

Abstract
Modern electron microscopy offers a wide variety of tools to investigate the ultrastructural organization of cells and tissues and to accurately pinpoint intracellular localizations of macromolecules of interest. New volumetric electron microscopy techniques and new instrumentation provide unique opportunities for high-throughput analysis of comparatively large volumes of tissue and their complete reconstitution in three-dimensional (3D) electron microscopy. However, due to a variety of technical issues such as the limited penetration of label into the tissue, low antigen preservation, substantial electron density of secondary detection reagents, and many others, the adaptation of immuno-detection techniques for use with such 3D imaging methods as focused ion beam–scanning electron microscopy (FIB-SEM) has been challenging. Here, we describe a sample preparation method for 3D FIB-SEM, which results in an optimal preservation and staining of ultrastructural details at a resolution necessary for tracing immunolabeled neuronal structures and detailed reconstruction of synapses. This technique is applicable to neuronal and non-neuronal cells, tissues, and a wide variety of antigens.
Keywords: brain, DAB, FIB-SEM, GSSP, labeling
Introduction
Focused ion beam–scanning electron microscopy (FIB-SEM) is a powerful method finding numerous applications in materials sciences and biology. The ability of FIB-SEM to subsequently remove nanometer-scale layers of the sample is used as a basis for imaging progressively deeper into the embedded specimen.1–3 The resulting data are used to reconstruct the sample in three-dimensional (3D) electron microscopy. Since the inception of the method and its adaptation to the analysis of biological ultrastructure, one critical limitation of FIB-SEM has been its limited compatibility with immunodetection methods, which are widely used in transmission electron microscopy (TEM) and light microscopy (LM) to visualize individual proteins or other antigens within their intracellular and tissue context. Recently, two immunodetection methods were used in combination with FIB-SEM. A number of studies have used combinations of FIB-SEM with LM techniques to correlate fluorescence in situ with ultrastructural information from FIB-SEM.4,5 Another approach is based on ectopic expression of the enzyme ascorbate peroxidase (APEX) and in situ detection of its electron-dense reaction products by FIB-SEM.6,7
We developed a protocol to directly integrate immunostaining with FIB-SEM without genetic manipulation of the source material, such as transfection of cells of interest with plasmids encoding APEX or similar enzymes. The protocol is based on the diaminobenzidine (DAB) reaction,8 a routinely used approach for TEM immunolabeling. For example, this method has recently been used for imaging dendritic spines and synapses in green fluorescent protein-labeled adult-generated neurons with FIB-SEM.9 However, the nature of DAB reaction significantly impedes precise high-resolution ultrastructural analysis of the label due to the formation of large, electron-dense deposits that may mask ultrastructural details of the immunopositive region. In addition, the large size and low uniformity of deposits often result in imprecise localization of the immunoreactivity. Clearly, a detection method based on highly uniform, small particles would offer significant advantages. Immunogold labeling is routinely used for TEM, but has a major limitation of poor penetration of the relatively large (2-15 nm) gold particles into the sample. Ultrasmall gold conjugates have greater labeling sensitivity and better penetration than larger gold particle conjugates, but ultrasmall gold cannot be seen in TEM without a “silver enhancement” step, which results in an increase in particle size and visibility. The most sensitive and reliable approach is the gold-substituted silver peroxidase (GSSP) technique,10 which solves both problems of gold particle visualization and penetration. GSSP is a comparatively labor-intensive and demanding technique, but it can be successfully integrated into routine protocols.11,12 Here, we endeavored to adapt the GSSP technique for use with FIB-SEM imaging. The initial DAB immunochemical reaction, as discussed above, results in a deposition of dark precipitate. DAB reaction is followed by a GSSP reaction, which first replaces DAB deposits with silver compounds, and then incorporates a further gold intensification of the silver products. This serves to replace the large DAB reaction deposits with smaller, more discreet gold particles, allowing for a clearer signal and ultimately more accurate protein localization.
