

Abstract
Abstract. Objective: A novel nanocomposite has recently been developed based on polyhedral oligomeric silsesquioxane attached by direct reaction onto a urethane segment, as a potential vascular graft material; its trade name is UCL‐Nano. The UCL‐Nano has been demonstrated to have similar viscoelastic properties to the walls of a natural artery, to be resistant to degradation and to be able to sustain endothelial cell seeding. Human peripheral blood contains both circulating endothelial cells and endothelial progenitor cells, which may be suitable for conduit seeding. The aim of this study was to develop a system with the potential to deliver an endothelial cell‐seeded bypass graft in a realistic time frame. Materials and methods: Endothelial progenitor cells and circulating endothelial cells were isolated from human peripheral blood and were characterized by fluorescent‐activated cell sorting, reverse transcriptase‐polymerase chain reaction and immunohistochemistry. Isolated cells were seeded on nanocomposite and were maintained in culture for 35 days. Results: The UCL‐Nano was successfully seeded with cells and a confluent cell layer was achieved after 14‐day culture. Cells remained viable and confluent on the nanocomposite for 35 days. Conclusion: In conclusion, these results suggest that this process has potential both for a realistic and achievable two‐stage seeding process for vascular bypass grafts and for the potential development of a device, with the aim of achieving in situ seeding once implanted.
INTRODUCTION
The use of prosthetic grafts in vascular surgery has increased since the introduction of fabric arterial grafts in the mid‐20th century. In the UK, it is estimated that one in three deaths could be attributed to diseases of the circulatory system or heart and this is likely to be a continuing problem in the future, as life expectancy of the population increases. In an ideal situation currently, an autologous vein or artery is the most suitable conduit choice for bypass operations (the saphenous vein being the best choice for lower limb arterial bypass), and these result in a 5‐year patency rate of around 75%. In around 30% of cases, a suitable autologous conduit is not available for lower limb bypass (Kannan et al. 2005a), leading to a requirement for the use of an artificial conduit (Rashid et al. 2004).
There are two major types of prosthetic graft in clinical usage at the moment, based on either expanded polytetrafluoroethylene or polyethylene terephthalate (Dacron®, Sulzer Vascutek Limited, Inchinnan, UK). Current prostheses employed have a low patency rate, however, due to low compliance and thrombogenicity of the materials used. Several approaches have been used to attempt to improve this patency, including surface modification of existing grafts, endothelial cell (EC) seeding and recently the development of tissue‐engineered bypass grafts (Baguneid et al. 2006; Isenberg et al. 2006; Sarkar et al. 2007), and development of new compliance prostheses with active biomolecules and endothelialization (Kidane et al. 2004).
The most interesting material our group has been working on is an novel nanocomposite based on polyhedral oligomeric silsesquioxane‐poly(carbonate‐urea)urethane, which has the trade name of UCL‐Nano; the potential advantages of UCL‐Nano have been reviewed previously (Kannan et al. 2005b). UCL‐Nano has been demonstrated to have similar viscoelastic properties to a natural artery and to be resistant to degradation (Kannan et al. 2005c). Several previous studies have also indicated that UCL‐Nano can sustain human umbilical vein endothelial cell (HUVEC) seeding (Punshon et al. 2005; Kannan et al. 2006b,c). A further aspect that has been investigated to overcome this problem is seeding the artificial material with ECs to improve blood compatibility of the device. Sources of material from which to obtain suitable cells to attempt seeding include the patient's own veins (Herring et al. 1978; Stanley et al. 1985), omentum, subcutaneous fat and peritoneal lavage (Tiwari et al. 2003), all of which have the disadvantages of being invasive to obtain (requiring an operation) and it is difficult to extract an adequate number of cells. These factors limit their potential in both research and clinical situations.
In a clinical context, two‐stage seeding (in which ECs are extracted from tissue then cultured for a period of time in the laboratory) has been demonstrated to be successful in a number of studies with a significant increase in graft patency. In one such study (Zilla et al. 1994), autologous ECs from the external jugular vein were seeded on to 70 cm long, 6 mm internal diameter, polytetrafluoroethylene grafts coated with fibrin glue. This was performed for 33 patients with either disabling claudication or with critical ischaemia. The average culture time required after vein incision was 25 days and growth failure occurred in 27% of the cases. Cases were followed up by angiography, platelet adhesion studies, assessment of the ankle–brachial index and duplex sonography. After 32 months, patency of the endothelialized grafts was 85% compared to 55% for the unseeded control grafts, thus demonstrating an improvement similar to that found when employing autologous grafts. These results were confirmed by further long‐term studies by the same group reporting similar results, when the failure rate reduced to 5% with a shorter growth period following optimization of cell extraction and growth process (Deutsch et al. 1999; Meinhart et al. 2001).
Despite demonstration of the potential of the two‐stage seeding technique developed by this group, such techniques have not, to date, been taken up in widespread practice. There are several reasons why this technique is been limited. First, it requires the patient to undergo an extra operation to collect the tissue required to extract ECs prior to culture in the laboratory. This exposes them to additional risk to obtain tissue and has considerable cost implications, over and above those for the cell culture stage where a dedicated facility is required. Second, the period required to grow up the cells to a sufficient number and to seed the device is considerable (3–4 weeks) and, in a number of cases, the cells fail to multiply sufficiently to seed the graft.
