‐CSF has a marked neuroprotective effect on neurodegenerative diseases, and the possible mechanisms include modulation of neuronal apoptosis, excitotoxicity, glial scar formation, neuroinflammation, trophic factors, stem cells, and neuronal plasticity. However, more information regarding the optimal site of administration and direct cellular mechanisms of G‐CSF in neuronal injury is needed. In the present study, we demonstrate for the first time that direct i.t. delivery of G‐CSF also exerts neuroprotective effects on SCI by attenuating glial scar formation. Administering i.t. G‐CSF suppressed spinal TGF‐β1 upregulation after SCI, possibly by restricting the expression of CSPGs and microglial activation at the spinal level, which might facilitate axonal regeneration, thus promoting neurological recovery after SCI.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgments
The study was supported, in part, by grants from the Ministry of Science and Technology, Taiwan (99‐2628‐B‐182A‐003‐MY2 and 101‐2314‐B‐182A‐023‐MY2), Chang Gung Memorial Hospital (CMRPG8A0151‐3) and National Research Program for Biopharmaceuticals, Taiwan (103‐2325‐B‐110‐001).
References
- 1. Bracken MB, Shepard MJ, Collins WF, et al. A randomized, controlled trial of methylprednisolone or naloxone in the treatment of acute spinal‐cord injury. Results of the Second National Acute Spinal Cord Injury Study. N Engl J Med 1990;322:1405–1411. [DOI] [PubMed] [Google Scholar]
- 2. Bracken MB, Shepard MJ, Holford TR, et al. Administration of methylprednisolone for 24 or 48 hours or tirilazad mesylate for 48 hours in the treatment of acute spinal cord injury. Results of the Third National Acute Spinal Cord Injury Randomized Controlled Trial. National Acute Spinal Cord Injury Study. JAMA 1997;277:1597–1604. [PubMed] [Google Scholar]
- 3. Lammertse DP. Clinical trials in spinal cord injury: Lessons learned on the path to translation. The 2011 International Spinal Cord Society Sir Ludwig Guttmann Lecture. Spinal Cord 2013;51:2–9. [DOI] [PubMed] [Google Scholar]
- 4. Molano Mdel R, Broton JG, Bean JA, Calancie B. Complications associated with the prophylactic use of methylprednisolone during surgical stabilization after spinal cord injury. J Neurosurg 2002;96:267–272. [DOI] [PubMed] [Google Scholar]
- 5. McCutcheon EP, Selassie AW, Gu JK, Pickelsimer EE. Acute traumatic spinal cord injury, 1993‐2000A population‐based assessment of methylprednisolone administration and hospitalization. J Trauma 2004;56:1076–1083. [DOI] [PubMed] [Google Scholar]
- 6. Eck JC, Nachtigall D, Humphreys SC, Hodges SD. Questionnaire survey of spine surgeons on the use of methylprednisolone for acute spinal cord injury. Spine (Phila Pa 1976) 2006;31:E250–E253. [DOI] [PubMed] [Google Scholar]
- 7. Hawryluk GW, Rowland J, Kwon BK, Fehlings MG. Protection and repair of the injured spinal cord: A review of completed, ongoing, and planned clinical trials for acute spinal cord injury. Neurosurg Focus 2008;25:E14. [DOI] [PubMed] [Google Scholar]
- 8. Schabitz WR, Kollmar R, Schwaninger M, et al. Neuroprotective effect of granulocyte colony‐stimulating factor after focal cerebral ischemia. Stroke 2003;34:745–751. [DOI] [PubMed] [Google Scholar]
- 9. Chen WF, Jean YH, Sung CS, et al. Intrathecally injected granulocyte colony‐stimulating factor produced neuroprotective effects in spinal cord ischemia via the mitogen‐activated protein kinase and Akt pathways. Neuroscience 2008;153:31–43. [DOI] [PubMed] [Google Scholar]
- 10. Chen WF, Sung CS, Jean YH, et al. Suppressive effects of intrathecal granulocyte colony‐stimulating factor on excessive release of excitatory amino acids in the spinal cerebrospinal fluid of rats with cord ischemia: Role of glutamate transporters. Neuroscience 2010;165:1217–1232. [DOI] [PubMed] [Google Scholar]
