

Abstract
Offspring exposed in utero to maternal diabetes exhibit long lasting insulin resistance, though the initiating mechanisms have received minimal experimental attention. Herein, we show that rat fetuses develop insulin resistance after only 2 days continuous exposure to isolated hyperglycemia starting on gestational day 18. Hyperglycemia-induced reductions in insulin-induced AKT phosphorylation localized primarily to fetal skeletal muscle. The skeletal muscle of hyperglycemia exposed fetuses also exhibited impaired in-vivo glucose uptake. To address longer term impacts of this short hyperglycemic exposure, neonates were cross-fostered and examined at 21 days postnatal age. Offspring formerly exposed to 2 days late gestation hyperglycemia exhibited mild glucose intolerance with insulin signaling defects localized only to skeletal muscle. Fetal hyperglycemic exposure has downstream consequences which include hyperinsulinemia and relative uteroplacental insufficiency. To determine whether these accounted for induction of insulin resistance, we examined fetuses exposed to late gestational isolated hyperinsulinemia or uterine artery ligation. Importantly, 2 days of fetal hyperinsulinemia did not impair insulin signaling in murine fetal tissues and 21 day old offspring exposed to fetal hyperinsulinemia had normal glucose tolerance. Similarly, fetal exposure to 2 days uteroplacental insufficiency did not perturb insulin stimulated AKT phosphorylation in fetal rats. We conclude that fetal exposure to hyperglycemia acutely produces insulin resistance. As hyperinsulinemia and placental insufficiency have no such impact, this occurs likely via direct tissue effects of hyperglycemia. Furthermore, these findings show that skeletal muscle is uniquely susceptible to immediate and persistent insulin resistance induced by hyperglycemia.
Keywords: Skeletal Muscle, Pregnancy, Diabetes, Developmental Origins of Health and Disease, Insulin Signaling, Glucose Metabolism, Ontogeny
Introduction
Offspring born from pregnancy complicated by diabetes have a lifelong increased risk of insulin resistance. In humans, such offspring have a 4–8 fold higher risk, beyond genetic risk, to develop type 2 diabetes, metabolic syndrome, and/or obesity (Dabelea et al. 2000, 2008; Franks et al. 2006; Clausen et al. 2008, 2009; Fraser & Lawlor 2014) . Thus, the diabetes-altered fetal environment induces a long-lasting change in offspring susceptibility to adverse metabolic outcomes. Likewise in animals, adult offspring that were exposed to maternal diabetes exhibit insulin resistance, as demonstrated in multiple models (Simmons et al. 2001a; Segar et al. 2009; Khalyfa et al. 2013; Kahraman et al. 2014; Latouche et al. 2014; Yokomizo et al. 2014; Blue et al. 2015). However, the underlying mechanisms that produce insulin resistance are not clearly understood. In fact, the immediate fetal molecular perturbations induced by maternal diabetes have received little experimental attention to date.
Diabetes during pregnancy exposes the fetus to multiple perturbations during a critical development period. These perturbations extend beyond hyperglycemia and include maternal ketosis, altered lipids and amino acids, altered placental hemodynamics, maternal inflammation, and systemic oxidative stress (Wijendran et al. 1999; Aerts & Van Assche 2001; Cetin et al. 2005; Gin et al. 2006; López-Tinoco et al. 2013; Ryckman et al. 2015; Shang et al. 2015; Zhu et al. 2015; Abdul Aziz et al. 2016; Khosrowbeygi et al. 2016; Saad et al. 2016). Although hyperglycemia is the defining aspect of diabetes, the role of maternal hyperglycemia itself in altering fetal metabolic programming has not been well isolated. In fact, some non-hyperglycemic factors independently modulate offspring insulin resistance risk (Mathias et al. 2014; Crume et al. 2015; Shomonov-Wagner et al. 2015; Umekawa et al. 2015). A longstanding obstacle to understanding the role of hyperglycemia has been lack of a technique to isolate glucose exposure from the other concurrent systemic maternal perturbations during diabetes. The localized fetomaternal hyperglycemia rat model circumvents these confounders by selectively delivering glucose to fetuses in the left uterine horn, while using right uterine horn fetuses as internal euglycemic controls (Yao et al. 2010; Baack et al. 2014; Gordon et al. 2015). Importantly, this model both isolates the effect of maternal hyperglycemia and allows precise temporal control of hyperglycemia exposure.
Nearly all animal studies to date examining fetally programmed insulin resistance have focused on the physiologic and molecular perturbations later rather than early in life (Simmons et al. 2001a; Segar et al. 2009; Khalyfa et al. 2013; Latouche et al. 2014; Yokomizo et al. 2014; Blue et al. 2015) . Although these later life findings are crucial to understanding the biology underlying offspring complications, they may not identify the primary molecular mechanisms, in that secondary and tertiary pathology from other postnatal factors may be at play. Understanding the earliest molecular perturbations may be essential to understanding the mechanisms that program offspring metabolic risk.
The aim of this study was to better understand the mechanisms by which diabetes exposure induces offspring insulin resistance, in part by characterizing the earliest impacts on insulin action and signaling. To accomplish our aim, we developed in vivo techniques to study rodent fetus glucose disposition, insulin sensitivity, and insulin signaling. We examined three distinct aspects of fetal diabetes exposure: fetal hyperglycemic exposure, uteroplacental insufficiency, and fetal hyperinsulinemia. Isolated uteroplacental insufficiency was examined because fetal hypoxia results from maternal diabetes, owing to a relative uteroplacental insufficiency via mechanisms that may include structural placental changes and/or increased fetal metabolic demand (Widness et al. 1981; Daskalakis et al. 2008; Taricco et al. 2009; Escobar et al. 2013) . Isolated fetal hyperinsulinism was studied because hyperglycemic exposure often increases fetal insulin (Gordon et al. 2015). We hypothesized that fetal exposure to late-gestational hyperglycemia would induce immediate tissue-specific insulin resistance which would persist into later life. We found that fetuses exposed to hyperglycemia rapidly developed skeletal muscle insulin resistance while still in utero. The insulin signaling defects persisted through postnatal life, localized to skeletal muscle. Uteroplacental insufficiency and fetal hyperinsulinism did not have these effects, suggesting that fetal hyperglycemia may directly and persistently impact fetal skeletal muscle.
Research Design and Methods
Animals and Breeding
All procedures were performed within the regulations of Animal Welfare Act and the National Institutes of Health Guide for the Care and Use of Laboratory Animals, and were approved by the University of Iowa Institutional Animal Care and Use Committee. Rodents were housed in a temperature controlled, 12-hour light-dark cycled animal care facility with free access to water and regular chow. Surgery was performed under inhalational isoflurane anesthesia after at least 24-hours of acclimation to the facility. Post-operative analgesia included topical bupivacaine applied immediately after wound closure and subcutaneous buprenorphine (0.05mg/kg) provided twice daily. For experiments conducted to study postnatal outcomes, 5 mg progesterone (mixed with 1cc of peanut oil) was administered intraperitoneally 24 hours after surgery to prevent preterm labor thus allowing Caesarean delivery at 21 days gestation. All procedures involving fetal manipulation or collection were performed in a fashion alternating between the two uterine horns to account for any time-dependent changes in environment.
Localized Fetomaternal Hyperglycemia:
A vascular catheter draining into the left uterine artery was placed in timed pregnant Hsd:Sprague Dawley SD rats (Envigo, Indianapolis, IN) as described (Yao et al. 2010) , including placement of ligatures on the iliac, superior gluteal and hypogastric trunk arteries. Catheters were secured within a Covance Infusion Harness (CIH95, Instech Lab, Plymouth Meeting PA) and attached to a single channel stainless steel swivel (375/22, Instech Lab, Plymouth Meeting PA). Glucose was infused through the catheter at the rate of 4 mg/min for 48 hours during late gestation to expose the left uterine horn fetuses to hyperglycemia. During infusion, the pregnant dams were awake and mobile, with unlimited access to water and food. For studies examining fetal outcomes, infusions were conducted on gestational days (GD) 18–20 in order to allow examination on GD20, prior to the time of parturition on GD21. For studies examining offspring, infusions were conducted on GD 19–21, to allow C-section delivery on GD21. Fetuses delivered by C-section were cross-fostered to healthy mothers.
Unilateral Uterine Artery Ligation:
The left or right lower abdominal quadrant were incised in GD18 rats to gain access to the left or right uterine artery respectively. The ipsilateral artery was ligated with 3–0 silk suture and the abdominal wall closed. The contralateral uterine artery was not manipulated and the contralateral fetuses served as paired controls. Fetuses from both uterine horns were collected 48 hours after uterine artery ligation.
Fetal Hyperinsulinemia:
Insulin detemir (Levemir, Novo Nordisk, Plainsboro, New Jersey) is a long acting insulin analog, and was administered to fetuses in order to induce late gestational fetal hyperinsulinemia. Pregnant C57BL/6J mice (Jackson Laboratory, Bar Harbor, ME) underwent laparotomy at E17.5 to exteriorize both uterine horns under warm, moist, sterile conditions. Fetuses were injected subcutaneously on their flank with either 0.1 unit of long acting insulin detemir diluted in saline containing 10% of ATS-3 General Rodent Tattoo System Pigment (AIMS Inc, Hornell, NY), or an equivalent amount of diluent and pigment, using an insulin syringe with a 31G X 5/16” needle (Easy Touch, Houston, Texas). Over 1–2 days of action, the detemir dose of 0.1 units amounts to an average insulin dose of 50–100 mU/kg/minute in a typical 0.7 gram fetus. This insulin exposure is hyperinsulinemic, less than a magnitude higher than what is used for high dose hyperinsulinemic clamps in adult rodents. Note that because the fetus is constantly supplied with glucose by the placenta, there is not concern for severe sustained hypoglycemia. In fact, much higher doses of insulin have been administered to rodent fetuses: for example, injection of 5 units insulin into gestational day 17 fetuses (Ogata et al. 1988) amounting to ~900 mU/kg/min. The experimental treatment administered (detemir or diluent) was encoded by the laterality of the flank tattoo. Both uterine horns were placed back in abdominal cavity and the abdominal wall closed. Fetuses were euthanized 48 hours after injection or allowed to be delivered spontaneously and cared for by the same mother.