There has been much interest in the potential for using human stem cells to treat a wide variety of medical conditions in the future (Sales et al. 2005). Much of this research is based on using embryonic stem cells, cells obtained from bone marrow or cells obtained from umbilical cord blood. While the potential for such work is great, it is associated with grave problems from the ethical viewpoint when working with human tissue in the case of embryonic stem cells, difficulty in obtaining adequate samples for research in the case of bone marrow or clinical practicality in the case of umbilical cord blood.
It has recently been demonstrated, however, that human peripheral blood contains both circulating endothelial cells (CEC) and endothelial progenitor cells (EPC), a type of stem cell with the potential to expand. An initial report by Asahara et al. (1997) used magnetic beads to separate CD34+ cells and studies by groups, such as Hill et al. (2003), have demonstrated that it was possible to extract progenitor cells without the use of magnetic beads. Further studies have demonstrated the potential of progenitor cells isolated from this source (Shirota et al. 2003; Hur et al. 2004; Ingram et al. 2004) and a previous pilot investigation carried out by our group has demonstrated the potential of UCL‐Nano to support initial formation of EPC colonies isolated from human peripheral blood (Alobaid et al. 2006).
In view of the above, the aim of this study was to develop a system with the potential to deliver an EC‐seeded bypass graft in a realistic time frame and in a manner such that general clinical uptake of the process would be possible. This could be achieved by building on the proven clinical success of two‐stage seeding, while attempting to overcome the potential pitfalls of some cell sources by employing peripheral blood as the origin of cells and using UCL‐Nano as the scaffold for seeding.
MATERIALS AND METHODS
Preparation of UCL‐Nano polymer films
Synthesis of the nanocomposite has been described in detail previously (Seifalian et al. 2005; Kannan et al. 2006a), but in brief, the inorganic urethane is made from 4,4′‐methylene‐bis(phenyl‐isocyanate) (MDI), poly(hexamethylene carbonate) diol, and silsesquioxane dissolved in tetrahydrofuran, here, bis[3‐(trimethoxysilyl)propyl]amine, and a chain extended with ethylene diamine in N,N′‐dimethylacetamide (DMAC). Glass Petri dishes (40 mm diameter) were then coated with 4 mL of nanocomposite (15% in DMAC) and were cured overnight at 55 °C. The resulting films were then thoroughly washed with sterile phosphate‐buffered saline (PBS) and were sterilized by immersion in 70% ethanol followed by air drying.
Blood collection
Blood samples were obtained with consent from healthy adult human volunteers. 20 mL samples were collected by venepuncture in ethylenediaminetetraacetic acid blood tubes (Sarstedt, Leicester, UK). Following collection, samples were mixed and were used for cell isolation within 1 h of collection.
Cell isolation
The mononuclear fraction of the blood was then isolated using Lymphoprep (Axis‐Shield, Huntingdon, UK). Briefly, 20 mL of blood was transferred to a 50‐mL centrifuge tube (Falcon; BD Biosciences, Cowley, UK) and was gently mixed with 20 mL Hanks’ balanced salt solution (HBSS) (Invitrogen, Paisley, UK). Five millilitres Lymphoprep was then added to each of eight 12‐mL polystyrene centrifuge tubes (Falcon; BD Biosciences) and 5 mL of the blood/HBSS mixture was then layered carefully on top. Tubes were then centrifuged at 280 g at 20 °C for 25 min. The mononuclear fraction was then separated from each and was placed equally into two 30‐mL universal tubes (Falcon; BD Biosciences). Ten millilitres of HBSS was then added slowly to each tube and the contents mixed. Tubes were then centrifuged at 280 g and 20 °C for 15 min. The supernatant was then discarded and cells were re‐suspended in 2 mL HBSS. The volume was made up to 10 mL and cells were centrifuged at 220 g at 20 °C for 15 min to remove red blood cells. The supernatant was again discarded and the cell pellet was re‐suspended in 10 mL HBSS and a final centrifugation step at 280 g at 20 °C for 15 min was performed. Finally, the supernatant was again discarded and the cells re‐suspended in 5 mL cell culture medium (CCM): RPMI 1640 supplemented with 20% foetal bovine serum and penicillin/streptomycin (Invitrogen). Cells were then counted using a haemocytometer.
Fluorescent‐activated cell sorting analysis for CD133+/VEGFR2+ EPC
One hundred microlitres of initial cell isolate was incubated with murine immunoglobulin G for 15 min at 4 °C to block non‐specific binding/specific binding, via Fragment crystallisable Region (FcR), followed by incubation with a novel cocktail of antibodies comprising fluorescein isothiocyanate (FITC)‐conjugated mouse antihuman CD2, CD13 and CD22 (BD Biosciences), phycoerythrin‐conjugated mouse antihuman VEGFR2 (R&D Systems, Abingdon, UK) and biotin‐conjugated mouse antihuman CD133 (Miltenyi Biotec, Surrey, UK) monoclonal antibodies for 10 min at 4 °C, then washed. Following this, streptavidin‐PeCy7 (Beckman Coulter, High Wycombe, UK) was added and samples were further incubated for 10 min at 4 °C. Red cells were lysed and the samples were fixed using the Coulter TQ‐prep system (Beckman Coulter). Samples were re‐suspended in HBSS and fluorescent particles [Sphero™ Accucount fluorescent particles (ACFP‐100‐3), 10 µm; Spherotech Inc., Lake Forest, USA] were added to calculate absolute cell numbers. The calculation was performed as follows:
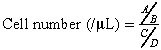 |
where, A is number of events for the test sample; B is number of events for the Accucount particles; C is number of Accucount particles per 50 µL and D is volume of test sample initially used in µL.
