

Abstract
Objectives
The aim of this study was to explore mechanisms by which salvianolic acid A (SAA) revealed its anti‐inflammatory activity, in lipopolysaccharide (LPS)‐stimulated RAW264.7 cells.
Materials and methods
Nitric oxide (NO) concentration was determined by the Griess reaction and cell viability was assessed by MTT assay. Interleukin‐6, TNFα and interleukin‐1β were determined by ELISA. The RAW264.7 cells were transfected with siRNA against p38 or HO‐1. Expressions of COX‐2, inducible NO synthase (iNOS), NF‐κB, HO‐1, p‐p38 and phosphorylation of IκB kinase α/β were detected by western blotting. Potential targets of SAA were analysed by homology modelling, target prediction, protein–protein interaction prediction and docking studies.
Results
Salvianolic acid A suppressed LPS‐triggered production of NO, TNFα and Interleukin‐6. It also reduced protein expression of inducible NO synthase and COX‐2, and reduced translocation of NF‐κB to nuclei. Moreover, SAA promoted expression of phosphorylated p38, and downstream HO‐1. Zn (II) protoporphyrin IX, a specific inhibitor of HO‐1, or siRNA against HO‐1 could effectively increase transfer of NF‐κB. SAA was predicted to target amyloid‐beta protein‐like protein and arachidonate 5‐lipoxygenase, that could regulate p38 and HO‐1.
Conclusions
In silico analysis and experimental validation together demonstrated that SAA exhibited its anti‐inflammatory effect via the p38‐HO‐1 pathway in LPS‐stimulated RAW264.7 cells, reduced transfer of NF‐κB to the nuclei and thus reduced production of inflammatory mediators.
Introduction
Acute and chronic inflammation are both multiple and complicated processes mediated by activating inflammatory and/or immune cells 1. Macrophages play a central role in the inflammatory response and serve as an essential interface between innate and adaptive immunity 2. Lipopolysaccharide (LPS), an activator of macrophages, can induce monocyte/macrophage differentiation, maturation and activation, which plays an important role in initiation and propagation of inflammatory responses by IL‐6, IL‐1β, TNF α, inducible NO synthase (iNOS), COX‐2 and other inflammatory mediators 3. Overexpression of inflammatory mediators in and around macrophages is involved in many inflammation‐related diseases, such as rheumatoid arthritis 4, atherosclerosis 5, chronic hepatitis 6 and pulmonary fibrosis 7. Thus, LPS‐stimulated mouse macrophage RAW264.7 cells have been used as an inflammatory cell model to study effects of anti‐inflammatory drugs.
Salvia miltiorrhiza Bge. is employed in traditional Chinese medicine and has been widely used in many Asian countries for treatment of various conditions including cerebrovascular diseases, coronary artery diseases and myocardial infarction, hepatitis and haemorrhage, for thousands of years 8, 9, 10, 11, 12. Salvianolic acid A (SAA) is one of the main active water soluble components of S. miltiorrhiza Bge., and demonstrates its powerful and multifunctional properties including antioxidative effects, free radical‐scavenging functions, anti‐platelet aggregation and antithrombotic activities 13, 14, 15, 16. However, there have been only few reports on anti‐inflammatory bioactivities of SAA; thus, in this study, we have explored molecular mechanisms by which SAA participates in its anti‐inflammatory activity, in LPS‐stimulated RAW264.7 cells, by a series of in silico analyses and experimental validation.
Materials and methods
Materials
Salvianolic acid A was obtained from Shenyang Pharmaceutical University and its chemical structure (Fig. 1a) was identified by comparing chemical and spectral data (1H‐NMR, 13C‐NMR) with those reported in the literature. Purity of the SAA was determined to be 98.2 by HPLC [Hitachi‐L‐7110 pump, Hitachi L‐7420 UV spectrophotometric detector at 286 nm, YMC C18 reversed‐phase column (5 μm, 4.6 × 250 mm), CH3OH‐CH3CN‐HCOOH‐H2O (30:10:1:59), flow rate 1.0 ml/min, Fig. 1b] and this was resolved in physiological saline to prepare a stock solution (50 mg/ml).