Biochemical analyses:
Fetal blood was collected via decapitation before placenta detachment. Tail blood was collected from 21 days old pups at 0, 15, 30, 60 and 120-minutes during glucose tolerance testing. Fetal and 21 day old pups blood glucose levels were measured using One Touch Ultra meter (LifeScan Inc., Milpitas, CA). Serum insulin concentrations were measured using Ultrasensitive Rat Insulin ELISA kit (#90060, Crystal Chem, Downers Grove, IL).
Radiolabeled glucose uptake:
Two hours after completion of glucose infusion, pregnant mothers were given 350 microcuries of 2-[1,2-3H(N)]-deoxy-D-glucose (3H-2DG) over 30 seconds via inferior vena cava injection while alternate fetuses in both uterine horns were injected with regular insulin (Novolin R, Novo Nordisk Inc, Plainsboro, New Jersey) or saline (negative control). Fetal tissues were collected 20 minutes thereafter and snap frozen immediately. Tissue samples were dissolved in 4N KOH overnight and the supernatants were neutralized. The 3H content was measured by liquid scintillation counting using a Beckman LS6000 Scintillation Counter (GMI, Ramsey, Minnesota). Tissue sample 3H content was also examined in supernatants after precipitation with Ba(OH)2 plus ZnSO4 (Halseth et al. 2000) to remove 2-deoxyglucose-6-phosphate. Tissue 2DG-6-P content was calculated by determining the difference of radioactivity with and without Ba(OH)2 : ZnSO4 precipitation, then normalized to tissue weight, and reported as that relative to normalized 2DG-6-P content in brain (Sawatzke et al. 2015) .
In-vivo insulin signaling:
Insulin administered at maximal stimulating doses, roughly 200 units/kg (Li et al. 2015), was used to acutely activate insulin signaling. To study tissue insulin stimulated responses, fetuses received 0.5 unit regular insulin (Novolin R, Novo Nordisk Inc, Plainsboro, New Jersey) or saline intraperitoneally while remaining in-utero. For postnatal studies, 21 days old pups were given 5 units regular insulin or saline via inferior vena cava injection. Skeletal muscle (muscle from whole leg for fetuses, soleus for 21 day old pups), liver and brown fat tissues were collected and snap frozen 15 minutes after injection.
Immunodetection:
Tissues were homogenized on ice and centrifuged at 10,000rpm at 4 degrees Celsius for 4 minutes. Supernatant content of AKT/phospho-AKT, PDK1/phosphor-PDK1, MTOR/phospho-MTOR, PTEN/phospho-PTEN was measured using western blotting as previously reported. (Baack et al. 2014; Gordon et al. 2015) Polyclonal antibodies recognizing AKT (9272, 1:500), phosphorylated AKT Ser 473 (9271, 1:500), PDK1 (3062, 1:1000), phosphorylated PDK1 Ser241 (3438, 1:1000), PTEN (9188, 1:1000), phosphorylated PTEN Ser380/Thr382/383 (9549, 1:1000), MTOR (2983, 1:1000), phosphorylated MTOR Ser2448 (5536, 1:1000) were purchased from Cell Signaling Technology, Inc, Danvers, MA. Anti-β-actin antibody was purchased from Sigma-Aldrich (A5441). Immunodetected protein quantity was determined by measuring pixel intensity of immunoblot bands using ImageJ software (NIH). In vivo insulin stimulated AKT-phosphorylation was calculated by dividing phosphorylated AKT over total AKT. Samples from the same mother were run and analyzed on the same blot so as to preserve the natural within-mother pairing of samples. Total IRS-1 and tyrosine phosphorylated IRS-1 protein content was quantified using MSD Insulin Signaling Panel Kit (Total protein - K15152C, phospho-protein panel: K15151C, Mesoscale Discovery, Rockville, MD). Phospho-AKT (Ser 473), GSK3-β (Ser 9), p70s6 kinase (Thr421/Ser424) were measured using MSD AKT Signaling Whole Cell Lysate Kit (K15115D-1). Fetal tissue lysates were prepared following manufacturer’s instruction and loaded to 96 well plates at a protein concentration of 20 ug/well. The 96-well plates were read with MESO QuickPlex SQ 120 (Mesoscale Discovery, Rockville, MD).
Statistical Analysis:
The statistical significance of differences between means of maternal measures was assessed by Student’s t-test. The impact of fetal interventions on fetal and offspring outcomes was assessed by paired t-test weighted by the total number of fetuses/offspring examined per mother, as implemented in the weights package in R (version 3.3.3, The R Foundation for Statistical Computing). This approach accounted for the natural pairing by mother and the inherently unbalanced number of replicates per mother per treatment since the number of fetuses per mother and per uterine horn varies naturally. The weighted paired t-test approach was used only for single interventions (i.e. simple comparison of two groups). For 2×2 experimental designs assessing fetal or offspring outcomes, linear mixed effects modeling was used treating the two interventions as mixed effects with potential interaction and treating mothers as a random effect thus effectively accounting for individual fetuses/offspring as within-mothers repeated measures. This was accomplished using the R package lme4, with p-values calculated using type-III analysis-of-variance tables as implemented in the car R package. Results of glucose tolerance tests were assessed by calculating the area under the curve (AUC) using the trapazoidal rule, followed by weighted, paired t-test comparing the two groups. Two-sided p values were computed and significance was defined as p<0.05. Confidence intervals, 95%, (CI95) were calculated from the standard errors estimated by the weighted paired t-test for single intervention experiment and from the lme4 package for 2×2 experiments. To help visualize within-mother pairing in figure panels which compare fetal treatments, data which originate from the same mother have the same color and are connected by a line. Thus, within-mother pairings are encoded by color and lines connecting the mean observed values. Note that colors are not conserved between figure panels and panels often represent different maternal cohorts. All data includes results from both genders. Bars represent means ± standard errors of measurement (SEM), calculated per mother for maternal outcomes and per fetus for fetal outcomes.
Results
Hyperglycemia Rapidly Induces Insulin Resistance in Fetal Skeletal Muscle
Rat fetuses were exposed to 48 hours of hyperglycemia via infusion of glucose into the left uterine artery on gestational days 18–20 (Figure 1A). Maternal serum glucose was unchanged during infusion (Figure 1B). By contrast, during infusion, glucose exposed fetuses experienced moderate hyperglycemia, whereas control fetuses remained euglycemic (Figure 1C, CI95 = +21–147 mg/dL). Note that the glucoses in Figures 1B and 1C should not be directly compared because they were obtained under differing conditions: 1B while the mothers were awake and ambulatory whereas 1C samples were collected during maternal general anesthesia and laparotomy which caused maternal glucose concentrations to rise. Previous work showed that fetal insulin is increased upon 24 hours hyperglycemic exposure using this model (Gordon et al. 2015) . However, by 48 hours of hyperglycemic infusion fetal insulin levels were not significantly different than in euglycemic fetuses (Figure 1D, CI95 = −1.2 – +2.1 ng/mL) despite fetal hyperglycemia.
Figure 1: Transient fetal hyperglycemia during late gestation.
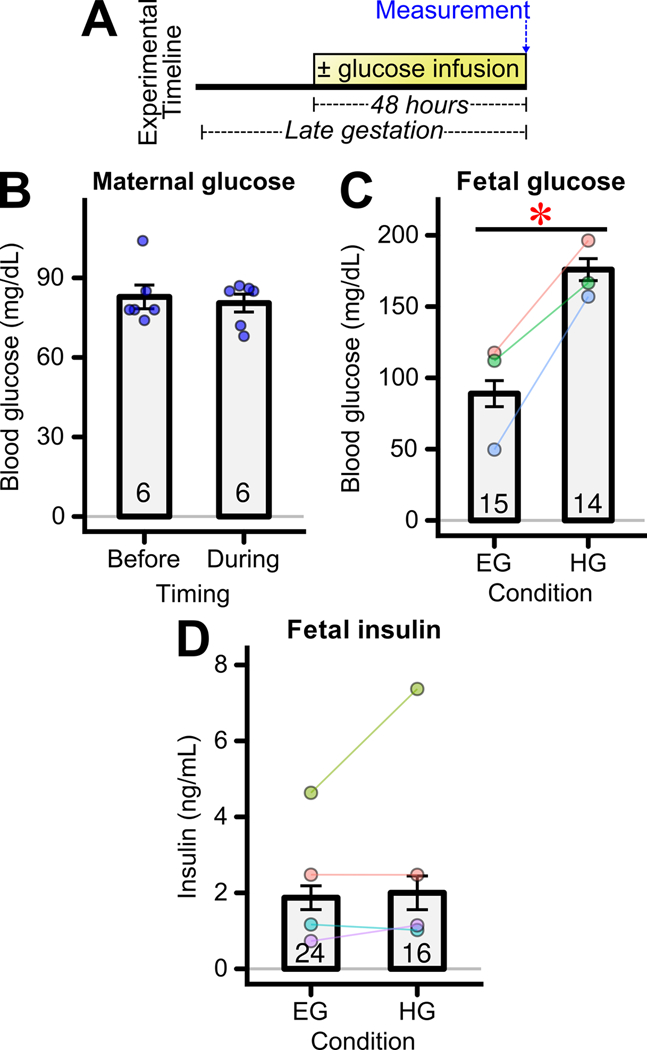
(A) Experimental timeline. Glucose was infused into the left uterine artery for 48 hours during late gestation. (B) Maternal blood glucose level before or just before the conclusion of 48 hours glucose infusion during late gestation (n=6). Maternal blood was collected during the awake state. Bars represent the mean±SEM for these maternal measures. (C) Fetal blood glucose level just before the conclusion of 48 hours exposure to euglycemia (EG) or hyperglycemia (HG) (n = 14–15 fetuses/groups from 3 mothers) * p<0.05 for difference between groups. Fetal blood was collected during isoflurane and maternal laparotomy. (D) Fetal serum insulin level just before the conclusion of 48 hours of infusion (n = 16–24 fetuses/group from 4 mothers). (C,D) In this and all subsequent plots for fetal measures, bars represent the mean±SEM across fetuses, with individual points of the same color representing the mean obtained from fetuses originating from a single mother. The data obtained from fetuses of the same mother are also connected by a solid line to aid visualization of this natural pairing and the trend of within-mother changes. The colors between panels are not necessarily conserved and often panels represent different cohorts. The total number of fetuses/offspring examined per group is indicated in each bar. For example, regarding Figure 1C there were 15 EG and 14 HG fetuses examined, obtained from 3 mothers shown in blue, pink, and green.