To test our protocol, we chose a highly specific and extensively characterized D28K (calbindin) antibody,13 which stains Purkinje cells, the principal neurons of the cerebellum. Calbindin is a calcium-binding protein specifically expressed in the cytoplasm of Purkinje cells,13 making it an ideal candidate to demonstrate the specificity of our technique. We endeavored to develop a robust protocol allowing for the best possible antigen preservation, antibody penetration, and preservation and visualization of the sample ultrastructure by FIB-SEM. As a proof of concept, we used two different FIB-SEM microscopes to visualize the resulting staining.
Materials and Methods
Animals
All animal experiments were done in accordance with a protocol approved by the Institutional Animal Care and Use Committee (IMCB, A*Star; Singapore) and in accordance with the Animal Welfare Committee guidelines. In total, the brains of five 3-week old male C57BL/6 mice were used. Animals were euthanized with a mixture of ketamine and xylazine (K113, Sigma-Aldrich; Singapore, Singapore) as per institutional guidelines.
Mice were transcardially perfused with 10 ml of 4% paraformaldehyde (#15714, EMS; Hatfield, PA) in 0.1 M phosphate buffer (PB), pH 7.4 for 10 min, and then the brains were postfixed overnight in the same solution at 4C. On the next day, after washing twice in PB, 80-µm-thick coronal vibratome sections of cerebellum were made.
DAB/GSSP Immunolabeling
The cerebellar sections are washed three times in PB on ice for 10 min and incubated in 0.3% hydrogen peroxide (#31642, Sigma-Aldrich) in the same buffer for 30 min on ice. After three washes with PB, sections are incubated in 0.5% sodium borohydride (#71320, Sigma-Aldrich) for 30 min at room temperature (RT), followed by three 10 min washes with PB. The sections are incubated in blocking buffer containing 1% BSA (A2153, Sigma-Aldrich), 0.01% glycine (G7126, Sigma-Aldrich), 0.01% lysine (L5501, Sigma-Aldrich), 1% normal goat serum (#25570, EMS), 0.05% Triton X-100 (#22146, EMS), 0.1% cold water fish gelatin (#25560, EMS) in PB for 2 hr on ice. The cerebellar sections are stained with primary antibody, mouse anti-calbindin-D28K (#AgCB10abs, Swant; Marly, Switzerland), at 1:5000 dilution in the same blocking buffer, at 4C overnight. The following day, slices are washed four times in PB on ice and probed with secondary antibody, goat anti-mouse conjugated to biotin (BA-9200, Vector Laboratories; Burlingame, CA), diluted 1:200 in blocking buffer, for 2 hr on ice.
Immunoreactivity is visualized with the avidin–biotin complex (ABC) method. While the slices are being labeled with secondary antibody, ABC complex (PK-4000, Vector Laboratories) is prepared according to the manufacturer’s instructions. The slices are washed twice in PB for 10 min and three times with PB with 0.01% Triton X-100 for 10 min each, on ice. Slices are then incubated with the ABC complex for 1 hr at room temperature. Immunoreactivity is visualized by incubation with 5 mg of 3.3′-diaminobenzidine (#13082, EMS) and 0.03% hydrogen peroxide in 10 ml of PB for 10 min at RT. The reaction is stopped by washing the sections in cold PB, and slices are fixed with 2% glutaraldehyde (#16100, EMS) in PB for 1 hr on ice. Several DAB-stained sections, without GSSP reaction, are collected after the 2% glutaraldehyde step and embedded in epon for use as GSSP controls. Slices are subsequently washed four times in PB for 10 min each and three times in 2% sodium acetate (#21120, EMS) in dH2O for 10 min each on ice and stored overnight in 10% sodium thioglycolate in dH2O (T0632, Sigma-Aldrich) at 4C.