Figure 1.
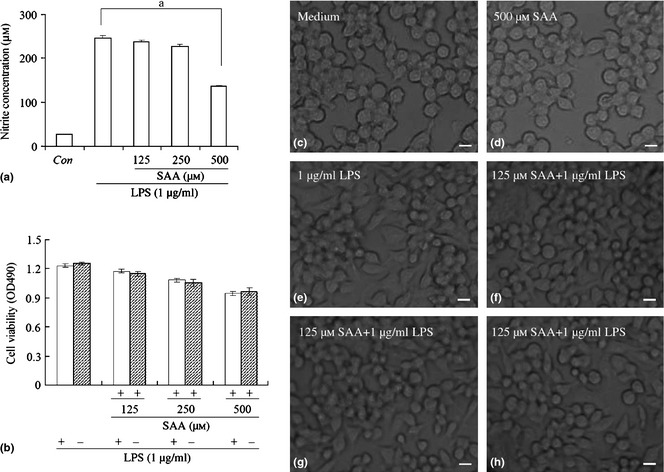
Effects of salvianolic acid A ( SAA ) on lipopolysaccharide ( LPS )‐induced production of nitric oxide and viability of RAW264.7 cells. Cells were treated with LPS (1 μg/ml) alone or LPS plus different concentrations of SAA (125, 250 and 500 μm) for 24 h. Amounts of nitric oxide were quantified using Griess reagent (a) and cell viability was measured by MTT assay (b). Cell morphological changes were observed using phase contrast microscopy (c: medium; d: 500 μm SAA; e: 1 μg/ml LPS; f: 125 μm SAA plus 1 μg/ml LPS; g: 250 μm SAA plus 1 μg/ml LPS; h: 500 μm SAA plus 1 μg/ml LPS, c–h ×400 magnification). Data are presented as mean ± SD of three independent experiments, a P < 0.01 versus LPS group. Con, control; Bar, 10 μm.
Foetal calf serum was obtained from Beijing Yuanhengshengma Biology Technology Research Institute (Beijing, China). Dulbecco's modified Eagle's medium (DMEM) was obtained from Gibco/BRL (Gaithersburg, MD, USA), 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT) was obtained from Sigma (St. Louis, MO, USA) and HO‐1 inhibitor ZnPP was obtained from Alfa Aesar (Jiangsu, China). Extracellular signal‐regulated kinase (ERK) inhibitor PD98059, p38 inhibitor SB203580 and c‐Jun N‐terminal kinase (JNK) inhibitor SP600125 were obtained from Beyotime Institute of Biotechnology (Jiangsu, China). ELISA kits for mouse TNF α, IL‐1β and IL‐6 were obtained from Sigma (St. Louis, MO, USA). Antibodies against iNOS, COX‐2, p38, phospho‐p38, NF‐κB, phospho‐IKK α/β, HO‐1, β‐actin and horseradish peroxidase‐conjugated secondary antibodies (goat anti‐rabbit, goat anti‐mouse and rabbit anti‐goat) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Cell culture
RAW264.7 cells, a murine monocyte/macrophage line, were cultured in DMEM supplemented with 10% FBS, 100 U/ml penicillin and 100 g/ml streptomycin. In all experiments, cells were grown to 80–90% confluence and subjected to no more than 20 cell passages. In all experiments, cells were left to acclimatize for 12 h before any procedures.