- 11. Chen CH, Huang SY, Chen NF, et al. Intrathecal granulocyte colony‐stimulating factor modulate glial cell line‐derived neurotrophic factor and vascular endothelial growth factor A expression in glial cells after experimental spinal cord ischemia. Neuroscience 2013;242:39–52. [DOI] [PubMed] [Google Scholar]
- 12. Guo Y, Zhang H, Yang J, et al. Granulocyte colony‐stimulating factor improves alternative activation of microglia under microenvironment of spinal cord injury. Neuroscience 2013;238:1–10. [DOI] [PubMed] [Google Scholar]
- 13. Dittgen T, Pitzer C, Plaas C, et al. Granulocyte‐colony stimulating factor (G‐CSF) improves motor recovery in the rat impactor model for spinal cord injury. PLoS ONE 2012;7:e29880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kamiya K, Koda M, Furuya T, et al. Neuroprotective therapy with granulocyte colony‐stimulating factor in acute spinal cord injury: A comparison with high‐dose methylprednisolone as a historical control. Eur Spine J 2015;24:963–967. [DOI] [PubMed] [Google Scholar]
- 15. Takahashi H, Yamazaki M, Okawa A, et al. Neuroprotective therapy using granulocyte colony‐stimulating factor for acute spinal cord injury: A phase I/IIa clinical trial. Eur Spine J 2012;21:2580–2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schabitz WR, Laage R, Vogt G, et al. AXIS: A trial of intravenous granulocyte colony‐stimulating factor in acute ischemic stroke. Stroke 2010;41:2545–2551. [DOI] [PubMed] [Google Scholar]
- 17. Kawabe J, Koda M, Hashimoto M, et al. Neuroprotective effects of granulocyte colony‐stimulating factor and relationship to promotion of angiogenesis after spinal cord injury in rats: Laboratory investigation. J Neurosurg Spine 2011;15:414–421. [DOI] [PubMed] [Google Scholar]
- 18. Xu J, Kim GM, Ahmed SH, et al. Glucocorticoid receptor‐mediated suppression of activator protein‐1 activation and matrix metalloproteinase expression after spinal cord injury. J Neurosci 2001;21:92–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Basso DM, Beattie MS, Bresnahan JC. A sensitive and reliable locomotor rating scale for open field testing in rats. J Neurotrauma 1995;12:1–21. [DOI] [PubMed] [Google Scholar]
- 20. Chen NF, Huang SY, Chen WF, et al. TGF‐beta1 attenuates spinal neuroinflammation and the excitatory amino acid system in rats with neuropathic pain. J Pain 2013;14:1671–1685. [DOI] [PubMed] [Google Scholar]
- 21. Chen NF, Huang SY, Lu CH, et al. Flexibilide obtained from cultured soft coral has anti‐neuroinflammatory and analgesic effects through the upregulation of spinal transforming growth factor‐beta1 in neuropathic rats. Mar Drugs 2014;12:3792–3817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Matthews JN, Altman DG, Campbell MJ, Royston P. Analysis of serial measurements in medical research. BMJ 1990;300:230–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bernards CM, Akers T. Effect of postinjury intravenous or intrathecal methylprednisolone on spinal cord excitatory amino‐acid release, nitric oxide generation, PGE2 synthesis, and myeloperoxidase content in a pig model of acute spinal cord injury. Spinal Cord 2006;44:594–604. [DOI] [PubMed] [Google Scholar]
- 24. Fitch MT, Silver J. Glial cell extracellular matrix: Boundaries for axon growth in development and regeneration. Cell Tissue Res 1997;290:379–384. [DOI] [PubMed] [Google Scholar]
- 25. Silver J, Miller JH. Regeneration beyond the glial scar. Nat Rev Neurosci 2004;5:146–156. [DOI] [PubMed] [Google Scholar]