Labeling is enhanced using the GSSP method.10 In brief, the solution for the GSSP reaction is a mixture of three solutions: 50 ml of solution (A) 5% sodium carbonate in dH2O (#21135, EMS); 50 ml of solution (B) 0.2% ammonium nitrate (A9642, Sigma-Aldrich), 0.2% silver nitrate (#21050, EMS), and 1% tungstosilicic acid hydrate (T2786, Sigma-Aldrich) sequentially dissolved in dH2O; and 40 µl of solution (C) 37% formalin (#252549, Sigma-Aldrich) in dH2O, passed through a 0.2 µm filter. All solutions must be prepared fresh and stored in the dark. First, 10 ml of RT solution A is mixed with 10 ml of RT solution B, without exposure to bright light. Solution B must be added to solution A dropwise while mixing, and the solution must stay transparent and clear without any precipitation. Dim light can be used to check that the solution remains clear but care must be taken to prevent prolonged exposure to light. Then, 40 µl of RT solution C slowly added to the mixture of A and B while gently stirring. Again, the solution must stay transparent and clear without precipitation. Sections are incubated in freshly made GSSP solution for 8 min, in the dark with agitation at RT. The reaction is stopped by incubating sections in 1% acetic acid (#10042-10, EMS) in dH2O at 4C, whereupon sections are washed four times for 10 min in 2% sodium acetate at 4C, followed by an incubation in 0.05% gold chloride (#RT16586, EMS) in dH2O at 4C in the dark with agitation for 8 min. Sections are washed again four times for 10 min in 2% sodium acetate at 4C, then fixed in 3% sodium thiosulfate (#RT21360, EMS) in dH2O at 4C with agitation two times for 2 min each and finally washed again four times for 10 min in 2% sodium acetate at 4C.
Embedding for EM
The GSSP-treated sections and DAB-only stained control sections are washed with 0.1 M sodium cacodylate buffer (#12300, EMS) pH 7.6, fixed in 0.2% glutaraldehyde in 0.1 M sodium cacodylate buffer for 10 min and washed four times for 10 min each in 0.1 M sodium cacodylate buffer on ice.
Samples are stained with 1% osmium tetroxide (#19152, EMS) and 1.5% potassium ferrocyanide (#455989, Sigma-Aldrich) in dH2O for 10 min, washed in dH2O two times each and then washed in 0.05 M maleate buffer pH 5.8-6.0 (#18150, EMS), five times for 7 min each. Next, samples are incubated in 1% uranyl acetate (#22400, EMS) in 0.05 M maleate buffer overnight at 4C and stained en bloc with Walton’s14 lead aspartate for 30 min at 60C. After washing with maleate buffer and then with dH2O, four times each for 5 min, samples are dehydrated in an ascending ethanol series (30%, 50%, 70%, 80%, 95%), 10 min per step, at 4C. Samples are treated twice for 15 min each with absolute ethanol at RT and infiltrated with hard Epon 812/ethanol mixtures overnight. The next day, sections are flat embedded in hard composition of Epon 812 (SERVA; Heidelberg, Germany) between two microscopic slides and ACLAR film (#50425, EMS) and polymerized for 2 days at 60C.
Imaging and Analysis
Polymerized blocks are first examined using LM (Fig. 1) and conventional TEM methods (Fig. 2) to identify blocks suitable for FIB-SEM analysis. LM imaging is performed using a Zeiss Axioimager z1 with an MRc5-camera (Carl Zeiss; Oberkochen, Germany). Ultrathin sections (70 nm) are generated manually using a Reichert Ultracut E ultramicrotome. All sections are collected as ribbons of four to five sections on formvar carbon-coated copper grids (Ted Pella; Redding, CA). Images are taken using a TEM microscope (JEM1010 with tungsten filament, JEOL; Tokyo, Japan) operated at 80 kV, equipped with a Kodak SIS camera. Photographs are taken at various magnifications (0.6–20k×) without any additional lead or uranyl acetate staining. Blocks containing appropriately labeled Purkinje cells are identified and subsequently used for FIB-SEM analysis.
Figure 1.

Light microscopy imaging of Purkinje cells labeled with calbindin-D28k antibody. (A) Sagittal vibratome sections (80 µm) of cerebellum obtained after labeling with calbindin followed by DAB/GSSP reactions display characteristic dark brown staining patterns corresponding to bodies of stained neurons bodies (arrows) and dendrite area (asterisks) in the cortex of cerebellum. (B) The same section (A, black box) at a higher magnification showing calbindin staining in bodies of Purkinje cells (arrows) and in the dendritic area (asterisk). (C) Purkinje cell somata (arrows) and dendrite area (asterisk) visualized in a resin block after embedding for electron microscopy. (D) Visualization of labeled Purkinje cell neuropil (asterisk) and bodies (arrows) in a semithick section (0.5 µm). Scale bar, A = 500 µm, B, D = 50 µm, C = 250 µm. Abbreviations: DAB, diaminobenzidine, GSSP, gold-substituted silver peroxidase.