To assess fetal insulin sensitivity, the uptake of maternally delivered tritiated 2-deoxyglucose (3H-2DG) into fetal tissues was measured with and without acute fetal insulin treatment (Figure 2A). Levels of 3H-2DG in maternal serum underwent the expected rapid peak after infusion and subsequent decay (Figure 2B). Fetal tissue content of tritiated 2-deoxyglucose-phosphate (3H-2DG-P) was measured 20 minutes after maternal 3H-2DG infusion. When considered across both hyperglycemia- and euglycemia-exposed fetuses, insulin had no statistically significant impact on tissue 3H-2DG-P content. However, there was a significant reduction in 3H-2DG-P content in hyperglycemia- compared to euglycemia-exposed fetuses when considered across both saline and insulin treatment groups (Figure 2C, CI95 = −0.49 – −0.01). The lower uptake of 3H-2DG-P into hyperglycemia-exposed fetal muscle suggested the possibility of impaired insulin sensitivity in this tissue.
Figure 2: Effects of transient hyperglycemia on fetal skeletal muscle insulin sensitivity.
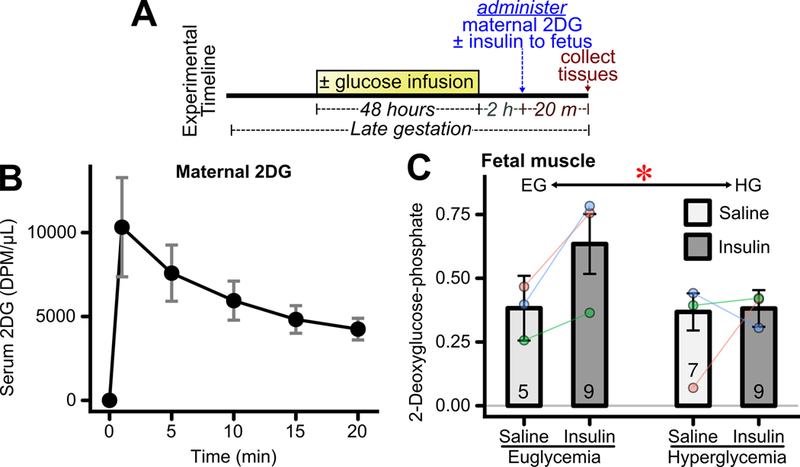
(A) Experimental timeline. Glucose was infused into the left uterine artery for 48 hours during late gestation. The hyperglycemic infusion was discontinued. Two hours later tritiated 2-deoxyglucose (2DG) was administered maternally and fetuses were administered insulin or saline. Fetal tissues were collected 20 minutes thereafter. (B) Maternal serum scintillation count after 2DG injection (n = 4 mothers), error bars represent per-mother SEM. (C) Fetal skeletal muscle insulin stimulated tritiated 2-deoxyglucose uptake, relative to that in fetal brain, after 48 hours exposure to euglycemia or hyperglycemia. (n=5–9 fetuses/group from 3 mothers). *p<0.05 for overall difference between euglycemia and hyperglycemia groups.
To better assess insulin actions in this tissue, the impact of hyperglycemic exposure on insulin-stimulated AKT phosphorylation was assessed; Figure 3A shows relevant experimental design. There was a significant impairment in insulin-stimulated AKT phosphorylation in hyperglycemia infused fetal skeletal muscle (Figure 3B, CI95 = −0.55 – −0.0007). Tyrosine phosphorylation of IRS-1 in skeletal muscle exhibited trends towards an increase in insulin-stimulated versus saline-treated fetuses across both the hyperglycemia- and euglycemia- exposed groups (Figure 3C, CI95 −0.002 – +0.03), though this increase did not reach statistical significance (p=0.096). However, the response to insulin did not differ between the euglycemia and hyperglycemia exposed groups (Figure 3C, CI95= −0.01 – +0.02). Furthermore, there were no changes among several proteins that influence upstream aspects of insulin signaling. Namely there were no changes in skeletal muscle PTEN, PDK-1, mTOR, or their phosphorylation states induced by fetal hyperglycemic exposure (Figure 3D).
Figure 3: Effects of transient hyperglycemia on fetal skeletal muscle insulin signaling.
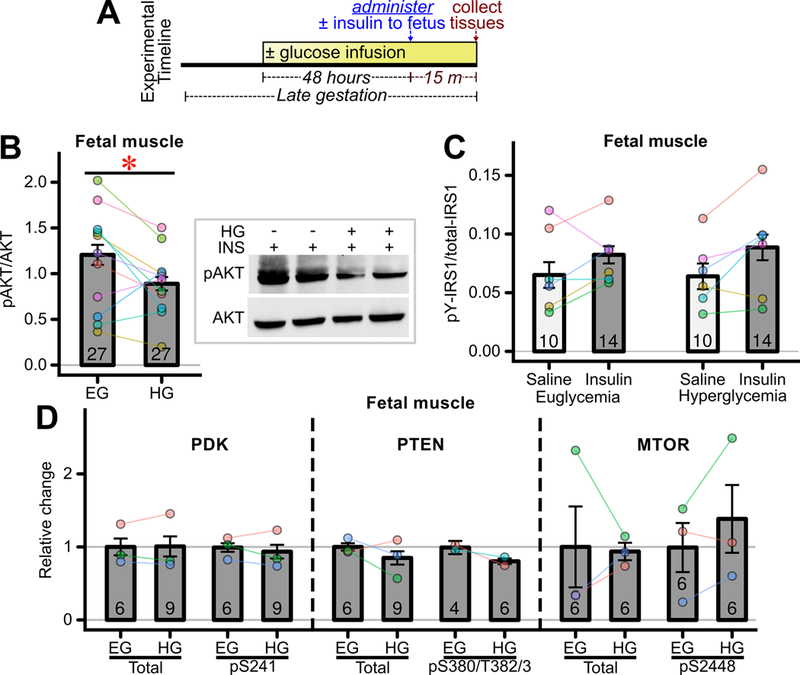
(A) Experimental timeline. Glucose was infused into the left uterine artery for 48 hours during late gestation. While the infusion was continued, regular insulin or saline was administered to fetuses and fetal tissues were collected 15 minutes thereafter. (B) Fetal skeletal muscle in-vivo insulin (INS) stimulated AKT phosphorylation in fetuses exposed to euglycemia (EG) or hyperglycemia (HG) (n=27 fetuses/group from 11 mothers). * p<0.05 for difference between the two groups. Representative western blots shown on right of panel. (C) Tyrosine phosphorylated IRS-1 expression measured using MSD Multiplex Assay (n = 10–14 fetuses/group from 6 mothers) in saline and insulin stimulated fetuses. (D) Relative protein expression of signaling molecules proximal to AKT measured using MSD Multiplex Assay. (Total PDK, phospho PDK, total MTOR, phospho MTOR and total PTEN n=6–9 fetuses/groups from 3 mothers, and phospho PTEN n = 4–6 fetuses/group from 2 mothers).
Hyperglycemia Rapidly Alters Insulin Signaling in Fetal Brown Fat and Liver
The impacts of fetal hyperglycemic exposure on insulin action in brown adipose tissue and liver were also examined. In fetal brown adipose tissue, hyperglycemic exposure produced similar trends in glucose uptake (Figure 4A) as observed in skeletal muscle, though there were no statistically significant differences between the groups (CI95 = −0.65 – +0.03) . Likewise, there was a trend towards impairment of insulin-stimulated AKT phosphorylation in brown adipose tissue induced by hyperglycemia exposure (Figure 4B, mean change −0.57, CI95= −1.2 – +0.01) similar to that observed in skeletal muscle (Figure 3B), though the change in brown adipose tissue did not quite reach statistical significance (p=0.055).
Figure 4: Effects of transient hyperglycemia on fetal brown adipose tissue (BAT) insulin action.
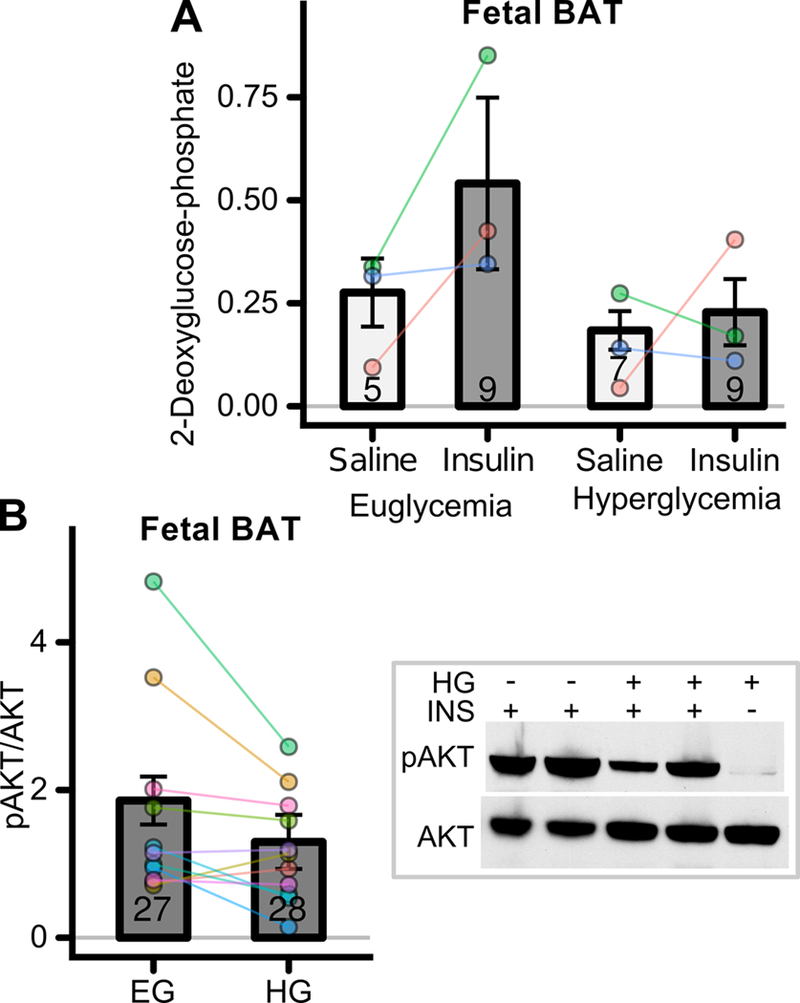
(A) See Figure 2A for experimental timeline. Fetal BAT insulin stimulated 2DG uptake in fetuses exposed to euglycemia or hyperglycemia (n=5–9 fetuses/group from 3 mothers). (B) See Figure 3A for experimental timeline. Fetal BAT insulin (INS) stimulated AKT phosphorylation in fetuses exposed to euglycemia (EG) or hyperglycemia (HG) (n=27–28 fetuses/group from 11 mothers). Representative western blots shown to right of panel.