- 26. McKeon RJ, Jurynec MJ, Buck CR. The chondroitin sulfate proteoglycans neurocan and phosphacan are expressed by reactive astrocytes in the chronic CNS glial scar. J Neurosci 1999;19:10778–10788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chung J, Kim MH, Yoon YJ, Kim KH, Park SR, Choi BH. Effects of granulocyte colony‐stimulating factor and granulocyte‐macrophage colony‐stimulating factor on glial scar formation after spinal cord injury in rats. J Neurosurg Spine 2014;21:966–973. [DOI] [PubMed] [Google Scholar]
- 28. Logan A, Berry M, Gonzalez AM, Frautschy SA, Sporn MB, Baird A. Effects of transforming growth factor beta 1 on scar production in the injured central nervous system of the rat. Eur J Neurosci 1994;6:355–363. [DOI] [PubMed] [Google Scholar]
- 29. Kohta M, Kohmura E, Yamashita T. Inhibition of TGF‐beta1 promotes functional recovery after spinal cord injury. Neurosci Res 2009;65:393–401. [DOI] [PubMed] [Google Scholar]
- 30. Yin J, Sakamoto K, Zhang H, et al. Transforming growth factor‐beta1 upregulates keratan sulfate and chondroitin sulfate biosynthesis in microglias after brain injury. Brain Res 2009;1263:10–22. [DOI] [PubMed] [Google Scholar]
- 31. Jahan N, Hannila SS. Transforming growth factor beta‐induced expression of chondroitin sulfate proteoglycans is mediated through non‐Smad signaling pathways. Exp Neurol 2015;263:372–384. [DOI] [PubMed] [Google Scholar]
- 32. Smith GM, Strunz C. Growth factor and cytokine regulation of chondroitin sulfate proteoglycans by astrocytes. Glia 2005;52:209–218. [DOI] [PubMed] [Google Scholar]
- 33. Yu Z, Yu P, Chen H, Geller HM. Targeted inhibition of KCa3.1 attenuates TGF‐beta‐induced reactive astrogliosis through the Smad2/3 signaling pathway. J Neurochem 2014;130:41–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. McTigue DM, Popovich PG, Morgan TE, Stokes BT. Localization of transforming growth factor‐beta1 and receptor mRNA after experimental spinal cord injury. Exp Neurol 2000;163:220–230. [DOI] [PubMed] [Google Scholar]
- 35. Streit WJ, Semple‐Rowland SL, Hurley SD, Miller RC, Popovich PG, Stokes BT. Cytokine mRNA profiles in contused spinal cord and axotomized facial nucleus suggest a beneficial role for inflammation and gliosis. Exp Neurol 1998;152:74–87. [DOI] [PubMed] [Google Scholar]
- 36. Flanders KC, Ren RF, Lippa CF. Transforming growth factor‐betas in neurodegenerative disease. Prog Neurobiol 1998;54:71–85. [DOI] [PubMed] [Google Scholar]
- 37. Moon LD, Fawcett JW. Reduction in CNS scar formation without concomitant increase in axon regeneration following treatment of adult rat brain with a combination of antibodies to TGFbeta1 and beta2. Eur J Neurosci 2001;14:1667–1677. [DOI] [PubMed] [Google Scholar]
- 38. Zhu Y, Yang GY, Ahlemeyer B, et al. Transforming growth factor‐beta 1 increases bad phosphorylation and protects neurons against damage. J Neurosci 2002;22:3898–3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Fitch MT, Doller C, Combs CK, Landreth GE, Silver J. Cellular and molecular mechanisms of glial scarring and progressive cavitation: In vivo and in vitro analysis of inflammation‐induced secondary injury after CNS trauma. J Neurosci 1999;19:8182–8198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rhodes KE, Raivich G, Fawcett JW. The injury response of oligodendrocyte precursor cells is induced by platelets, macrophages and inflammation‐associated cytokines. Neuroscience 2006;140:87–100. [DOI] [PubMed] [Google Scholar]
- 41. Boneberg EM, Hartung T. Molecular aspects of anti‐inflammatory action of G‐CSF. Inflamm Res 2002;51:119–128. [DOI] [PubMed] [Google Scholar]
- 42. Heard SO, Fink MP. Counterregulatory control of the acute inflammatory response: Granulocyte colony‐stimulating factor has anti‐inflammatory properties. Crit Care Med 1999;27:1019–1021. [DOI] [PubMed] [Google Scholar]