Figure 2.

TEM imaging of Purkinje cells labeled with calbindin-D28k antibody. (A) TEM micrograph of a DAB-labeled dendrite (dd) of a Purkinje cell. (B) Micrograph of DAB/ GSSP-labeled dendrite. Electron-dense DAB deposits mask the ultrastructure of a Purkinje cell dendrite (2A), compared with a GSSP-stained dendrite displaying much smaller labeling and ultimately clearer details (2B). (C) Low-magnification TEM micrograph of the cerebellum showing DAB/GSSP labeling in the soma (S) of a Purkinje cell (white asterisk) and in a dendrite (black asterisk), displaying punctate black staining characteristic for the GSSP reaction. (D) High-magnification TEM micrograph corresponding to an area within the black box in 2C showing a diffuse GSSP staining pattern corresponding to a dendritic tree of a Purkinje cell (black asterisk) and its soma (white asterisk). (E) Presynaptic calbindin GSSP labeling (asterisks) in synapses (Syn) localized in the molecular layer of the cerebellum. Note the preservation and resolution of membranous structures and organelle such as mitochondria (arrows). Specimens were not poststained with lead or uranyl acetate (see text for details). Scale bars: A, B, D = 0.5 µm, C = 5 µm, E = 1 µm. Abbreviations: DAB, diaminobenzidine, GSSP, gold-substituted silver peroxidase; TEM, transmission electron microscopy.
For FIB-SEM, the embedded samples are mounted onto aluminum SEM stubs (diameter = 12 mm) and samples are coated with ~8 nm of platinum. FIB-SEM imaging is performed using a Zeiss Auriga Crossbeam (Carl Zeiss) system (Fig. 3 and Supplemental Video 1) with Atlas 3D software or with a Scios DualBeam (FEI; Eindhoven, the Netherlands) system (Fig. 4 and Supplemental Video 2) with Amira software.
Figure 3.

FIB-SEM imaging of calbindin GSSP-labeled Purkinje cells. (A-B) A single block-face image from an FIB-imaged stack, showing a fragment of the Purkinje cell layer (S, Purkinje cell soma) with GSSP-positive areas in somata of Purkinje cells (white box) and in dendrites (dds; black box). (B) Higher magnification images of areas highlighted in 3A. Top, fragment of a body of a Purkinje cell showing calbindin immunolabeling as distinct round deposits that are darker than any elements of cellular ultrastructure, white arrowheads. Bottom, fragment of a dendrite of a Purkinje cell, rotated image. Note the highly contrasted, regularly shaped deposits corresponding to calbindin immunoreactivity. (C-D) The size and density of GSSP particles allows to easily follow and segment immunopositive structures. Individual GSSP particles shown in yellow and manual segmentation of a Purkinje cell dendritic segment in purple (dd). Dendrite indicates the same dendrite in 3A, 3B (bottom), 3C, and 3D. Imaging was performed using a ZEISS Auriga Crossbeam FIB-SEM. Scale bar = 200 µm. Abbreviations: FIB-SEM, focused ion beam–scanning electron microscopy; GSSP, gold-substituted silver peroxidase.
Figure 4.