Samples required a count of 10 000 fluorescent particles to give a total of 0.5–1 × 106 mononuclear events. They were analysed immediately by FACScan using CellQuest 3.1 software (Becton Dickinson). The instrument was standardized daily with calibration beads (FluoroSpheres 6‐Peak; DakoCytomation, Ely, UK) and was thoroughly cleaned before data acquisition, to exclude any trace amounts of cellular debris or residual cells from a previous tube. The cleaning process composed of sequential tubes with water, bleach and water again, for about 30 min.
Cell culture
Isolated cells were cultured for an initial 2 days in a 6‐well plate (Falcon; BD Biosciences) at a seeding density of 9.2 × 106 cells/well, following which the supernatant (containing the non‐attached cells) from two wells was transferred to a UCL‐Nano film‐coated Petri dish. Cells were then cultured in CCM for a further 5 days. The supernatant was then removed from the UCL‐Nano film‐coated dishes and remaining adherent cells were cultured in CCM with the medium being changed every 2–3 days for a total of 35 days. Cells were also cultured on 8‐well slides (Falcon; BD Biosciences) for characterization studies. Experiments were repeated between three and five times.
Measurement of cell metabolism
Cell metabolism was assessed by Alamar blue™ (AB) (Serotec, Kidlington, UK) assay at days 7, 14, 21, 28 and 35 as described previously (Seifalian et al. 2001). In brief, medium was removed from the UCL‐Nano film‐coated dishes and 4 mL 10% AB in CCM was added. After a 4‐h incubation, samples of AB/CCM were removed and were measured on a Fluroskan Ascent FL (Thermo Labsystems, Basingstoke, UK) fluorescent plate reader (excitation 530 nm, emission at 620 nm). Unseeded UCL‐Nano film‐coated Petri dishes were used as controls.
RNA isolation and investigation
RNA was extracted from initial cell isolates, confluent cell cultures on 6‐well plates, and day 35 confluent cell cultures seeded on UCL‐Nano as described previously (Vara et al. 2006), by using an ‘RNeasy™’ kit (Qiagen Ltd., Crawley, UK). The RNA concentration and purity were calculated by measuring the absorbance at 260 and 280 nm by Eppendorf Biophotometer (EppendorfAG, Hamburg, Germany). Quality of the RNA was assessed by 2% agarose gel electrophoresis and mRNA obtained was used for polymerase chain reactions (PCR) for CD14, CD34, CD133, PECAM‐1 and von Willebrand factor (vWF). Reverse transcriptase (RT)‐PCR was performed using a One‐Step PCR kit (Qiagen Ltd.). Template RNA (0.25 µg) and 0.5 µm of the appropriate primers (Table 1) were used for each PCR. Cycle conditions were as follows: CD14, 30 cycles at 94 °C, 57 °C and 72 °C; CD34, 35 cycles at 94 °C, 54 °C and 72 °C; CD133, 35 cycles at 94 °C, 52 °C and 72 °C; VEGFR2, 35 cycles at 94 °C, 59 °C and 72 °C; vWF, 35 cycles at 94 °C, 50 °C and 72 °C; PECAM‐1, 35 cycles at 94 °C, 50 °C and 72 °C, and amplification was carried out using MasterCycler Gradient PCR apparatus (EppendorfAG). In the case of vWF and PECAM‐1, RNA extracted from HUVEC was used as a positive control. PCR products were then analysed by 2% agarose gel electrophoresis with a GeneGenius darkroom, ‘GeneSnap’ version 6.02 (Syngene, Cambridge, UK).
Table 1.
Primer sequences for CD14, CD34, CD133, VEGFR2, CD31 and von Willebrand factor (vWF) gene expression analysis
| Marker | Sense | Antisense |
|---|---|---|
| CD14 | 5′‐CCTCCCAAGTTTTAGGACAA‐3′ | 5′‐CAGCTGGTGATAAGGGTTAG‐3′ |
| CD34 | 5′‐CCTCCCAAGTTTTAGGACAA‐3′ | 5′‐CAGCTGGTGATAAGGGTTAG‐3′ |
| CD133 | 5′‐CAGTCTGACCAGCGTGAAAA‐3′ | 5′‐GGCCATCCAAATCTGTCCTA‐3′ |
| VEGFR2 | 5′‐GTGACCACATGGAGTCGTG‐3′ | 5′‐CCAGAGATTCCATGCCACTT‐3′ |
| CD31 | 5′‐GCTGTTGGTGGAAGGAGT‐3′ | 5′‐GAAGTTGGCTGGAGGTGCTC‐3′ |
| vWF | 5′‐TGCTGACACCAGAAAAGTGC‐3′ | 5′‐AGTCCCCAATGGACTCACAG‐3′ |
Giemsa staining
Cells were washed with PBS and ice‐cold methanol was then added to fix the cells. Two hundred microlitres Giemsa stain (Invitrogen) were added and the cells were left to stain for 15 min at room temperature. Cells were destained with PBS then were examined microscopically. This staining was performed on days 5, 14, 21 and 35.