Measurement of NO release
The RAW264.7 cells were seeded in 96‐well plates (2 × 105/ml) and incubated at 37 °C in a humidified atmosphere with 5% CO2, overnight. After 12 h, cells were treated with different concentrations of SAA (125, 250 and 500 μm) for 1 h, then, LPS (1 μg/ml) was added for 24 h. Nitric oxide (NO) levels were determined by measuring nitrite levels using supernatant (100 μl) mixed with the same volume of Griess reagent (1% sulphanilamide, 0.1% N‐1‐naphthylenediamine dihydrochloride and 2.5% phosphoric acid); absorbance was measured at 570 nm 17. Nitrite concentration was determined by using a standard curve of sodium nitrite made up in DMEM, free of phenol red.
Measurement of RAW264.7 cell viability
Cell viability was measured using the MTT assay. The cells were seeded in 96‐well plates (2 × 105/ml) and incubated at 37 °C in a humidified atmosphere containing 5% CO2, overnight. After 12 h, they were treated with different concentrations of SAA (125, 250 and 500 μm) for 1 h, then LPS (1 μg/ml) was added for 24 h. MTT (5 mg/ml) was subsequently applied and incubated for 4 h. Culture medium was removed and crystals were dissolved in DMSO. Optical densities were measured at 490 nm, using a microplate reader.
Determination of TNF α, IL‐6 and IL‐1β levels by ELISA
Levels of TNF α, IL‐6 and IL‐1β present in each sample were determined with the use of a commercially available ELISA kit from R&D; assays were performed according to the manufacturer's instructions. Briefly, cells were cultured in 24‐well plates (5 × 105) for 12 h, pre‐incubated for 1 h with different concentrations of SAA, then stimulated with LPS (1 μg/ml) for 24 h. Cell culture supernatants were collected and immediately quantified using ELISA kits according to the manufacturer's instructions.
SiRNA transfection
Small interfering RNA (SiRNAs) against mouse p38, HO‐1 and control siRNA were purchased from Invitrogen (Carlsbad, CA, USA). Cells were transfected with siRNAs at 33 nm final concentration using Lipofectamine 2000 (Invitrogen), according to the manufacturer's instructions. Transfected cells were used for subsequent experiments 24 h later.
Preparation of nuclear and cytosolic extracts
Cells were collected then washed twice in ice‐cold PBS and spun. Pellets were re‐suspended in ice‐cold HMKEE buffer (250 mm sucrose, 20 mm Hepes, 10 mm KCl, 1.5 mm MgCl2, 1 mm EDTA, 1 mm EGTA, 1 mm dithiothreitol, 0.1 mm phenylmethanesulphonyl fluoride, 10 μg/ml pepstatin and 10 μg/ml leupeptin) and cells were homogenized and centrifuged at 14 000 g at 4 °C for 60 min. Supernatant was used as cytosol fraction and pellets were re‐suspended in lysis buffer as the membrane fraction.
Western blot analysis
Cells were treated with SAA and LPS for 24 h then were collected by centrifugation and washed once in PBS. Washed pellets were lysed in lysis buffer and protein concentration was measured using a BCA protein assay kit (Beyotime Institute of Biotechnology). Protein (10 μg) was separated on 10% SDS‐polyacrylamide gels and transferred to nitrocellulose membranes which were incubated for 1 h in blocking solution (5% skim milk) at room temperature, then 2‐h incubation in 1:1000 dilution primary polyclonal antibodies against COX‐2, p‐p38, NF‐κB, phospho‐IKK α/β, iNOS, HO‐1 and monoclonal β‐actin antibody. Membranes were washed three times in Tween‐20/Tris buffered saline and incubated in 1:1000 dilution horseradish‐conjugated anti‐rabbit, anti‐goat or anti‐mouse IgG secondary antibody, for 4 h at room temperature. Membranes were again washed three times in Tween‐20/Tris buffered saline then developed using ECL™ detection reagents.
Homology modelling
Three‐dimensional structures of amyloid‐beta protein‐like protein long isoform (Q53ZT3_MOUSE) and arachidonate 5‐lipoxygenase (LOX5_MOUSE) were acquired by homology modelling 18, with structure of E2 domain of human amyloid precursor protein (PDB ID: 3UMH) and stable‐5‐lipoxygenase (PDB ID: 3O8Y) as template.