Liver insulin action is not readily amenable to study by measuring 3H-2-DG uptake or phosphorylation. Instead we measured not only AKT phosphorylation but also GSK-3β serine phosphorylation, a distal aspect of insulin signaling in liver. Insulin stimulated phosphorylation of AKT and of GSK in fetal liver, both in euglycemia and hyperglycemia exposed fetuses (Figure 5A/B). However, overall GSK phosphorylation levels were reduced in hyperglycemia compared to euglycemia-exposed fetuses across conditions (Figure 5B, CI95 −0.17 – −0.02). Likewise, phosphorylation of AKT in liver showed similar reductions (Figure 5A, CI95 −0.67 – +0.06), though this did not reach statistical significance (p=0.10).
Figure 5: Effects of transient hyperglycemia on fetal liver insulin signaling.
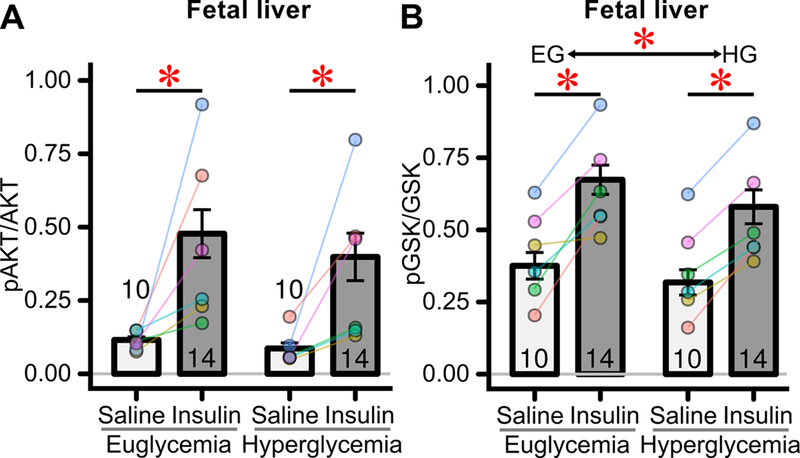
See Figure 3A for relevant experimental timeline. (A) Fetal liver insulin stimulated AKT phosphorylation measured by MSD multiplex panel (n=10–14 fetuses/group, 6 mothers) just before the conclusion of exposure to euglycemia or hyperglycemia. The increase induced by insulin was significant for each group (euglycemia and hyperglycemia). * p<0.05 for increase induced by insulin across experimental groups. (B) Fetal liver insulin stimulated GSK phosphorylation measured by MSD multiplex panel (n=10–14 fetuses/group, 6 mothers). The increase induced by insulin was significant for each group (euglycemia and hyperglycemia). Lower *: p<0.05 for increase induced by insulin across experimental groups. Upper *: p<0.05 for overall difference between euglycemia and hyperglycemia exposed fetuses.
Fetal Hyperinsulinemia Has No Acute Impact on Insulin Signaling
As previously reported, hyperglycemia exposed fetuses develop hyperinsulinemia after 24 hours of infusion (Gordon et al. 2015) though this is not sustained at 48 hours (Figure 1D). Maternal diabetes and hyperglycemia can also induce fetal hyperinsulinemia in humans and other animal models (de Ménibus & Mallet 1980; White et al. 2015) , though the impact of isolated fetal hyperinsulinemia on development of insulin resistance is unknown. It is possible that transient hyperinsulinemia could modulate fetal tissue insulin sensitivity. For example, hyperinsulinemia induces insulin resistance in a variety of cell culture models (Andreozzi et al. 2011). To test this directly in vivo during fetal maturation, mouse fetuses were administered, in utero, detemir, a long acting neutral-pH insulin, or diluent as control (Figure 6A). Insulin sensitivity was then assessed 48 hours after insulin detemir administration, by measuring the acute response to regular insulin fetal injection. In both detemir and diluent administered fetuses, regular insulin produced a rapid and significant reduction in fetal blood glucose (Figure 6B), indicative of intact insulin action. There were no statistically significant differences in glucoses between detemir and diluent treated fetuses, nor was there a statistical difference in their responses to acute insulin (Figure 6B). Consistent with this, insulin-stimulated AKT phosphorylation was not statistically altered in detemir- versus diluent- injected fetuses when examined in fetal skeletal muscle (Figure 6C), brown adipose tissue (Figure 6D), or liver (Figure 6E).
Figure 6: Impact of fetal hyperinsulinemia on acute insulin signaling.
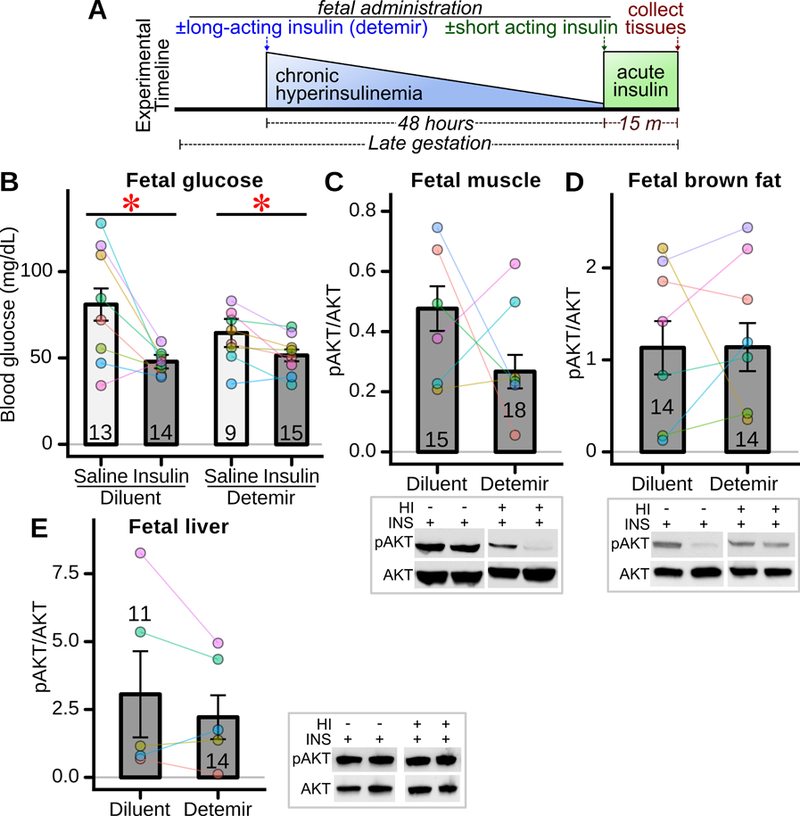
(A) Experimental timeline. Detemir, a long-acting insulin, or control diluent, was administered to fetuses during late gestation to induce chronic hyperinsulinemia (HI). Forty-eight hours later, regular insulin (short acting) or saline was administered to the fetuses, and fetal blood / tissues collected 15 minutes thereafter to assess insulin signaling. (B) Fetal blood glucose in response to exogenous insulin stimulation (n=9–15 fetuses/group, 8 mothers) after 48 hours exposure to long-acting insulin (detemir) or control (diluent). * p<0.05 for decrease in glucose induced by insulin across groups. (C) In-vivo insulin stimulated AKT phosphorylation in fetal skeletal muscle (n=15–18 fetuses/group from 6 mothers), (D) fetal brown adipose tissue (n=14 fetuses/group, 7 mothers), and (E) fetal liver (n=11–14 fetuses/group, 5 mothers). Representative western blots shown adjacent to panels.
Fetal Uteroplacental Insufficiency Does Not Acutely Impair Fetal Insulin Signaling
Fetoplacental exposure to maternal diabetes and hyperglycemia results in fetal hypoxia, owing to a relative uteroplacental insufficiency via mechanisms that may include structural placental changes and/or increased fetal metabolic demand (Widness et al. 1981; Daskalakis et al. 2008; Taricco et al. 2009; Escobar et al. 2013). Thus, relative uteroplacental insufficiency and fetal hypoxia may have been induced in our rat model of hyperglycemia exposure and thus could have been a mechanism of fetal perturbation. Relevantly, uteroplacental insufficiency induces subsequent offspring insulin resistance, which is thought to involve skeletal muscle (Simmons et al. 2001b; Selak et al. 2003; Gatford et al. 2010) . It was thus of interest to determine the impact of uteroplacental insufficiency on fetal skeletal muscle insulin sensitivity. Uteroplacental insufficiency was induced in rats by unilateral uterine artery ligation and fetuses examined 48 hours thereafter (Figure 7A). As expected, fetal rats exposed to uterine artery ligation were significantly smaller (Figure 7B). However, unlike hyperglycemia exposed fetuses, uterine artery ligation exposed fetuses had no impairment in insulin-stimulated AKT phosphorylation (Figure 7C), and instead exhibited the opposite trend towards enhanced AKT phosphorylation (CI95 = −0.15 – +0.54).
Figure 7: Acute fetal effects of 48h placental insufficiency.