Immunohistochemistry for CD34, CD133, vWF and PECAM‐1 (CD31) staining of initial cell isolations and confluent cells
Cell samples were collected from the initial cell isolation and were cytospun on to polylysine‐coated glass microscope slides (VWR, Lutterworth, UK). Confluent cell cultures were grown on 8‐well slides; cells were then fixed in ice‐cold methanol (VWR), air‐dried and stored at 4 °C until immunostained.
Staining for CD34 was carried out using an FITC‐conjugated CD34 antibody (Milteyni, Bisley, UK), for CD133 staining was carried out using a biotin‐conjugated CD133 primary antibody and visualized using antibiotin allophycocyanin (APC) secondary antibody (both Miltenyi). For vWF, immunostaining was carried out using a mouse monoclonal primary antibody to vWF and visualization using a goat polyclonal antimouse immunoglobulin G H&L (FITC‐conjugated) secondary antibody (ABCAM, Cambridge, UK). CD31 immunostaining was carried out using an APC‐conjugated anti‐CD31 antibody (Milteyni). All staining was carried out following manufacturers’ protocols and all preparations were mounted with mounting medium (Vector, Peterborough, UK) before being examined microscopically.
RESULTS
Number of cells isolated
The average number of cells per isolation was 37.5 × 106 ± 2.41 (standard error of the mean) cells/20 mL blood. Cells were successfully isolated from all blood samples taken.
Fluorescent‐activated cell sorting analysis for CD133+/VEGFR2+ EPC
The number of CD133+ cells per mL of initial cell isolation (all isolations 5 mL) as determined by fluorescent‐activated cell sorting (FACS) analysis was 131 ± 13.53 cells per mL of cell isolation. For CD133+/VEGFR2+ (EPC) cells, 13.65 ± 6.21 were produced per mL of cell isolation.
Assessment of metabolism of cells seeded on UCL‐Nano
Metabolism of cells seeded on the nanocomposite films was measured over a 35‐day period. Cells were present at all time‐points measured and there was no significant reduction in cell metabolism over the course of the investigation (Fig. 1).
Figure 1.
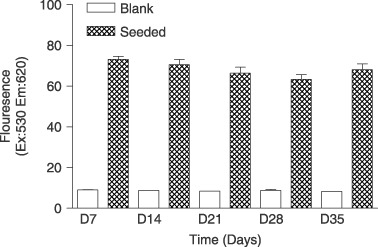
Cell metabolism determined by Alamar blue™ assay measuring fluorescence (excitation: 530; emission: 620) up to 25 days (n = 5).
RNA isolation and investigation of initial cell isolations, confluent cells and day 35 confluent cells seeded on UCL‐Nano
Figure 2a demonstrates a 2% agarose gel of three initial cell isolations (A, B and C) examined by RT‐PCR for CD14, CD34, CD133 and VEGFR2. All isolations had CD14, CD34, CD133 and VEGFR2 expression. Figure 2b similarly shows a 2% agarose gel of three confluent cell cultures (A, B and C) examined in the same way. CD14, CD34 and VEGFR2 were still detected while CD133 was not found to be present. All negative controls showed no PCR product present. Figure 2c shows a 2% agarose gel of an initial cell isolation (A) and two day 35 nanocomposite‐seeded confluent cell samples (B and C). Results are similar to those obtained from confluent cells on 6‐well plates. Figure 2d shows a 2% agarose gel of two HUVEC cell samples (A and B), two initial cell isolations (C and D) and two day 35 nanocomposite‐seeded confluent cell samples (E and F) examined for PECAM‐1 and vWF. All isolations showed expression of both genes.
Figure 2.
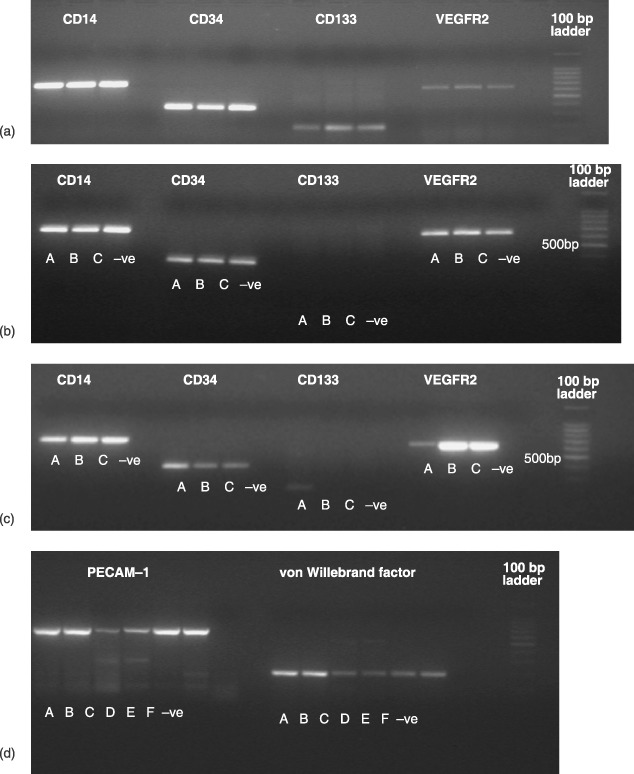
A 2% agarose gel of reverse transcript‐polymerase chain reaction (RT‐PCR) for (a) three initial cell isolations (A, B and C) with CD14, CD34, CD133 and VEGFR2, (b) three confluent cell isolations (A, B and C) with CD14, CD34, CD133 and VEGFR2; (c) an initial cell isolation A and two day 35 nanocomposite confluent seeded samples (B and C) with CD14, CD34, CD133 and VEGFR2; and (d) two human umbilical vein endothelial cell (HUVEC) isolations (A and B), two initial cell isolations (C and D) and two day 35 nanocomposite confluent seeded samples (E and F) for PECAM‐1 and von Willebrand factor.