Predictive targets of salvianolic acid A
Similarities of library molecules to sets of known ligands of the targets, in WOMBAT (Version 2006.2), were assessed using the similarity ensemble approach 19. This method yields a predicted target for every query molecule based on similarity of the query to sets of known ligands of the respective target. Library bias was assessed by counting numbers of molecules with a certain target class assignment, that is, significant similarity of a molecule to the target ligand set. Criterion for significant assignment was E value of <1 × 10−10.
Protein–protein interaction (PPI) prediction
Primary global human protein–protein interaction (PPI) network was constructed with diverse PPIs from mouse PPI databases 20, 21. Potential connections between amyloid‐beta protein‐like protein long isoform together with arachidonate 5‐lipoxygenase and HO‐1 as well as p38, were predicted within the naïve Bayesian model, with modifications 22, 23.
Molecular docking
Molecular docking was performed using the UCSF DOCK6.5 program, which utilized DOCK algorithm to address rigid body docking by superimposing the ligand on to a negative image of the binding pocket 24, 25. DOCK6.5 program is described in a three‐step process: (i) Rigid portion of ligand (anchor) is docked by geometric methods; (ii) Non‐rigid segments added in layers; energy minimized; (iii) Resulting configurations ‘pruned’ and energy re‐minimized, yielding docked configurations. First, we rolled a small probe (default radius = 1.4 Å = size of a water molecule) over the surface of the receptor and calculated surface normal at each point. By this step, we were able to calculate receptor surface as many spheres. Next, all spheres were selected within a user‐defined radius of a ligand, our radius of SAA being 10 angstroms. The selected sphere is a binding site that SAA can dock amyloid‐beta protein‐like protein and arachidonate 5‐lipoxygenase, respectively. SAA is taken as fully flexible. Only one amino‐acid side chain was taken to catch the in silico docking experiment and ligand and receptors were taken as fully flexible.
Statistical analysis
Results are summarized from three independent experiments and presented as mean ± SD. Statistical comparisons were made using Student's t‐test. P < 0.05 was considered statistically significant.
Results
SAA inhibited NO release from LPS‐stimulated RAW264.7 cells
Nitric oxide is known to be a pro‐inflammatory factor in many different acute and chronic inflammatory diseases 26. Thus, inhibition of NO production is a major target for anti‐inflammatory agent development. In this study, murine macrophage RAW 264.7 cells, which can produce NO on stimulation with LPS, were used for determination of anti‐inflammatory effects of SAA. Cells were pre‐incubated in 0, 125, 250 and 500 μm SAA for 1 h, then stimulated with 1 μg/ml LPS for 24 h; as controls, cells were untreated with LPS. After culture media were collected, nitrite levels were determined. Results showed that 125, 250 and 500 μm SAA markedly reduced NO production in a dose‐dependent manner. Moreover, 500 μm SAA reduced the level of NO from 246.9 to 136.4 μm (Fig. 1a).
To avoid the possibility that SAA reduced NO level by being toxic to the cells was determined by MTT assay. Results indicated that compared to the control group, 125, 250 and 500 μm SAA in the presence or absence of LPS was not toxic (Fig. 1b). To further confirm this result, cell morphology was observed by microscopy. In 125, 250 and 500 μm SAA‐treated cells, in the presence or absence of LPS, cells grew equally well as those of the control group and were distributed evenly across surfaces of culture plates (Fig. 1c–h). These results indicate that SAA significantly inhibited NO production in LPS‐stimulated cells, probably by inactivating inflammatory‐related signal pathways or activating anti‐inflammation transduction signals, but not by influencing cell viability.