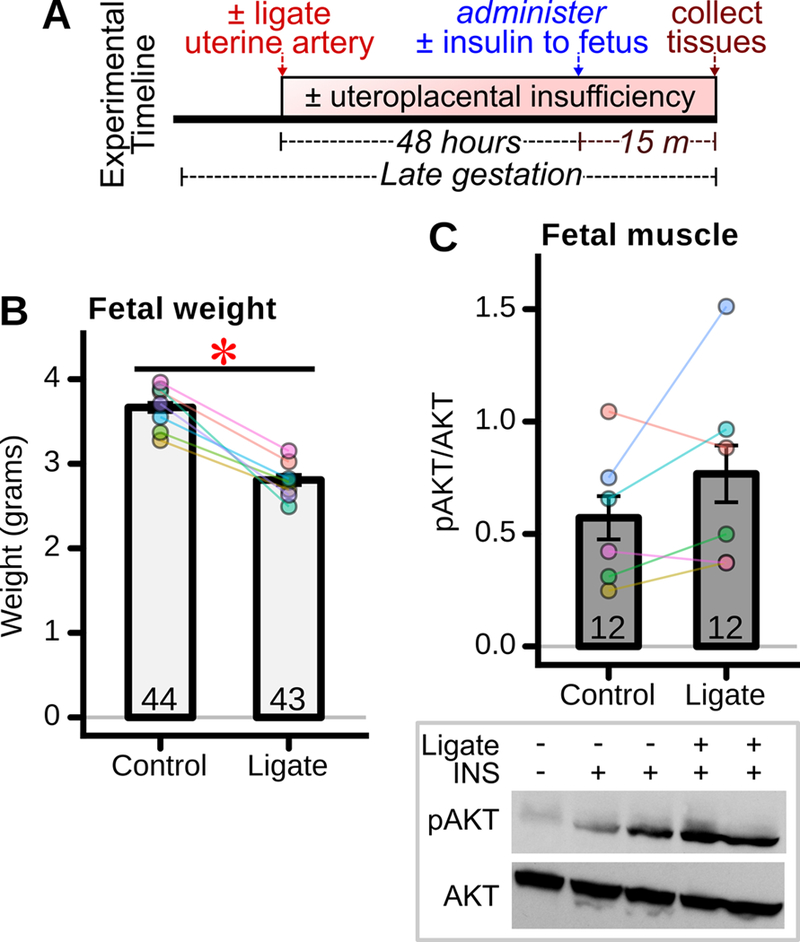
(A) Experimental timeline. One uterine artery was ligated during late gestation while the contralateral artery was left intact (control). Forty-eight hours later, insulin or saline was administered to fetuses, and fetal tissues harvested 15 minutes thereafter. (B) Fetal weight 48 hours after induction of placental insufficiency by uterine artery ligation (“ligate” group) (n= 43–44 fetuses/group from 7 mothers). *p<0.05 for difference between groups. (C) Fetal skeletal muscle insulin stimulated AKT phosphorylation 48 hours after ligation (n = 12 fetuses/group from 6 mothers). Representative western blot shown below panel.
Impaired Skeletal Muscle Insulin Signaling and Glucose Intolerance in Offspring Exposed to Fetal Hyperglycemia but not Fetal Hyperinsulinism.
Given the rapid induction of fetal insulin resistance during hyperglycemic exposure, it was thus of considerable interest as to whether insulin sensitivity normalized after birth and cessation of the hyperglycemic exposure. To test this, rat pups were birthed by C-section, reared by foster mothers, and their insulin responsiveness or glucose tolerance tested at 21 days of age (Figure 8A). Interestingly, skeletal muscle insulin sensitivity was impaired at 21 days of age in offspring formerly exposed to hyperglycemia as assessed by insulin-stimulated AKT phosphorylation (Figure 8B, CI95 = −0.38 – −0.02). By contrast, there was no impairment in insulin stimulated AKT phosphorylation at 21 days of age in hyperglycemic exposed offspring brown adipose tissue (Figure 8C) or liver (Figure 8D). Importantly, offspring exposed to fetal hyperglycemia exhibited a modest but statistically robust degree of glucose intolerance, as among all six paired groups the glucose area under the curve during glucose tolerance testing was higher in the hyperglycemia exposed offspring (Figure 8F, p=0.03).
Figure 8: Offspring insulin signaling and glucose tolerance after late gestation fetal exposure to hyperglycemia.
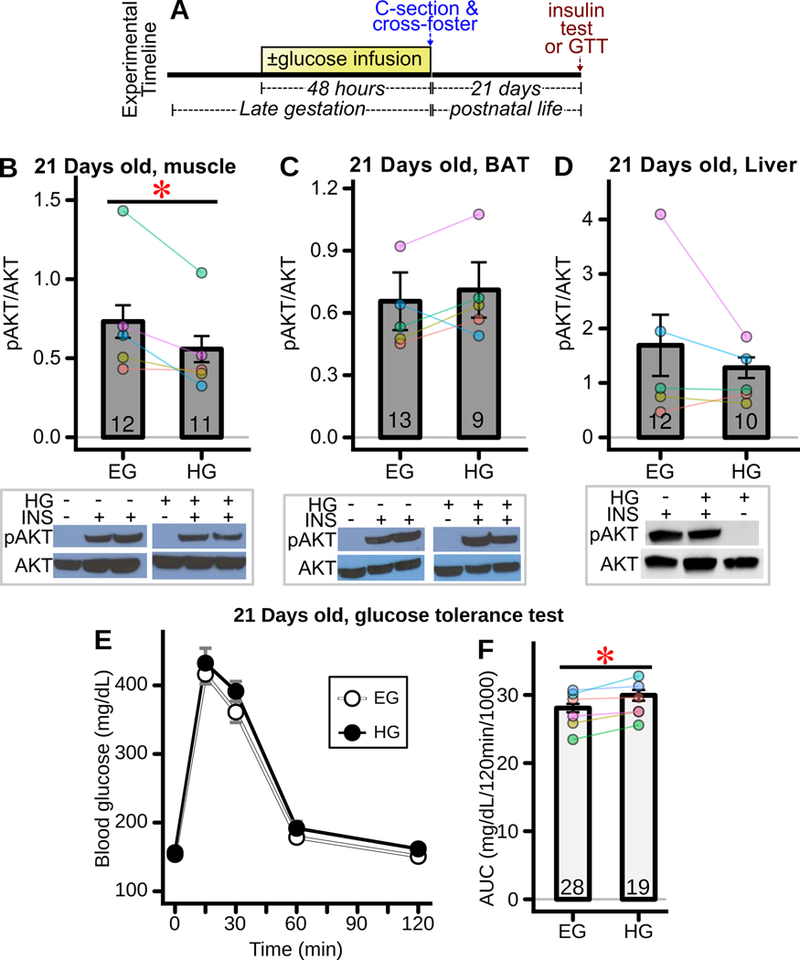
(A) Experimental timeline. Glucose was infused for 48 hours into the left uterine artery. Pups were delivered by C-section and cross-fostered to surrogate mothers. Twenty-one days later insulin signaling and glucose tolerance were interrogated. (B-D) Insulin (INS) stimulated AKT phosphorylation in 21 day old weanlings assessing (B) soleus muscle (11–12 offspring/group, from 5 mothers) * p<0.05 for difference between groups, (C) BAT (n=9–13 offspring/group, 5 mothers), and (D) liver (n=10–12 offspring/group, 5 mothers) in offspring exposed as fetuses to euglycemia (EG) or hyperglycemia (HG). Representative western blots shown below panels. (E) Intraperitoneal glucose tolerance test (GTT) of weanling offspring exposed in utero to euglycemia (EG) versus hyperglycemia (HG) (all genders included, 19–28 offspring/group from 6 mothers) and (F) corresponding glucose area under the curve (AUC) (n=19–28 offspring/group from 6 mothers) *p<0.05 for difference between groups. For all offspring measures, bars represent the mean±SEM across offspring.
Mice offspring exposed to fetal hyperinsulinism were also studied at 21 days of age (Figure 9A). Interestingly, there was a significant impairment in insulin-stimulated AKT phosphorylation in the liver (Figure 9C, CI95 = −0.66 – −0.07) but not skeletal muscle (Figure 9B) of fetally detemir- versus diluent- treated offspring when examined at 21 days of age. These offspring exhibited normal glucose tolerance as indicated by no overall change in area under the curve (Figure 9D-E).
Figure 9: Offspring insulin signaling and glucose tolerance after late gestation fetal exposure to hyperinsulinemia.
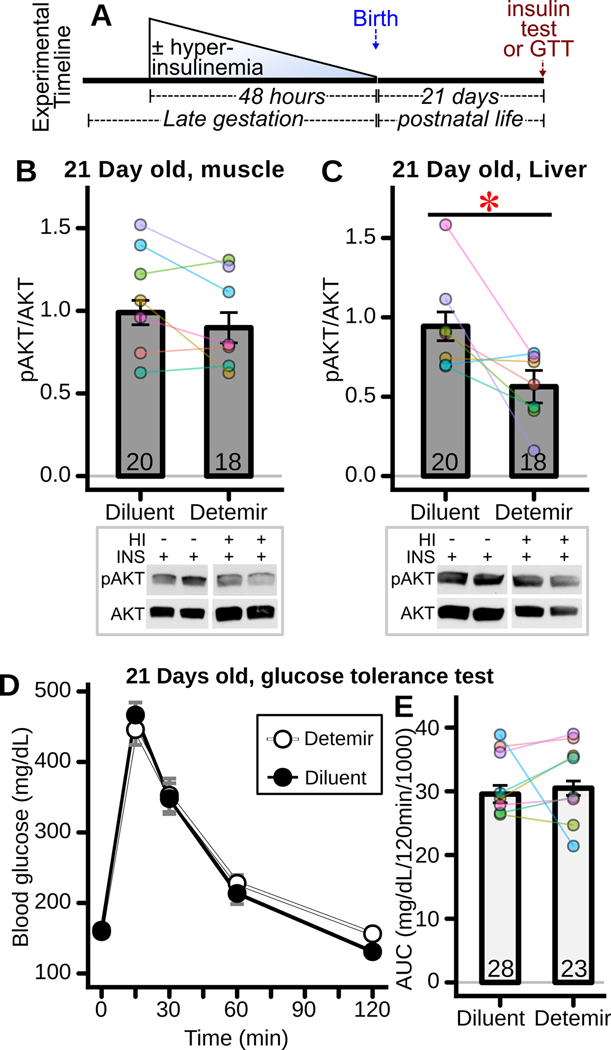
(A) Experimental timeline. Hyperinsulinemia was induced in fetuses using detemir, whereas controls received diluent. Forty-eight hours thereafter, pups were delivered and raised by their mothers. Twenty-one days later insulin signaling and glucose tolerance were interrogated. (B-C) Insulin (INS) stimulated AKT phosphorylation in 21 day old weanlings assessing insulin stimulated AKT phosphorylation in (B) skeletal muscle (18–20 fetuses/group, 7 mothers) and (C) liver (18–20 fetuses/group, 7 mothers), * p<0.05 for difference between groups, in offspring exposed as fetuses to hyperinsulinemia (detemir) versus control (diluent). Representative western blots shown below panels. (D) Intraperitoneal glucose tolerance test (GTT) of weanling offspring exposed in utero to hyperinsulinemia (detemir) versus control (diluent) (n=23–28 offspring/group from 8 mothers), and (E) corresponding glucose area AUC (n=23–28 offspring/group from 8 mothers).