Giemsa staining of cells seeded on UCL‐Nano
Cells seeded on nanocomposite were stained with Giemsa stain at days 5, 14, 21 and 35. At day 5, several Hill colonies were present. By day 14, the majority of the nanocomposite was covered by cells with some dense areas where Hill colonies had developed; this was maintained to day 35 (Fig. 3).
Figure 3.
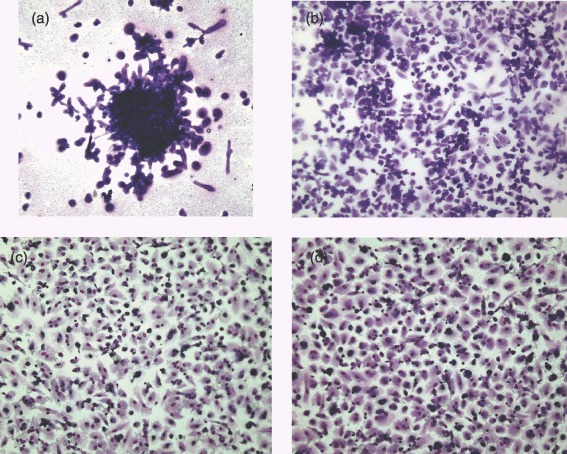
Cells seeded on nanocomposite and stained with Giemsa stain at (a) day 5, (b) day 14, (c) day 21 and (d) day 35 (×10 magnification).
CD34 and CD133 staining of initial cell isolations and confluent cells
Figure 4 shows staining for CD34 and CD133 on an initial cell isolation sample. All views are of the same cell population. Figure 4a demonstrates a large mixed cell population present, with two basic cell types, small round cells and larger round cells. Figure 4b illustrates cells staining expressing CD34 [FITC (green)]. All larger cells present were CD34 positive whereas only a small proportion of the smaller cells were CD34+ve. Figure 4c shows cells positive for CD133 [allophycocyanin (red)]. All of the larger CD34+ve cells appeared to be CD133−ve, whereas, again, a small proportion of the smaller cells appeared to be CD133+ve. Figure 4d shows dual staining for CD34 and CD133. A low proportion of the smaller cells can clearly be seen to be both CD34+ve and CD133+ve.
Figure 4.

Initial cell isolation stained for CD34 (FITC) and CD133 (allophycocyanin): (a) unstained, (b) CD34 stained, (c) CD133 stained and (d) dual stained. All views are of the same cell population (×100 magnification).
Figure 5 shows staining for CD34 and CD133 on a confluent cell culture sample. All views are of the same cell population (Fig. 5a), indicating a fairly consistent cell population. Figure 5b demonstrates cells with positive staining for CD34 [FITC (green)]. The majority of cells present were CD34 positive; Fig. 5c shows cells positive for CD133 [allophycocyanin (red)]. Again the majority of cells expressed CD133, with a few of them expressing high levels. Figure 5d shows dual staining for CD34 and CD133; the majority of them appearing to be both CD34+ve and CD133+ve at confluence.
Figure 5.

Confluent cell culture stained for CD34 (FITC) and CD133 (allophycocyanin): (a) unstained, (b) CD34 stained, (c) CD133 stained and (d) dual stained. All views are of the same cell population (×100 magnification).
von Willebrand factor staining of initial cell cultures and confluent cells
Staining for vWF is demonstrated in Fig. 6. Figure 6a,c shows an initial cell isolation stained for vWF, with a mixed cell population of small and larger cells, larger cells being strongly positive for vWF. There was, in addition, some staining in the population of smaller cells. Figure 6b,d shows confluent cell cultures stained for vWF and demonstrate that the majority of the cells were vWF positive.
Figure 6.
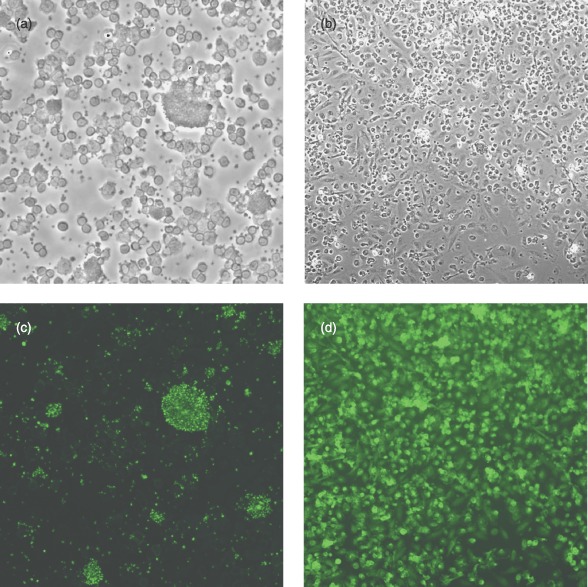
Initial cell isolation and confluent cells stained for von Willebrand factor (vWF): (a) initial isolation unstained, (b) confluent cells unstained, (c) initial isolation stained for vWF and (d) confluent cells stained for vWF (×100 magnification).