Effects of SAA on production of LPS‐induced pro‐inflammatory cytokines
Activated macrophages take part in inflammatory responses by releasing TNF α, IL‐6 and IL‐1β and more, which are well‐known pro‐inflammatory cytokines of many immune cells such as macrophages, monocytes and T cells; they have various pro‐inflammatory effects in acute and chronic inflammatory diseases 27. To examine the effect of SAA on production of those pro‐inflammatory cytokines in LPS‐stimulated RAW 264.7 cells, TNF α, IL‐6 and IL‐1β concentrations in culture supernatants were measured by the sandwich ELISA method. Cells treated with LPS produced significant levels of all cytokines examined. Concentration of TNF α, IL‐1β and IL‐6 increased up to 6870, 1215 and 1032 ng/l after LPS stimulation, respectively. However, when cells were added to 500 μm SAA in advance for 1 h, concentrations of TNF α and IL‐6 in supernatants were significantly lower to 4727 ng/l and 727 ng/l (Fig. 2a,b). In contrast, concentrations of IL‐1β did not change significantly (Fig. 2c). These results demonstrate that SAA inhibited inflammation development by reducing production of TNF α and IL‐6.
Figure 2.
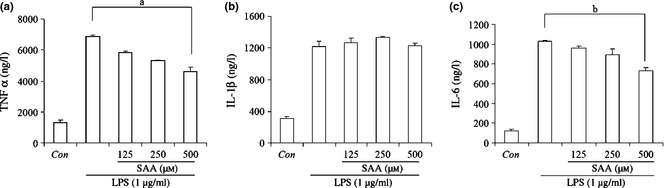
Effect of salvianolic acid A ( SAA ) on lipopolysaccharide ( LPS )‐induced production of TNF α, IL‐1β and IL‐6. Cells were treated with 1 μg/ml LPS alone or LPS plus different concentrations of SAA (125, 250 and 500 μm) for 24 h, and levels of TNF α (a), IL‐1β (b) and IL‐6 (c) were measured by ELISA. Data are presented as mean ± SD of three independent experiments, a P < 0.01, b P < 0.01 versus LPS group. Con, control.
SAA inhibited iNOS and COX‐2 protein expression in LPS‐stimulated RAW264.7 cells
Nitric oxide has been shown to be released from arginine as a result of activation of iNOS, expressed in response to a variety of inflammatory stimuli, and generates high levels of NO in macrophages during the inflammatory process 28, 29. To confirm that concentration‐dependent NO reduction triggered by SAA was due to regulation of iNOS, influence of SAA on iNOS expression was measured by western blot analysis. iNOS protein was fully induced on LPS stimulation for 12 h and SAA reduced synthesis of iNOS in a dose‐dependent manner (Fig. 3a,b).
Figure 3.
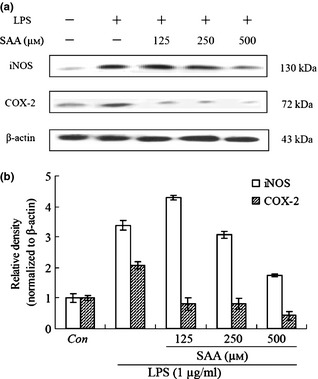
Effect of salvianolic acid A ( SAA ) on iNOS and COX‐2 expression in lipopolysaccharide ( LPS )‐treated cells. Cells were treated with SAA (125, 250 and 500 μm) in the presence of 1 μg/ml LPS for 24 h. Whole cell lysates were prepared and analysed for expressions of iNOS, COX‐2 and β‐actin by western blot analysis (a); relative density of bands normalized to β‐actin was assayed by Bio‐Rad Quantity One 4.62 (b). Data are presented as mean ± SD of three independent experiments. Con, control.
COX‐2, a cyclooxygenase, catalyses production of prostaglandins, which represents an important step in the inflammatory process. Also, prostaglandin production in LPS‐treated macrophages is primarily due to expression of COX‐2 30. In our study, the effect of SAA on expression of COX‐2 was examined by means of western blot analysis. Results reveal that COX‐2 protein expression was high 12 h after LPS stimulation. SAA similarly reduced the level of COX‐2 protein (Fig. 3a,b).