Discussion
Human offspring exposed to maternal diabetes during fetal life have increased risk of insulin resistance in adulthood, beyond genetic risks. (Dabelea et al. 2000, 2008; Clausen et al. 2008, 2009) Likewise, animal models of diabetes during pregnancy produce offspring that exhibit insulin resistance. (Segar et al. 2009; Blue et al. 2015) Due to the complex pathology induced by diabetes, the role of hyperglycemia alone in this process has been difficult to define. In fact, the direct role of hyperglycemia in producing offspring diabetes risk has been recently questioned (Donovan & Cundy 2015) . To our best knowledge, this is the first experimental design examining offspring insulin sensitivity that isolates hyperglycemia’s effects alone in absence of other systemic maternal alterations that otherwise accompany diabetes in pregnancy (Baack et al. 2014) . We found most strikingly that fetal exposure to hyperglycemia during late gestation induces immediate and lasting insulin resistance in skeletal muscle, whereas isolated fetal hyperinsulinism or uteroplacental insufficiency did not exert this effect.
The detrimental impact of fetal hyperglycemic exposure on skeletal muscle insulin sensitivity was evident even before birth, at the conclusion of 48 hours of hyperglycemic exposure. Similarly, human fetuses exposed to maternal glucose intolerance have diminished insulin sensitivity as calculated from umbilical cord blood insulin and glucose (Luo et al. 2010) . The induction of skeletal muscle insulin resistance by hyperglycemia is not entirely surprising, as in-vivo (Oku et al. 2001; Haber et al. 2003) and in-vitro (Kurowski et al. 1999) experiments demonstrate that hyperglycemia acutely induces insulin resistance in mature skeletal muscle. However, fetal insulin signaling in skeletal muscle has received little experimental attention (Anand et al. 2002; Dunlop et al. 2015) such that even the molecular ontogeny of fetal insulin signaling is not well characterized. Fetal skeletal muscle may be at special risk for development of permanent insulin resistance. Importantly, the majority of lifelong muscle cells are formed during fetal myogenesis (Du et al. 2010) . By contrast, brown adipose tissue (Rehnmark & Nedergaard 1989) and liver (Moreno-Carranza et al. 2018) experience ongoing cellular formation after birth. Thus, it has been hypothesized that adverse exposure during fetal development could exert long lasting effects on adult muscle insulin sensitivity (Du et al. 2010) . Our findings are consistent with this hypothesis, in that impaired insulin signaling persisted in skeletal muscle for at least several weeks after fetal hyperglycemic exposure, whereas insulin signaling remained normal in liver and brown adipose tissue over the same time frame. Our studies herein only address insulin sensitivity but do not address its impact on the observed mild defect in glucose tolerance. It is possible that beta cell defects might contribute to the impaired glucose tolerance we observed in hyperglycemia exposed offspring at 21 days of age. In fact, exposure to maternal diabetes has also been shown to induce long-lasting beta cell dysfunction and impaired insulin secretion (Gauguier et al. 1991; Frost et al. 2012) .
Fetal exposure to hyperglycemia alone has been also achieved in ovine pregnancy models which have studied its impact on leucine oxidation, insulin secretion, and glucose transporter expression (Liechty et al. 1993; Anderson et al. 2001; Frost et al. 2012) . However, we are unaware of studies examining the impact of hyperglycemia alone on insulin signaling in these models. In our model, 48 hours of hyperglycemic exposure reduced glucose uptake into skeletal muscle and diminished insulin stimulated AKT phosphorylation (Figures 2–3). However, insulin stimulated IRS1 tyrosine phosphorylation was not affected, indicating that the hyperglycemia-induced defect in the insulin signaling pathway lies between IRS and AKT. This is consistent with findings in mature rat muscle acutely exposed ex vivo to hyperglycemia in which there were impairments in insulin stimulated AKT phosphorylation but not insulin stimulated phosphatidylinositol 3-kinase activity (Kurowski et al. 1999) . We found no changes in the expression of PDK, PTEN, and mTOR, which are proteins that mediate aspects of insulin signaling upstream of AKT. These findings suggest the presence of other molecular mechanisms that mediate the negative impact of hyperglycemia on fetal skeletal muscle insulin signaling. The magnitude of the defect in skeletal muscle insulin signaling at 21 days of age was mild (~25% reduction), though this is similar to other rodent studies of offspring exposed to systemic maternal diabetes / hyperglycemia (Holemans et al. 1997; Nasu-Kawaharada et al. 2013). Likewise, the impairment in glucose tolerance was mild, which is likewise similar to prior studies in rodent offspring from diabetic pregnancy (Gauguier et al 1990;). We chose 48 hours of hyperglycemic exposure in part due to practical considerations related to a non-trivial day-to-day failure rate of these infusions but also because even brief periods of maternal system hyperglycemia induce metabolic effects which persist at least through the neonatal period (Ktorza A et al. 1981). However, it is possible that stronger lasting effects may have been observed after a longer infusion. Furthermore, it is uncertain how these defects would have evolved over time had the offspring been allowed to further mature.
In our model, hyperglycemia induces transient fetal hyperinsulinemia at twenty-four (Gordon et al. 2015) hours. This fetal hyperinsulinemia is not sustained by forty-eight (Figure 1D) hours of infusion, consistent with prior observation that maternal diabetes exposure can produce glucotoxicity and degranulation of fetal islets (Aerts & van Assche 1977) . Since hyperinsulinemia induces insulin resistance in a variety of cell culture models (Andreozzi et al. 2011), this raised the possibility that hyperinsulinism may have contributed to fetal skeletal muscle insulin resistance. To address this directly, we used a model of chronic hyperinsulinism in the fetal rat through use of a long acting insulin analog. Interestingly, no acute impact on fetal insulin signaling was observed, whereas postnatal hepatic insulin signaling was impacted at 3 weeks of age. Glucose tolerance area under the curve was normal. Importantly for the present focus, these results suggest that transient induction of fetal hyperinsulinism is not the mechanism by which hyperglycemia induces acute or chronic skeletal muscle insulin resistance.
Fetuses developing in the context of diabetic pregnancy often exhibit hypoxia and markers of chronic hypoxia (Taricco et al. 2009; Daskalakis et al. 2008; Escobar et al. 2013; Widness et al. 1981) even when fetal weight is increased (Taricco et al. 2009). The origins of the fetal hypoxia are not perfectly understood but are thought to reflect a degree of relative placental insufficiency, related to structural placental abnormalities (Taricco et al. 2009; Daskalakis et al. 2008;), increased fetal oxidative metabolism and oxygen consumption induced by hyperglycemia (Philipps et al. 1984; Crandell et al. 1985), fetal overgrowth relative to the placenta (Surányi et al. 2016; Gloria-Bottini et al. 2016), and/or diminished uteroplacental blood flow (Reed et al. 2018). We did not examine the impact of uteroplacental insufficiency or hyperglycemia on placental glucose transport to the fetus. Nonetheless, we thus tested whether uteroplacental insufficiency incurred by uterine artery ligation induces the same acute defects in skeletal muscle insulin signaling as caused by hyperglycemia. We found that uteroplacental insufficiency induced by unilateral uterine artery ligation did not induce insulin resistance, but instead induced a trend towards increased insulin sensitivity consistent with findings in related models from large (Limesand et al. 2007) animals. This is interesting because uterine artery ligation is well described to eventually induce offspring skeletal muscle insulin resistance in the long run (Simmons et al. 2001b; Selak et al. 2003; Gatford et al. 2010) and suggests that the proximal mechanisms by which hyperglycemia versus uteroplacental insufficiency induce offspring skeletal muscle insulin resistance differ.
In conclusion, we find that fetal exposure to isolated maternal hyperglycemia is sufficient to induce insulin resistance, especially in skeletal muscle where insulin signaling was disrupted for at least several weeks after birth when the hyperglycemic exposure ceased. Our results imply that the mechanism by which uteroplacental insufficiency programs later dysmetabolism and diabetes risk differs from the mechanisms induced by hyperglycemia. Finally, our results raise the possibility that fetal skeletal muscle is uniquely at risk for programming of long-lasting insulin resistance.
Acknowledgements
We thank Philip Gruppuso, MD, Brown University, for helpful discussion.
Funding
This project was funded by the American Academy of Pediatrics (Marshall Klaus Award to KLK), the University of Iowa Children’s Miracle Network (research grant to KLK), the Fraternal Order of Eagle Diabetes Research Center (scholar award to AWN),and the National Institutes of Health through R01-DK115791 (to AWN), R01-DK081548 (to AWN), and R24-DK096518 (to AWN).