PECAM‐1 (CD31) staining of initial cell isolations and confluent cells
Initial cell isolations stained for CD31 had a small minority of cells that were positive (Fig. 7a,c). Confluent cultures demonstrated low overall level of staining for this protein (Fig. 7b,d).
Figure 7.

Initial cell isolations and confluent cells stained for PECAM‐1: (a) initial isolation unstained, (b) confluent cells unstained, (c) initial isolation stained for PECAM‐1 and (d) confluent cells stained for PECAM‐1 (×100 magnification).
DISCUSSION
Recent research on adult stems cells has focused on differentiating bone marrow progenitor cells into a variety of cell types. This is a promising area of study, but has many potential pitfalls and the mechanisms involved are complex. More practical at this time from the viewpoint of developing a functional EC‐seeded vascular prosthesis may be the use of the EPC, and here we have looked at isolating these cells and their culture on a novel nanocomposite material (UCL‐Nano), previously shown to be an excellent candidate for vascular graft matrix. The results obtained demonstrate that not only can this material support colonies of EPC, but also that they will differentiate into EC, grow to near confluence on the graft and remain viable for a considerable period of time on the UCL‐Nano surface.
To characterize the initial cell extract, a variety of techniques was employed. FACS analysis for CD133+/VEGFR2+ cells [the current standard for FACS analysis of EPC; Khan et al. (2005)] demonstrated that EPCs were present in the initial cell isolation in numbers similar to those obtained in other studies (George et al. 2006). RNA analysis confirmed that in the initial cell extract the monocyte marker CD14 and the EPC/mature EC marker CD34 were strongly expressed and the stem cell marker CD133 was also expressed to a lesser extent. PECAM‐1, VEGFR2 and vWF were also expressed in the initially isolated cells. These results were confirmed by immunohistochemical studies for CD34, CD133, vWF and PECAM‐1. These findings indicate that the isolation of EPC from peripheral blood was successfully achieved, which is in line with reports from other groups.
The process for culturing EPCs once isolated is currently a matter for debate and experimentation. Many groups use a ‘pre‐plating’ stage, after which non‐adherent cells are collected and cultured to remove rapidly adhering cells (such as CECs, monocytes and macrophages), the period for which varies from 1 h (Asahara et al. 1997; Shirota et al. 2003) to 48 h (Hill et al. 2003; George et al. 2006). Others omit a ‘pre‐plating’ stage and allow cells to seed for a variety of times ranging from 24 h (Ingram et al. 2004) to 3 days (Zhang et al. 2006) or 6 days (Hur et al. 2004). In this study, a 48‐h pre‐plating period was employed. Some samples of the initially adherent cells were continued in culture, but failed to prosper (data not shown). Following the pre‐plating period, cells were left for 5 days to adhere after which non‐adherent cells were disposed of. Again, some samples of the non‐adherent cells after 5‐day seeding were continued in culture but did not grow to any extent (data not shown).
All isolations cultured produced viable cell populations and developed a number of colonies with Hill colony characteristics when seeded on the UCL‐Nano. Cultures on UCL‐Nano achieved confluence in 14 days and resulted in cell populations with a cobblestone appearance by day 21. Cultures were maintained at confluence for a total of 35 days. Alamar blue™ studies confirmed that metabolically active cells were present over the 35‐day period of the study. The level of cell metabolism did not increase greatly in the initial period, which may be due to a combination of monocytes dying off, with EPC and CEC proliferating at the same time leading to little net change in the overall level of cell metabolism.
RNA analysis of confluent cells confirmed that CD14, CD34 and VEGFR2 expression was maintained, although CD133 expression was lost, which may be attributed to the conversion of EPC to EC over time. PECAM‐1 and vWF expression was also maintained and increased in comparison to that in the initial cell isolations. Immunostaining for CD34 and CD133 again showed that the majority of confluent cells expressed both markers and, in addition, also expressed vWF and PECAM‐1. Thus, the conditions employed in this study may be optimal for differentiation (due to the presence of growth factors in the foetal bovine serum), but not for prolonged division. An area of further investigation to be pursued, based on this study, will be to investigate cell proliferation in this model under dynamic conditions (such as shear stress), as this has been shown previously to be a trigger for cell proliferation.