Inhibitory effects of SAA onTNF α, IL‐6, iNOS and COX‐2 production mediated by suppressing transfer of NF‐κB, in LPS‐stimulated RAW264.7 cells
NF‐κB is known to play a critical role in controlling most inflammatory responses, by induction of iNOS and COX‐2, pro‐inflammatory cytokines (IL‐1, IL‐2, IL‐6 and TNF α), chemokines, cell adhesion molecules, growth factors, acute phase proteins and immune receptors 31. Activation of NF‐κB results from phosphorylation formed of IKK α and IKK β, and proteasome‐mediated degradation of Iκ B 32, 33. As SAA inhibited production of TNF α, IL‐6, iNOS and COX‐2 in LPS‐stimulated RAW 264.7 cells, western blot analysis was carried out to investigate the effect of SAA on nuclear translocation of p65, one subunit of NF‐κB, and its upstream IKK α/β activation. As shown in Fig. 4a,b, when cells were treated with LPS alone for 24 h, cytosolic levels of p‐IKK α/β and nuclear levels of p65 increased significantly, and when cells were pre‐cultured in 125, 250, 500 μm SAA, reduced expression of cytosolic phosphorylation of IKK α/β and nuclear levels of p65 occurred. From these results, it is suggested that SAA inhibited inflammatory mediator (TNF α, IL‐6, iNOS and COX‐2) production by inactivation of signal transduction, related to the NF‐κB pathway.
Figure 4.
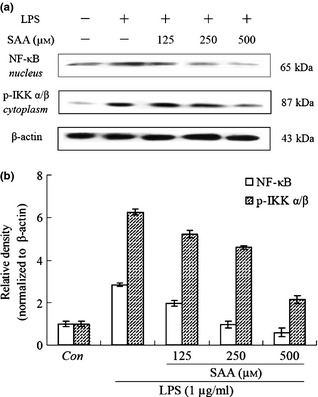
Inhibitory effects of salvianolic acid A ( SAA ) on the protein levels of p‐IKK α/β and NF‐κB (p65) in lipopolysaccharide ( LPS )‐treated cells. Cells were treated with SAA (125, 250 and 500 μm) in the presence of 1 μg/ml LPS for 24 h. Western blot analysis was used to determine levels NF‐κB (p65) in nuclei and p‐IKK α/β in cytoplasm (a); relative density of bands normalized to β‐actin was assayed by Bio‐Rad Quantity One 4.62 (b). Data are presented as mean ± SD of three independent experiments. Con, control.
SAA triggered phosphorylation of p38, which reduced NO level in LPS‐stimulated RAW 264.7 cells
Mitogen‐activated protein kinase (MAPK) molecules provide one of the most the important signalling pathways controlling synthesis and release of pro‐inflammatory mediators by activated macrophages during the inflammatory response 34 and here, it has been investigated whether MAPK was involved in the anti‐inflammatory process of SAA in LPS‐stimulated RAW 264.7 cells. We examined effects of MAPK on the level of NO by using MAPK specific inhibitors for p38 (SB203580), JNK (SP600125) and ERK (PD98059). Cells were pre‐treated in 1, 5 and 10 μm inhibitor for 1 h, stimulated with 500 μm SAA for 1 h and then 1 μg/ml LPS was added to for 24 h. Concentration of NO was determined by use of Griess reagent. ERK inhibitor PD98059 and JNK inhibitor SP600125 had almost no influence on the level (Fig. 5b,c). However, p38 inhibitor SB203580 significantly increased NO production compared to the SAA and LPS‐treated group (Fig. 5a). Based on this result, western blot analysis was carried out to detect phosphorylation of p38 and showed that after treatment of with either 250 or 500 μm SAA in the presence of 1 μg/ml LPS for 24 h, expression of phosphorylated p38 increased to a higher level compared to LPS‐treated group (Fig. 5d,e). These results indicate that p38 was activated by SAA and participated in anti‐inflammatory process in LPS‐stimulated cells.