- 43. Mu Y, Gudey SK, Landstrom M. Non‐Smad signaling pathways. Cell Tissue Res 2012;347:11–20. [DOI] [PubMed] [Google Scholar]
- 44. Mueller BK, Mack H, Teusch N. Rho kinase, a promising drug target for neurological disorders. Nat Rev Drug Discov 2005;4:387–398. [DOI] [PubMed] [Google Scholar]
- 45. Vardouli L, Moustakas A, Stournaras C. LIM‐kinase 2 and cofilin phosphorylation mediate actin cytoskeleton reorganization induced by transforming growth factor‐beta. J Biol Chem 2005;280:11448–11457. [DOI] [PubMed] [Google Scholar]
- 46. Dergham P, Ellezam B, Essagian C, Avedissian H, Lubell WD, McKerracher L. Rho signaling pathway targeted to promote spinal cord repair. J Neurosci 2002;22:6570–6577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gopalakrishnan SM, Teusch N, Imhof C, et al. Role of Rho kinase pathway in chondroitin sulfate proteoglycan‐mediated inhibition of neurite outgrowth in PC12 cells. J Neurosci Res 2008;86:2214–2226. [DOI] [PubMed] [Google Scholar]
- 48. Choi JK, Park SY, Kim KH, Park SR, Lee SG, Choi BH. GM‐CSF reduces expression of chondroitin sulfate proteoglycan (CSPG) core proteins in TGF‐beta‐treated primary astrocytes. BMB Rep 2014;47:679–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Monnier PP, Sierra A, Schwab JM, Henke‐Fahle S, Mueller BK. The Rho/ROCK pathway mediates neurite growth‐inhibitory activity associated with the chondroitin sulfate proteoglycans of the CNS glial scar. Mol Cell Neurosci 2003;22:319–330. [DOI] [PubMed] [Google Scholar]
- 50. Schneider A, Kruger C, Steigleder T, et al. The hematopoietic factor G‐CSF is a neuronal ligand that counteracts programmed cell death and drives neurogenesis. J Clin Invest 2005;115:2083–2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kleinschnitz C, Schroeter M, Jander S, Stoll G. Induction of granulocyte colony‐stimulating factor mRNA by focal cerebral ischemia and cortical spreading depression. Brain Res Mol Brain Res 2004;131:73–78. [DOI] [PubMed] [Google Scholar]
- 52. Liberto CM, Albrecht PJ, Herx LM, Yong VW, Levison SW. Pro‐regenerative properties of cytokine‐activated astrocytes. J Neurochem 2004;89:1092–1100. [DOI] [PubMed] [Google Scholar]
- 53. Faulkner JR, Herrmann JE, Woo MJ, Tansey KE, Doan NB, Sofroniew MV. Reactive astrocytes protect tissue and preserve function after spinal cord injury. J Neurosci 2004;24:2143–2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ladeby R, Wirenfeldt M, Garcia‐Ovejero D, et al. Microglial cell population dynamics in the injured adult central nervous system. Brain Res Brain Res Rev 2005;48:196–206. [DOI] [PubMed] [Google Scholar]
- 55. Prewitt CM, Niesman IR, Kane CJ, Houle JD. Activated macrophage/microglial cells can promote the regeneration of sensory axons into the injured spinal cord. Exp Neurol 1997;148:433–443. [DOI] [PubMed] [Google Scholar]
- 56. Rapalino O, Lazarov‐Spiegler O, Agranov E, et al. Implantation of stimulated homologous macrophages results in partial recovery of paraplegic rats. Nat Med 1998;4:814–821. [DOI] [PubMed] [Google Scholar]
- 57. Bessis A, Bechade C, Bernard D, Roumier A. Microglial control of neuronal death and synaptic properties. Glia 2007;55:233–238. [DOI] [PubMed] [Google Scholar]
- 58. Kempermann G, Neumann H. Neuroscience. Microglia: The enemy within? Science 2003;302:1689–1690. [DOI] [PubMed] [Google Scholar]
- 59. Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms Underlying Inflammation in Neurodegeneration. Cell 2010;140:918–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Philips T, Robberecht W. Neuroinflammation in amyotrophic lateral sclerosis: Role of glial activation in motor neuron disease. Lancet Neurol 2011;10:253–263. [DOI] [PubMed] [Google Scholar]