Results obtained in this study suggest that it is possible to obtain a confluent layer of cells with endothelial‐like properties on UCL‐Nano after 14 days of culture, following extraction from normal human blood; all the extractions produced viable cells. Previous work has demonstrated that HUVECs attach to UCL‐Nano and (particularly with the employment of preconditioning) survive on it when exposed to physiological blood flow conditions (Vara et al. 2006). Thus, the cell extraction method employed compares favourably with initial findings of a 25‐day time period and a 27% failure rate quoted from clinical studies in two‐stage seeding, prior to their optimization, with the added advantage of the ease of blood collection compared to the difficulty of obtaining cells from other sources and requirement for an additional operation for patients. Further investigation will be carried out to improve and optimize the cell extraction and growth methods, perhaps employing magnetic beads to isolate CD34+ or CD133+ cells (Asahara et al. 1997; Salven et al. 2003). While it is possible that a mixed population of cells may ensue from the extraction method employed (including monocytes, EPCs and CECs), this should not be a major problem in the application envisaged, as long‐term studies carried out in humans suggest that with 9‐year patencies, ECs must be renewing on the graft either by infiltration from the anastomosis site or by EPCs or CECs from the circulation, attaching. Thus, the initial presence of a seeded layer of cells would seem to be the significant factor in producing a successful graft and improving patency (Zilla et al. 1994; Meinhart et al. 2001).
In conclusion, these results suggest that the process investigated has potential both for a realistic and achievable two‐stage seeding process for vascular bypass grafts or for the potential development of a device with the aim of achieving in situ seeding once implanted. Further optimization and development of the technique may result in a clinically successful method for improving the patency of vascular bypass grafts in the future.
ACKNOWLEDGEMENTS
The authors would like to thank Mr. Arnold Derbyshire, Biomaterial and Tissue Engineering Centre, University College London, for the nanocomposite synthesis; Ms. Natalie Saunders for carrying out FACS analysis and Mr. Innes Clatworthy for his assistance with cell characterization. We would like to acknowledge financial support for the development of nanomaterials for medical devices, provided by Engineering and Physical Sciences Research Council and University College London Business PLC.
REFERENCES
- Alobaid N, Salacinski HJ, Sales KM, Ramesh B, Kannan RY, Hamilton G, Seifalian AM (2006) Nanocomposite containing bioactive peptides promote endothelialisation by circulating progenitor cells: an in vitro evaluation. Eur. J. Vasc. Endovasc. Surg. 32, 76–83. [DOI] [PubMed] [Google Scholar]
- Asahara T, Murohara T, Sullivan A, Van Der Silver MZR, Li T, Witzenbichler B, Schatteman G, Isner JM (1997) Isolation of putative progenitor endothelial cells for angiogenesis. Science 275, 964–967. [DOI] [PubMed] [Google Scholar]
- Baguneid MS, Seifalian AM, Salacinski HJ, Murray D, Hamilton G, Walker MG (2006) Tissue engineering of blood vessels. Br. J. Surg. 93, 282–290. [DOI] [PubMed] [Google Scholar]
- Deutsch M, Meinhart J, Fischlein T, Preiss P, Zilla P (1999) Clinical autologous in vitro endothelialization of infrainguinal ePTFE grafts in 100 patients: a 9‐year experience. Surgery 126, 847–855. [PubMed] [Google Scholar]
- George J, Shmilovich H, Deutsch V, Miller H, Keren G, Roth A (2006) Comparative analysis of methods for assessment of circulating endothelial progenitor cells. Tissue Eng. 12, 331–335. [DOI] [PubMed] [Google Scholar]
- Herring M, Gardner A, Glover J (1978) A single‐staged technique for seeding vascular grafts with autogenous endothelium. Surgery 84, 498–504. [PubMed] [Google Scholar]
- Hill JM, Zalos G, Halcox JP, Schenke WH, Waclawiw MA, Quyyumi AA, Finkel T (2003) Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. N. Engl. J. Med. 348, 593–600. [DOI] [PubMed] [Google Scholar]
- Hur J, Yoon CH, Kim HS, Choi JH, Kang HJ, Hwang KK, Oh BH, Lee MM, Park YB (2004) Characterization of two types of endothelial progenitor cells and their different contributions to neovasculogenesis. Arterioscler. Thromb. Vasc. Biol. 24, 288–293. [DOI] [PubMed] [Google Scholar]
- Ingram DA, Mead LE, Tanaka H, Meade V, Fenoglio A, Mortell K, Pollok K, Ferkowicz MJ, Gilley D, Yoder MC (2004) Identification of a novel hierarchy of endothelial progenitor cells using human peripheral and umbilical cord blood. Blood 104, 2752–2760. [DOI] [PubMed] [Google Scholar]
- Isenberg BC, Williams C, Tranquillo RT (2006) Small‐diameter artificial arteries engineered in vitro . Circ. Res. 98, 25–35. [DOI] [PubMed] [Google Scholar]