Figure 5.
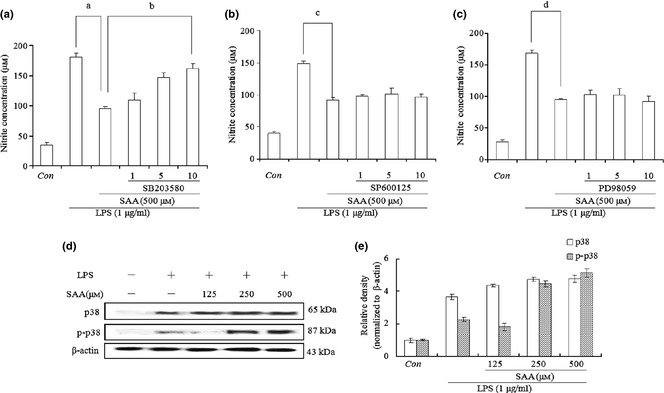
Effects of mitogen‐activated protein kinase ( MAPK ) specific inhibitors on salvianolic acid A ( SAA ) suppression of nitric oxide production in lipopolysaccharide ( LPS )‐treated cells. Cells were treated with LPS (1 μg/ml) alone or LPS plus different concentrations of inhibitors (1, 5 and 10 μm) for p38 (SB203580, a), JNK (SP600125, b) and ERK (PD98059, c) for 1 h, then SAA (500 μm) was added for 24 h. Levels of nitric oxide were quantified using Griess reagent. Data are presented as mean ± SD of three independent experiments, a P < 0.01, c P < 0.01, d P < 0.01 versus LPS group, b P < 0.01 versus LPS + 500 μm SAA group. Con, control. Cells were treated with SAA (125, 250, 500 μm) in the presence of 1 μg/ml LPS for 24 h, and western blot analysis was carried out to determine levels of p38 and phosphorylated p38 proteins (d); relative density of bands normalized to β‐actin was assayed by Bio‐Rad Quantity One 4.62 (e).
Activation of p38 upregulated HO‐1 expression and consequently suppressed translocation of NF‐κB in LPS‐stimulated cells
HO‐1 is induced by various stimuli via activation of the p38 MAPK signalling pathway and development of inflammation is thus suppressed 35; therefore, expression of HO‐1 was investigated. Cells were pre‐treated with 500 μm SAA for 1 h, then cultured in 1 μg/ml LPS for 24 h. Results indicate that 500 μm SAA promoted expression of HO‐1 in comparison to the LPS alone group. However, when cells were added to 10 μm p38 inhibitor SB203580, HO‐1 expression was markedly inhibited. To further validate this result, cells were transfected with p38 siRNA, which effectively reversed the level of HO‐1 compared to 500 μm SAA and 1 μg/ml LPS groups (Fig. 6a,b).
Figure 6.
