
Figure 2.
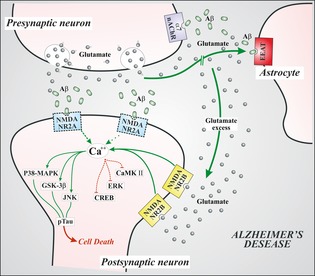
This figure represents the glutamate‐mediated transmission at synaptic level in Alzheimer's disease. Aβ oligomers interfere with NMDA signaling, inducing an internalization of postsynaptic NMDANR2A subunits (dotted). Due to interference of Aβ with EAAT, glutamate concentration in synaptic cleft increases. Moreover, Abeta oligomers form complexes with alpha7‐nicotinic receptors at presynaptic sites. This interaction induces increased levels of glutamate release. Glutamate spillover would activate extrasynaptic NMDANR2B receptors, with increased calcium levels and activation of metabolic pathways (green path) responsible for neuronal shrinkage and synaptic loss, associated with inhibition of prosurvival pathways (red path). The more this condition persists, the more the activated pathologic pathways lead to hyperphosphorylation of cytoskeletal tau protein, with neuronal degeneration and cell death. VGlut (vesicular glutamate transporter); EAAT (excitatory amino acid transporter); α7‐nAchR (alpha‐7 nicotinic acetylcholine receptor); NMDANR2A (N‐methyl‐d‐aspartate NR2A subunit); NMDANR2B (N‐methyl‐d‐aspartate NR2B subunit); ERK (extracellular signal‐related kinase); CaMKII (calcium calmodulin‐dependent kinase II); CREB (cyclic AMP response element binding protein); GSK3β (glycogen synthase kinase 3β); p38‐MAPK (p38 mitogen‐activated protein kinase), JNK (c‐Jun N‐terminal kinase), pTau (hyperphosphorylated tau protein).