Footnotes
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
References
- Abdul Aziz SH, John CM, Mohamed Yusof NIS, Nordin M, Ramasamy R, Adam A & Mohd Fauzi F 2016. Animal Model of Gestational Diabetes Mellitus with Pathophysiological Resemblance to the Human Condition Induced by Multiple Factors (Nutritional, Pharmacological, and Stress) in Rats. BioMed Research International 2016 9704607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aerts L & van Assche FA 1977. Rat foetal endocrine pancreas in experimental diabetes. The Journal of Endocrinology 73 339–346. [DOI] [PubMed] [Google Scholar]
- Aerts L & Van Assche FA 2001. Low taurine, gamma-aminobutyric acid and carnosine levels in plasma of diabetic pregnant rats: consequences for the offspring. Journal of Perinatal Medicine 29 81–84. [DOI] [PubMed] [Google Scholar]
- Anand P, Boylan JM, Ou Y & Gruppuso PA 2002. Insulin signaling during perinatal liver development in the rat. American Journal of Physiology. Endocrinology and Metabolism 283 E844–E852. [DOI] [PubMed] [Google Scholar]
- Anderson MS, Flowers-Ziegler J, Das UG, Hay WW Jr & Devaskar SU 2001. Glucose transporter protein responses to selective hyperglycemia or hyperinsulinemia in fetal sheep. American Journal of Physiology. Regulatory, Integrative and Comparative Physiology 281 R1545–R1552. [DOI] [PubMed] [Google Scholar]
- Andreozzi F, Procopio C, Greco A, Mannino GC, Miele C, Raciti GA, Iadicicco C, Beguinot F, Pontiroli AE, Hribal ML et al. 2011. Increased levels of the Akt-specific phosphatase PH domain leucine-rich repeat protein phosphatase (PHLPP)-1 in obese participants are associated with insulin resistance. Diabetologia 54 1879–1887. [DOI] [PubMed] [Google Scholar]
- Baack ML, Wang C, Hu S, Segar JL & Norris AW 2014. Hyperglycemia induces embryopathy, even in the absence of systemic maternal diabetes: an in vivo test of the fuel mediated teratogenesis hypothesis. Reproductive Toxicology 46 129–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blue EK, Ballman K, Boyle F, Oh E, Kono T, Quinney SK, Thurmond DC, Evans-Molina C & Haneline LS 2015. Fetal hyperglycemia and a high-fat diet contribute to aberrant glucose tolerance and hematopoiesis in adult rats. Pediatric Research 77 316–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cetin I, de Santis MSN, Taricco E, Radaelli T, Teng C, Ronzoni S, Spada E, Milani S & Pardi G 2005. Maternal and fetal amino acid concentrations in normal pregnancies and in pregnancies with gestational diabetes mellitus. American Journal of Obstetrics and Gynecology 192 610–617. [DOI] [PubMed] [Google Scholar]
- Clausen TD, Mathiesen ER, Hansen T, Pedersen O, Jensen DM, Lauenborg J & Damm P 2008. High prevalence of type 2 diabetes and pre-diabetes in adult offspring of women with gestational diabetes mellitus or type 1 diabetes: the role of intrauterine hyperglycemia. Diabetes Care 31 340–346. [DOI] [PubMed] [Google Scholar]
- Clausen TD, Mathiesen ER, Hansen T, Pedersen O, Jensen DM, Lauenborg J, Schmidt L & Damm P 2009. Overweight and the metabolic syndrome in adult offspring of women with diet-treated gestational diabetes mellitus or type 1 diabetes. The Journal of Clinical Endocrinology and Metabolism 94 2464–2470. [DOI] [PubMed] [Google Scholar]
- Crandell SS, Fisher DJ, & Morriss FH Jr 1985. Effects of ovine maternal hyperglycemia on fetal regional blood flows and metabolism. American Journal of Physiology 249 E454–460. [DOI] [PubMed] [Google Scholar]
- Crume TL, Shapiro AL, Brinton JT, Glueck DH, Martinez M, Kohn M, Harrod C, Friedman JE & Dabelea D 2015. Maternal fuels and metabolic measures during pregnancy and neonatal body composition: the healthy start study. The Journal of Clinical Endocrinology and Metabolism 100 1672–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabelea D, Hanson RL, Lindsay RS, Pettitt DJ, Imperatore G, Gabir MM, Roumain J, Bennett PH & Knowler WC 2000. Intrauterine exposure to diabetes conveys risks for type 2 diabetes and obesity: a study of discordant sibships. Diabetes 49 2208–2211. [DOI] [PubMed] [Google Scholar]
- Dabelea D, Mayer-Davis EJ, Lamichhane AP, D’Agostino RB Jr, Liese AD, Vehik KS, Narayan KMV, Zeitler P & Hamman RF 2008. Association of intrauterine exposure to maternal diabetes and obesity with type 2 diabetes in youth: the SEARCH Case-Control Study. Diabetes Care 31 1422–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daskalakis G, Marinopoulos S, Krielesi V, Papapanagiotou A, Papantoniou N, Mesogitis S & Antsaklis A 2008. Placental pathology in women with gestational diabetes. Acta Obstetricia et Gynecologica Scandinavica 87 403–407. [DOI] [PubMed] [Google Scholar]
- Donovan LE & Cundy T 2015. Does exposure to hyperglycaemia in utero increase the risk of obesity and diabetes in the offspring? A critical reappraisal. Diabetic Medicine: A Journal of the British Diabetic Association 32 295–304. [DOI] [PubMed] [Google Scholar]
- Du M, Tong J, Zhao J, Underwood KR, Zhu M, Ford SP & Nathanielsz PW 2010. Fetal programming of skeletal muscle development in ruminant animals. Journal of Animal Science 88 E51–E60. [DOI] [PubMed] [Google Scholar]
- Dunlop K, Cedrone M, Staples JF & Regnault TRH 2015. Altered fetal skeletal muscle nutrient metabolism following an adverse in utero environment and the modulation of later life insulin sensitivity. Nutrients 7 1202–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escobar J, Teramo K, Stefanovic V, Andersson S, Asensi MA, Arduini A, Cubells E, Sastre J & Vento M 2013. Amniotic fluid oxidative and nitrosative stress biomarkers correlate with fetal chronic hypoxia in diabetic pregnancies. Neonatology 103 193–198. [DOI] [PubMed] [Google Scholar]
- Franks PW, Looker HC, Kobes S, Touger L, Tataranni PA, Hanson RL & Knowler WC 2006. Gestational glucose tolerance and risk of type 2 diabetes in young Pima Indian offspring. Diabetes 55 460–465. [DOI] [PubMed] [Google Scholar]
- Fraser A & Lawlor DA 2014. Long-term health outcomes in offspring born to women with diabetes in pregnancy. Current Diabetes Reports 14 489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost MS, Zehri AH, Limesand SW, Hay WW Jr & Rozance PJ 2012. Differential effects of chronic pulsatile versus chronic constant maternal hyperglycemia on fetal pancreatic β-cells. Journal of Pregnancy 2012 812094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatford KL, Simmons RA, De Blasio MJ, Robinson JS & Owens JA 2010. Review: Placental programming of postnatal diabetes and impaired insulin action after IUGR. Placenta 31 Suppl S60–S65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauguier D, Bihoreau MT, Ktorza A, Berthault MF, & Picon L 1990. Inheritance of diabetes mellitus as consequence of gestational hyperglycemia in rats. Diabetes 39 734–739. [DOI] [PubMed] [Google Scholar]
- Gauguier D, Bihoreau MT, Picon L & Ktorza A 1991. Insulin secretion in adult rats after intrauterine exposure to mild hyperglycemia during late gestation. Diabetes 40 Suppl 2 109–114. [DOI] [PubMed] [Google Scholar]
- Gin H, Vambergue A, Vasseur C, Rigalleau V, Dufour P, Roques A, Romon M, Millet D, Hincker P, Fontaine P et al. 2006. Blood ketone monitoring: a comparison between gestational diabetes and non-diabetic pregnant women. Diabetes & Metabolism 32 592–597. [DOI] [PubMed] [Google Scholar]
- Gloria-Bottini F, Neri A, Coppeta L, Magrini A, Bottini E 2016. Correlation between birth weight and placental weight in healthy and diabetic puerperae. Taiwanese Journal of Obstetrics and Gynecology 55 697–699. [DOI] [PubMed] [Google Scholar]
- Gordon EE, Reinking BE, Hu S, Yao J, Kua KL, Younes AK, Wang C, Segar JL & Norris AW 2015. Maternal Hyperglycemia Directly and Rapidly Induces Cardiac Septal Overgrowth in Fetal Rats. Journal of Diabetes Research 2015 479565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haber CA, Lam TKT, Yu Z, Gupta N, Goh T, Bogdanovic E, Giacca A & Fantus IG 2003. N-acetylcysteine and taurine prevent hyperglycemia-induced insulin resistance in vivo: possible role of oxidative stress. American Journal of Physiology. Endocrinology and Metabolism 285 E744–E753. [DOI] [PubMed] [Google Scholar]
- Halseth AE, Bracy DP & Wasserman DH 2000. Limitations to basal and insulin-stimulated skeletal muscle glucose uptake in the high-fat-fed rat. American Journal of Physiology. Endocrinology and Metabolism 279 E1064–E1071. [DOI] [PubMed] [Google Scholar]
- Holemans K, Van Bree R, Verhaeghe J, Meurrens K, & Van Assche FA 1997. Maternal semistarvation and streptozotocin-diabetes in rats have different effects on the in vivo glucose uptake by peripheral tissues in their female adult offspring. Journal of Nutrition 127 1371–1376. [DOI] [PubMed] [Google Scholar]
- Kahraman S, Dirice E, De Jesus DF, Hu J & Kulkarni RN 2014. Maternal insulin resistance and transient hyperglycemia impact the metabolic and endocrine phenotypes of offspring. American Journal of Physiology. Endocrinology and Metabolism 307 E906–E918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalyfa A, Carreras A, Hakim F, Cunningham JM, Wang Y & Gozal D 2013. Effects of late gestational high-fat diet on body weight, metabolic regulation and adipokine expression in offspring. International Journal of Obesity 37 1481–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khosrowbeygi A, Shiamizadeh N & Taghizadeh N 2016. Maternal circulating levels of some metabolic syndrome biomarkers in gestational diabetes mellitus. Endocrine 51 245–255. [DOI] [PubMed] [Google Scholar]