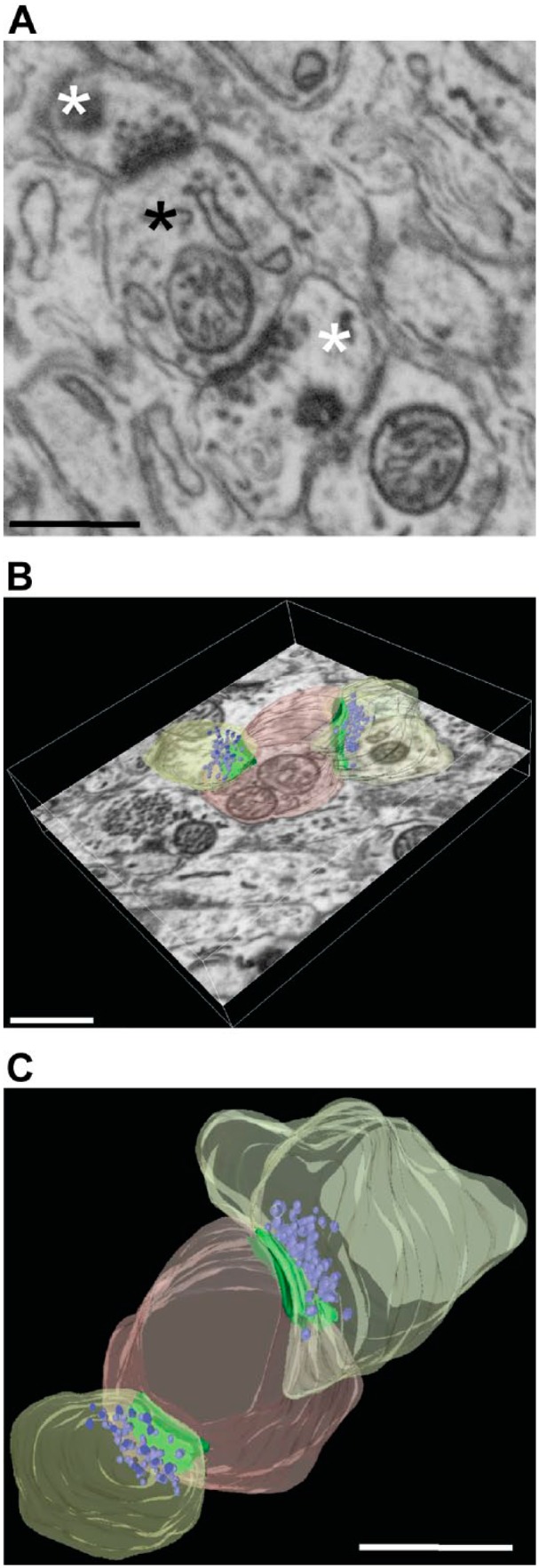
FIB-SEM imaging of Purkinje cells synapses. (A) Details of synaptic ultrastructure after FIB-SEM imaging of an unlabeled area of a calbindin-stained specimen. A sample image of a postsynaptic bouton (black asterisk) making contact with two presynaptic elements (white asterisks), showing good preservation of ultrastructure. (B-C) Same synapse manually segmented and reconstructed; postsynapse is shown in pink, presynaptic profiles in yellow, AZ-PSD complex in green, and vesicles in purple. Imaging was performed using a FEI Scios DualBeam system. Scale bar = 100 µm. Abbreviations: FIB-SEM, focused ion beam–scanning electron microscopy; AZ, active zone; PSD, postsynaptic densities.
The FIB-SEM (Zeiss) is set to remove 20-nm-thick layers of specimen by propelling gallium ions at the surface of the specimen. Image acquisition with the Zeiss microscope for segmentation, tracing, and 3D reconstruction is done at 1.5 kV using a backscattered electron detector, at 5k× magnification. The FIB-SEM (FEI) is set to remove 5-nm-thick layers of specimen. Imaging with FEI microscope for synaptic imaging is done at 1.7 kV using a backscattered electron detector at 7.6k× magnification. The imaging for neuronal tracing is performed at 0.005 µm/pixel (Zeiss) or for synaptic imaging at 0.00488 µm/pixel (FEI). Images are then aligned using Imaris (Bitplane; Zurich, Switzerland) or ImageJ software. Individual dendrites are segmented, and synapses are reconstructed and visualized using Imaris and Amira software.