- Kannan RY, Salacinski HJ, Butler PE, Hamilton G, Seifalian AM (2005a) Current status of prosthetic bypass grafts: a review. J. Biomed. Mater. Res. B Appl. Biomater. 74, 570–581. [DOI] [PubMed] [Google Scholar]
- Kannan RY, Salacinski HJ, Butler PE, Seifalian AM (2005b) Polyhedral oligomeric silsesquioxane nanocomposites: the next generation material for biomedical applications. Acc. Chem. Res. 38, 879–884. [DOI] [PubMed] [Google Scholar]
- Kannan RY, Salacinski HJ, Odlyha M, Butler PE, Seifalian AM (2005c) The degradative resistance of polyhedral oligomeric silsesquioxane nanocore integrated polyurethanes: an in vitro study. Biomaterials 27, 1971–1979. [DOI] [PubMed] [Google Scholar]
- Kannan RY, Salacinski HJ, Clatworthy I, Bozec L, Horton M, Butler PE, Seifalian AM (2006a) The antithrombogenic potential of a polyhedral oligomeric silsesquioxane (POSS) nanocomposite. Biomacromolecules 7, 215–223. [DOI] [PubMed] [Google Scholar]
- Kannan RY, Salacinski HJ, Edirisinghe MJ, Hamilton G, Seifalian AM (2006b) Polyhedral oligomeric silsequioxane‐polyurethane nanocomposite microvessels for an artificial capillary bed. Biomaterials 27, 4618–4626. [DOI] [PubMed] [Google Scholar]
- Kannan RY, Salacinski HJ, Sales KM, Butler PE, Seifalian AM (2006c) The endothelialization of polyhedral oligomeric silsesquioxane nanocomposites: an in vitro study. Cell Biochem. Biophys. 45, 129–136. [DOI] [PubMed] [Google Scholar]
- Khan SS, Solomon MA, McCoy JP Jr (2005) Detection of circulating endothelial cells and endothelial progenitor cells by flow cytometry. Cytometry B Clin. Cytom. 64, 1–8. [DOI] [PubMed] [Google Scholar]
- Kidane AG, Salacinski H, Tiwari A, Bruckdorfer KR, Seifalian AM (2004) Anticoagulant and antiplatelet agents: their clinical and device application (s) together with usages to engineer surfaces. Biomacromolecules 5, 798–813. [DOI] [PubMed] [Google Scholar]
- Meinhart JG, Deutsch M, Fischlein T, Howanietz. N , Froschl A, Zilla P (2001) Clinical autologous in vitro endothelialization of 153 infrainguinal ePTFE grafts. Ann. Thorac. Surg. 71, S327–S331. [DOI] [PubMed] [Google Scholar]
- Punshon G, Vara DS, Sales KM, Kidane AG, Salacinski HJ, Seifalian AM (2005) Interactions between endothelial cells and a poly (carbonate‐silsesquioxane‐bridge‐urea) urethane. Biomaterials 26, 6271–6279. [DOI] [PubMed] [Google Scholar]
- Rashid ST, Salacinski HJ, Fuller BJ, Hamilton G, Seifalian AM (2004) Engineering of bypass conduits to improve patency. Cell Prolif. 37, 351–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sales KM, Salacinski HJ, Alobaid N, Mikhail M, Balakrishnan V, Seifalian AM (2005) Advancing vascular tissue engineering: the role of stem cell technology. Trends Biotechnol. 23, 461–467. [DOI] [PubMed] [Google Scholar]
- Salven P, Mustjoki S, Alitalo R, Alitalo K, Rafii S (2003) VEGFR‐3 and CD133 identify a population of CD34+ lymphatic/vascular endothelial precursor cells. Blood 101, 168–172. [DOI] [PubMed] [Google Scholar]
- Sarkar S, Schmitz‐Rixen T, Hamilton G, Seifalian AM (2007) Achieving the ideal properties for vascular bypass grafts using a tissue engineered approach: a review. Med. Biol. Eng. Comput. 45, 327–336. [DOI] [PubMed] [Google Scholar]
- Seifalian AM, Handcock S, Salacinski HJ (2005) Polymer for use in conduits and medical devices. Patent Number: WO2005070998.
- Seifalian AM, Salacinski HJ, Punshon G, Krijgsman B, Hamilton G (2001) A new technique for measuring the cell growth and metabolism of endothelial cells seeded on vascular prostheses. J. Biomed. Mater. Res. 55, 637–644. [DOI] [PubMed] [Google Scholar]
- Shirota T, He H, Yasui H, Matsuda T (2003) Human endothelial progenitor cell‐seeded hybrid graft: proliferative and antithrombogenic potentials in vitro and fabrication processing. Tissue Eng. 9, 127–136. [DOI] [PubMed] [Google Scholar]
- Stanley JC, Burkel WE, Graham LM, Lindblad B (1985) Endothelial cell seeding of synthetic vascular prostheses. Acta Chir. Scand. Suppl. 529, 17–27. [PubMed] [Google Scholar]
- Tiwari A, Kidane A, Punshon G, Hamilton G, Seifalian AM (2003) Extraction of cells for single‐stage seeding of vascular‐bypass grafts. Biotechnol. Appl. Biochem. 38, 35–41. [DOI] [PubMed] [Google Scholar]
- Vara DS, Punshon G, Sales KM, Hamilton G, Seifalian AM (2006) The effect of shear stress on human endothelial cells seeded on cylindrical viscoelastic conduits: an investigation of gene expression. Biotechnol. Appl. Biochem. 45, 119–130. [DOI] [PubMed] [Google Scholar]
- Zhang SJ, Zhang H, Wei YJ, Su WJ, Liao ZK, Hou M, Zhou JY, Hu SS (2006) Adult endothelial progenitor cells from human peripheral blood maintain monocyte/macrophage function throughout in vitro culture. Cell Res. 16, 577–584. [DOI] [PubMed] [Google Scholar]
- Zilla P, Deutsch M, Meinhart J, Puschmann R, Eberl T, Minar E, Dudczak R, Lugmaier H, Schmidt P, Noszian I (1994) Clinical in vitro endothelialization of femoropopliteal bypass grafts: an actuarial follow‐up over three years. J. Vasc. Surg. 19, 540–548. [DOI] [PubMed] [Google Scholar]