- Ktorza A, Girard JR, Kinebanyan MF, Picon L 1981. Hyperglycaemia induced by glucose infusion in the unrestrained pregnant rat during the last three days of gestation: metabolic and hormonal changes in the mother and the fetuses. Diabetologia 21 569–574 [DOI] [PubMed] [Google Scholar]
- Kurowski TG, Lin Y, Luo Z, Tsichlis PN, Buse MG, Heydrick SJ & Ruderman NB 1999. Hyperglycemia inhibits insulin activation of Akt/protein kinase B but not phosphatidylinositol 3-kinase in rat skeletal muscle. Diabetes 48 658–663. [DOI] [PubMed] [Google Scholar]
- Latouche C, Heywood SE, Henry SL, Ziemann M, Lazarus R, El-Osta A, Armitage JA & Kingwell BA 2014. Maternal overnutrition programs changes in the expression of skeletal muscle genes that are associated with insulin resistance and defects of oxidative phosphorylation in adult male rat offspring. The Journal of Nutrition 144 237–244. [DOI] [PubMed] [Google Scholar]
- Li M, Vienberg SG, Bezy O, O’Neill BT, & Kahn CR 2015. Role of PKCδ in Insulin Sensitivity and Skeletal Muscle Metabolism. Diabetes 64 4023–4032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liechty EA, Boyle DW, Moorehead H, Liu YM & Denne SC 1993. Increased fetal glucose concentration decreases ovine fetal leucine oxidation independent of insulin. The American Journal of Physiology 265 E617–E623. [DOI] [PubMed] [Google Scholar]
- Limesand SW, Rozance PJ, Smith D & Hay WW Jr 2007. Increased insulin sensitivity and maintenance of glucose utilization rates in fetal sheep with placental insufficiency and intrauterine growth restriction. American Journal of Physiology. Endocrinology and Metabolism 293 E1716–E1725. [DOI] [PubMed] [Google Scholar]
- López-Tinoco C, Roca M, García-Valero A, Murri M, Tinahones FJ, Segundo C, Bartha JL & Aguilar-Diosdado M 2013. Oxidative stress and antioxidant status in patients with late-onset gestational diabetes mellitus. Acta Diabetologica 50 201–208. [DOI] [PubMed] [Google Scholar]
- Luo Z-C, Delvin E, Fraser WD, Audibert F, Deal CI, Julien P, Girard I, Shear R, Levy E & Nuyt A-M 2010. Maternal glucose tolerance in pregnancy affects fetal insulin sensitivity. Diabetes Care 33 2055–2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathias PCF, Elmhiri G, de Oliveira JC, Delayre-Orthez C, Barella LF, Tófolo LP, Fabricio GS, Chango A & Abdennebi-Najar L 2014. Maternal diet, bioactive molecules, and exercising as reprogramming tools of metabolic programming. European Journal of Nutrition 53 711–722. [DOI] [PubMed] [Google Scholar]
- de Ménibus CH & Mallet E 1980. [Hyperinsulinism of the newborn infant of diabetic mothers]. Bulletin de l’Academie nationale de medecine 164 834–838. [PubMed] [Google Scholar]
- Moreno-Carranza B, Bravo-Manríquez M, Baez A, Ledesma-Colunga MG, Ruiz-Herrera X, Reyes-Ortega P, De Los Ríos EA, Macotela Y, Martínez de la Escalera G & Clapp C 2018. Prolactin regulates liver growth during postnatal development in mice. American Journal of Physiology. Regulatory, Integrative and Comparative Physiology (doi: 10.1152/ajpregu.00003.2018) [DOI] [PubMed] [Google Scholar]
- Nasu-Kawaharada R, Nakamura A, Kakarla SK, Blough ER, Kohama K, & Kohama T 2013. A maternal diet rich in fish oil may improve cardiac Akt-related signaling in the offspring of diabetic mother rats. Nutrition 29 688–692. [DOI] [PubMed] [Google Scholar]
- Ogata ES, Collins JW Jr, & Finley S 1988. Insulin injection in the fetal rat: accelerated intrauterine growth and altered fetal and neonatal glucose homeostasis. Metabolism 37 649–655. [DOI] [PubMed] [Google Scholar]
- Oku A, Nawano M, Ueta K, Fujita T, Umebayashi I, Arakawa K, Kano-Ishihara T, Saito A, Anai M, Funaki M et al. 2001. Inhibitory effect of hyperglycemia on insulin-induced Akt/protein kinase B activation in skeletal muscle. American Journal of Physiology. Endocrinology and Metabolism 280 E816–E824. [DOI] [PubMed] [Google Scholar]
- Philipps AF, Porte PJ, Stabinsky S, Rosenkrantz TS, Raye JR. 1984. Effects of chronic fetal hyperglycemia upon oxygen consumption in the ovine uterus and conceptus. Journal of Clinical Investigation 74 279–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed LC, Estrada SM, Walton RB, Napolitano PG, Ieronimakis N 2018. Evaluating maternal hyperglycemic exposure and fetal placental arterial dysfunction in a dual cotyledon, dual perfusion model. Placenta 69 109–116. [DOI] [PubMed] [Google Scholar]
- Rehnmark S & Nedergaard J 1989. DNA synthesis in mouse brown adipose tissue is under beta-adrenergic control. Experimental Cell Research 180 574–579. [DOI] [PubMed] [Google Scholar]
- Ryckman KK, Spracklen CN, Smith CJ, Robinson JG & Saftlas AF 2015. Maternal lipid levels during pregnancy and gestational diabetes: a systematic review and meta-analysis. BJOG: An International Journal of Obstetrics and Gynaecology 122 643–651. [DOI] [PubMed] [Google Scholar]
- Saad MI, Abdelkhalek TM, Saleh MM, Haiba MM, Tawfik SH & Kamel MA 2016. Maternal diabetes impairs oxidative and inflammatory response in murine placenta. SpringerPlus 5 532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawatzke AB, Norris AW, Spyropoulos F, Walsh SA, Acevedo MR, Hu S, Yao J, Wang C, Sunderland JJ & Boles Ponto LL 2015. PET/CT imaging reveals unrivaled placental avidity for glucose compared to other tissues. Placenta 36 115–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segar EM, Norris AW, Yao J- R, Hu S, Koppenhafer SL, Roghair RD, Segar JL & Scholz TD 2009. Programming of growth, insulin resistance and vascular dysfunction in offspring of late gestation diabetic rats. Clinical Science 117 129–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selak MA, Storey BT, Peterside I & Simmons RA 2003. Impaired oxidative phosphorylation in skeletal muscle of intrauterine growth-retarded rats. American Journal of Physiology. Endocrinology and Metabolism 285 E130–E137. [DOI] [PubMed] [Google Scholar]
- Shang M, Zhao J, Yang L & Lin L 2015. Oxidative stress and antioxidant status in women with gestational diabetes mellitus diagnosed by IADPSG criteria. Diabetes Research and Clinical Practice 109 404–410. [DOI] [PubMed] [Google Scholar]
- Shomonov-Wagner L, Raz A & Leikin-Frenkel A 2015. Alpha linolenic acid in maternal diet halts the lipid disarray due to saturated fatty acids in the liver of mice offspring at weaning. Lipids in Health and Disease 14 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons RA, Templeton LJ & Gertz SJ 2001a. Intrauterine growth retardation leads to the development of type 2 diabetes in the rat. Diabetes 50 2279–2286. [DOI] [PubMed] [Google Scholar]
- Simmons RA, Templeton LJ & Gertz SJ 2001b. Intrauterine growth retardation leads to the development of type 2 diabetes in the rat. Diabetes 50 2279–2286. [DOI] [PubMed] [Google Scholar]
- Surányi A, Kozinszky Z, Molnár A, Németh G 2016. Placental volume relative to fetal weight estimated by sonography in diabetic pregnancies. Journal of Maternal-Fetal and Neonatal Medicine 29 1229–1232. [DOI] [PubMed] [Google Scholar]
- Taricco E, Radaelli T, Rossi G, Nobile de Santis MS, Bulfamante GP, Avagliano L & Cetin I 2009. Effects of gestational diabetes on fetal oxygen and glucose levels in vivo. BJOG: An International Journal of Obstetrics and Gynaecology 116 1729–1735. [DOI] [PubMed] [Google Scholar]
- Umekawa T, Sugiyama T, Du Q, Murabayashi N, Zhang L, Kamimoto Y, Yoshida T, Sagawa N & Ikeda T 2015. A maternal mouse diet with moderately high-fat levels does not lead to maternal obesity but causes mesenteric adipose tissue dysfunction in male offspring. The Journal of Nutritional Biochemistry 26 259–266. [DOI] [PubMed] [Google Scholar]
- White V, Jawerbaum A, Mazzucco MB, Gauster M, Desoye G & Hiden U 2015. Diabetes-associated changes in the fetal insulin/insulin-like growth factor system are organ specific in rats. Pediatric Research 77 48–55. [DOI] [PubMed] [Google Scholar]
- Widness JA, Susa JB, Garcia JF, Singer DB, Sehgal P, Oh W, Schwartz R & Schwartz HC 1981. Increased erythropoiesis and elevated erythropoietin in infants born to diabetic mothers and in hyperinsulinemic rhesus fetuses. The Journal of Clinical Investigation 67 637–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijendran V, Bendel RB, Couch SC, Philipson EH, Thomsen K, Zhang X & Lammi-Keefe CJ 1999. Maternal plasma phospholipid polyunsaturated fatty acids in pregnancy with and without gestational diabetes mellitus: relations with maternal factors. The American Journal of Clinical Nutrition 70 53–61. [DOI] [PubMed] [Google Scholar]
- Yao J, Wang C, Walsh SA, Hu S, Sawatzke AB, Dang D, Segar JL, Ponto LLB, Sunderland JJ & Norris AW 2010. Localized fetomaternal hyperglycemia: spatial and kinetic definition by positron emission tomography. PloS One 5 e12027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokomizo H, Inoguchi T, Sonoda N, Sakaki Y, Maeda Y, Inoue T, Hirata E, Takei R, Ikeda N, Fujii M et al. 2014. Maternal high-fat diet induces insulin resistance and deterioration of pancreatic β-cell function in adult offspring with sex differences in mice. American Journal of Physiology. Endocrinology and Metabolism 306 E1163–E1175. [DOI] [PubMed] [Google Scholar]
- Zhu C, Yang H, Geng Q, Ma Q, Long Y, Zhou C & Chen M 2015. Association of oxidative stress biomarkers with gestational diabetes mellitus in pregnant women: a case-control study. PloS One 10 e0126490. [DOI] [PMC free article] [PubMed] [Google Scholar]