Results and Discussion
DAB/GSSP Reaction and Light Microscopy
Calbindin-D28k (calbindin) is one of the major calcium-binding proteins and is important for the maintenance of calcium homeostasis. In the cerebellum, calbindin antibody staining is exclusive to Purkinje cells.13 We used a well-characterized commercial calbindin antibody for labeling the cerebellar Purkinje cells for the validation of our development of the labeling protocol for FIB-SEM (Fig. 1). As detection reagents, we used a biotinylated secondary antibody followed by the DAB reaction. Once a brown staining clearly coinciding with Purkinje neurons against a perfectly clear background was observed, the reaction was stopped and the sections were submitted to the silver gold substitution (GSSP) protocol. As a result of the GSSP reaction, the staining became dark brown or black. Labeled Purkinje cells could be easily recognized in vibratome sections before embedding into the resin for electron microscopy (Fig. 1A and B), after embedding (Fig. 1C), and in the semithin sections imaged after cutting with LM (Fig. 1D). In sagittal sections of the cerebellum, numerous immunopositive cells were evident in the Purkinje cell layer, and the reaction products were observed in Purkinje cells somata and dendritic trees (Fig. 1B and C) under the light microscope.
Epon Embedding and TEM Analysis
The next step was to determine the effect of the developed protocol on the preservation of cellular structure at TEM level. First, we compared side by side calbindin DAB and DAB/GSSP staining at TEM level (Fig. 2A vs B). The DAB calbindin staining (Fig. 2A) resulted in dark reaction products that blocked ultrastructural details, whereas the GSSP reaction products were observed as small black particles of variable size which allowed identification of underlying details (Fig 2B and C–E). The GSSP reaction product at TEM level was localized in bodies (Fig. 2C and D), dendritic profiles (Fig. 2B and D), and synaptic terminals of Purkinje cells (Fig. 2E), which is in agreement with literature data13 and our LM observations (see Fig. 1). Overall, the samples displayed good preservation of the ultrastructure and contrast sufficient to resolve organelles such as mitochondria, even without additional postcutting contrasting with uranyl acetate/lead compounds (Fig. 2E). This is important, as the subsequent FIB-SEM step does not allow for further postembedding contrasting.
Focused Ion Beam–Scanning Electron Microscopy
After examination with TEM, the same blocks were used for imaging by FIB-SEM. FIB-SEM imaging was performed using two different instruments, as a proof of concept: the Auriga Crossbeam system (Zeiss) and the Scios DualBeam microscope (FEI). The block area for FIB-SEM containing the labeled Purkinje cell layer was sectioned at 20-nm thickness to enable dendritic tracing (Fig. 3, Zeiss), and a total volume of 7128 μm3 of the cerebellar Purkinje cells area was imaged. To resolve complete synapses at high resolution, the FEI Scios was used to image the cerebellar neuropil area with 5 nm “slicing” (i.e., depth of optical milling; Fig. 4, FEI), followed by 3D reconstruction. The total volume of the imaged cerebellar neuropil was 154 μm3. Both sets of samples were investigated using the two different FIB-SEM systems, generating backscattered electron images of the block-face (Figs. 3A and 4A). We found that the contrast of the resulting images was better than that in images obtained with TEM (compare Figs. 3A and 4A with Fig. 2B–E), with easily detectable calbindin labeling observed throughout the entire stack, facilitating manual or semiautomatic tracing (Fig. 3 and Supplemental Video 1).
After FIB-SEM imaging, Z-stack image series for tracing (Fig. 3 and Supplemental Video 1) were manually segmented using the Imaris software, and calbindin-positive structures were segmented via semiautomatic thresholding in the same program (Fig. 3B and C). The labeling within the Purkinje cells somata and dendritic trees was easily followed throughout the entire stack volume (Supplemental Video 1).
We endeavored to image and fully reconstruct synaptic boutons (i.e., those boutons that could be followed in their entirety in consecutive serial sections) as examples of highly complex synaptic elements visualized at high resolution (Fig. 4). The resolution used for scanning the neuropil volume in the molecular layer of the cerebellum was sufficient to define with confidence dendritic and axonal membranes, as well as synaptic components (Fig. 4A). Purkinje cells synapses in the molecular layer of cerebellum were identified on the basis of morphological criteria, such as the presence of large, spheroid vesicles and a prominent postsynaptic density of the postsynaptic profile. A reference area without any labeling was specifically chosen to fully resolve the synaptic ultrastructural details without potential interference from staining.
In this study, we used 80-µm-thick vibratome sections, which are thin enough to allow substantial, albeit incomplete and decreasing with depth, penetration of the antibody into the tissue slice. Milling deep into the specimen yielded images with progressively reduced label intensity. We were able to continue imaging synaptic boutons of Purkinje cells well beyond the labeled area of the specimen, and fully reconstructed such structures in 3D. Individual synaptic elements, such as the active zone (AZ), postsynaptic densities (PSD), mitochondria and the vesicle pool within individual boutons, and the presynaptic profiles contacted by the postsynapse were reconstructed (Fig. 4B–C and Supplemental Video 2). From the resulting contours, 3D volumetric reconstructions can be easily assembled, and numeric parameters of individual synapses such as volume measurements, sizes and numbers of vesicles, and area of AZ-PSD can be calculated.
We developed a robust immunolabeling protocol for FIB-SEM imaging. Labeled samples can be imaged at the electron microscopic level at high resolution using various FIB-SEM instrumentation, resulting in easily detectable immunoreactivity in the context of well-preserved tissue. Our method contributes to the repertoire of detection techniques compatible with modern 3D imaging techniques utilizing FIB-SEM principles. We thoroughly tested our application using calbindin staining of cerebellar Purkinje cells. However, our technique does not involve any steps specific to this particular antibody or this anatomical structure, and is therefore applicable to any cells or tissues. In stark contrast to previously reported FIB-SEM immunodetection techniques, our method does not require any genetic manipulations, making it substantially more adaptable for routine use.
Acknowledgments
We thank all members of the Gounko laboratory for helpful discussions and comments. Light imaging was performed with the help of the Light Imaging facilities and Dr. Graham Wright (A*STAR, IMB; Singapore). We thank Dr. Anna Brichkina (A*STAR, IMCB; Singapore) for valuable suggestions on the article and for sharing reagents. We thank Edmund Wee and Rebecca Poh from the Application Laboratories of Carl Zeiss Pte Ltd Singapore Zeiss Center, Singapore, and Andy Ong from the FEI, Singapore, as well as the FEI Application Laboratories in Eindhoven, the Netherlands, for access to their microscopes for exploratory experiments.
Footnotes
Competing Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions: NVG conceived the project and designed the experiments. AB, DK, and KV performed the experiments. AB, VR, DK, and KV analyzed the data. VR and NVG wrote the article.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The study was supported by internal funds of the Joint IMB-IMCB core facility (A*Star; Singapore) and of the Electron Microscopy Platform Facility (VIB-KU Leuven, Belgium).
ORCID iD: NV Gounko  https://orcid.org/0000-0003-0791-9477
https://orcid.org/0000-0003-0791-9477
Contributor Information
Adrian Boey, Institute of Molecular and Cell Biology-Institute of Medical Biology Joint Electron Microscopy Suite, Agency for Science, Technology and Research, Singapore, Singapore.
Vasily Rybakin, Laboratory of Immunobiology, Department of Microbiology and Immunology, Rega Institute, KU Leuven, Leuven, Belgium.
Dharamdajal Kalicharan, Institute of Molecular and Cell Biology-Institute of Medical Biology Joint Electron Microscopy Suite, Agency for Science, Technology and Research, Singapore, Singapore.
Katlijn Vints, VIB-KU Leuven Center for Brain & Disease Research, Electron Microscopy Platform & VIB-Bioimaging Core, Leuven, Belgium; Department of Neurosciences, Leuven Brain Institute, KU Leuven, Leuven, Belgium.
Natalia V. Gounko, Institute of Molecular and Cell Biology-Institute of Medical Biology Joint Electron Microscopy Suite, Agency for Science, Technology and Research, Singapore, Singapore; VIB-KU Leuven Center for Brain & Disease Research, Electron Microscopy Platform & VIB-Bioimaging Core, Leuven, Belgium; Department of Neurosciences, Leuven Brain Institute, KU Leuven, Leuven, Belgium.
Literature Cited
- 1. Bushby AJ, P’ng KM, Young RD, Pinali C, Knupp C, Quantock AJ. Imaging three-dimensional tissue architectures by focused ion beam scanning electron microscopy. Nat Protoc. 2011;6(6):845–58. [DOI] [PubMed] [Google Scholar]
- 2. Kizilyaprak C, Bittermann AG, Daraspe J, Humbel BM. FIB-SEM tomography in biology. Methods Mol Biol. 2014;1117:541–58. [DOI] [PubMed] [Google Scholar]
- 3. Vidavsky N, Akiva A, Kaplan-Ashiri I, Rechav K, Addadi L, Weiner S, Schertel A. Cryo-FIB-SEM serial milling and block face imaging: large volume structural analysis of biological tissues preserved close to their native state. J Struct Biol. 2016;196(3):487–95. [DOI] [PubMed] [Google Scholar]
- 4. Luckner M, Wanner G. Precise and economic FIB/SEM for CLEM: with 2 nm voxels through mitosis. Histochem Cell Biol. 2018;150(2):149–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Russell MR, Lerner TR, Burden JJ, Nkwe DO, Pelchen-Matthews A, Domart MC, Durgan J, Weston A, Jones ML, Peddie CJ, Carzaniga R, Florey O, Marsh M, Gutierrez MG, Collinson LM. 3D correlative light and electron microscopy of cultured cells using serial blockface scanning electron microscopy. J Cell Sci. 2017;130(1):278–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Martell JD, Deerinck TJ, Sancak Y, Poulos TL, Mootha VK, Sosinsky GE, Ellisman MH, Ting AY. Engineered ascorbate peroxidase as a genetically encoded reporter for electron microscopy. Nat Biotechnol. 2012;11:1143–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shi Y, Wang L, Zhang J, Zhai Y, Sun F. Determining the target protein localization in 3D using the combination of FIB-SEM and APEX2. Biophys Rep. 2017;3(4):92–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Graham RC, Jr, Karnovsky MJ. The early stages of absorption of injected horseradish peroxidase in the proximal tubules of mouse kidney: ultrastructural cytochemistry by a new technique. J Histochem Cytochem. 1966;4:291–302. [DOI] [PubMed] [Google Scholar]
- 9. Bosch C, Martínez A, Masachs N, Teixeira CM, Fernaud I, Ulloa F, Pérez-Martínez E, Lois C, Comella JX, DeFelipe J, Merchán-Pérez A, Soriano E. FIB/SEM technology and high-throughput 3D reconstruction of dendritic spines and synapses in GFP-labeled adult-generated neurons. Front Neuroanat. 2015;9:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. van den Pol AN, Gorcs T. Synaptic relationships between neurons containing vasopressin, gastrin-releasing peptide, vasoactive intestinal polypeptide, and glutamate decarboxylase immunoreactivity in the suprachiasmatic nucleus: dual ultrastructural immunocytochemistry with gold-substituted silver peroxidase. J Comp Neurol. 1986;252(4):507–21. [DOI] [PubMed] [Google Scholar]
- 11. Swinny JD, Kalicharan D, Brouwer N, Biber K, Shi F, Gramsbergen A, van der Want JJ. The postnatal developmental expression pattern of urocortin in the rat olivocerebellar system. J Comp Neurol. 2004;472(1):40–51. [DOI] [PubMed] [Google Scholar]
- 12. Gounko NV, Kalicharan D, Rybakin V, Gramsbergen A, van der Want JJ. The dynamic developmental localization of the full-length corticotropin-releasing factor receptor type 2 in rat cerebellum. Eur J Neurosci. 2006,12:3217–24. [DOI] [PubMed] [Google Scholar]
- 13. Maeda H, Ellis-Davies GC, Ito K, Miyashita Y, Kasai H. Supralinear Ca2+ signaling by cooperative and mobile Ca2+ buffering in Purkinje neurons. Neuron. 1999;4:989–1002. [DOI] [PubMed] [Google Scholar]
- 14. Walton J. Lead aspartate, an en bloc contrast stain particularly useful for ultrastructural enzymology. J Histochem Cytochem. 1979;10:1337–42. [DOI] [PubMed] [Google Scholar]