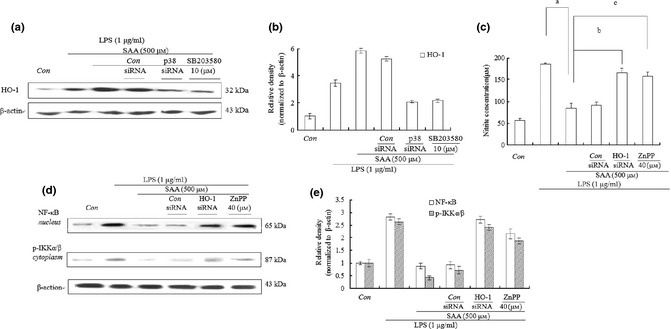
Effects of salvianolic acid A ( SAA ) on production of haem oxygenase‐1 (HO‐1) in lipopolysaccharide ( LPS )‐treated cells. After cells were cultured in LPS (1 μg/ml) alone or LPS plus SB203580 (10 μm) or LPS plus different concentrations of ZnPP (40 μm) for 1 h, they were treated with SAA (500 μm) for 24 h. Cells were then transfected with p38, HO‐1 or control siRNA for 24 h, and cultured in LPS (1 μg/ml) for 1 h. They were then treated with SAA (500 μm) for 24 h. HO‐1 level was examined by western blot analysis in (a) and relative density of bands normalized to β‐actin, was assayed by Bio‐Rad Quantity One 4.62 in (b). Level of nitric oxide was tested using Griess reagent in (c). Western blot analysis was also used to examine levels of NF‐κB (p65) in nuclei and p‐IKK α/β in cytoplasm in (d); relative density of bands normalized to β‐actin was assayed by Bio‐Rad Quantity One 4.62 in (e). Data are presented as mean ± SD of three independent experiments, a P < 0.01 versus LPS group, b P < 0.01, c P < 0.01 versus LPS group + 500 μm SAA, Con, control.
To examine effects of HO‐1 on production of NO and translocation of NF‐κB, HO‐1 inhibitor protoporphyrin IX (ZnPP) was applied to our RAW264.7 macrophages. After the cells were pre‐treated with 40 μm ZnPP or pre‐transfected with HO‐1 siRNA, and treated with 500 μm SAA for 1 h, then stimulated with 1 μg/ml LPS for 24 h, the level of NO was tested by use of the Griess reagent. As expected, HO‐1 siRNA or HO‐1 inhibitor ZnPP almost completely reversed reduction of NO production from 86 to 165 or 158 μm, respectively, in contrast to 500 μm SAA and 1 μg/ml LPS group (Fig. 6b). In addition, effects of HO‐1 on translocation of NF‐κB and phosphorylation of IKK α/β were examined. HO‐1 siRNA or ZnPP significantly improved cytosolic levels of p‐IKKα/β and nuclear levels of p65, compared to 500 μm SAA and 1 μg/ml LPS group (Fig. 6c,d). These results demonstrate that SAA played an anti‐inflammatory role on LPS‐treated cells through activation of the p38/HO‐1 signalling pathway, which inhibited transfer of NF‐κB to nuclei, and production of inflammatory mediators.
Identification of potential targets of SAA and related signalling pathways involved in p38 and HO‐1
We calculated cumulated predictions for searching novel targets of SAA. We showed that amyloid‐beta protein‐like protein long isoform and arachidonate 5‐lipoxygenase could be predicted as potential targets of SAA. Then, we used the flexible ligand docking method with all setting as default. We were able to show that energies for SAA binding to amyloid‐beta protein‐like protein and arachidonate 5‐lipoxygenase were −45.828728 KJ/mol and −40.654358 KJ/mol, respectively (Fig. 7). In addition, we found the above‐mentioned targets, amyloid‐beta protein‐like protein long isoform and arachidonate 5‐lipoxygenase, indirectly modulated p38 and HO‐1 via four other proteins AKT‐1, TNFA, CSF2 and LYAM3(Fig. 8).
Figure 7.
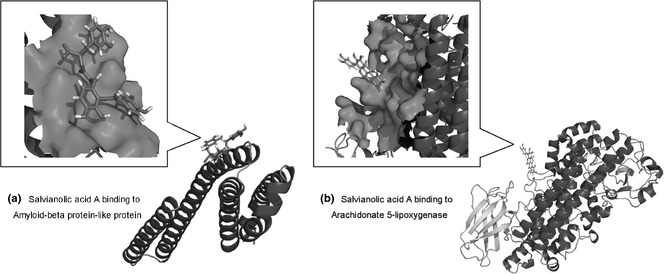
Identification of two potential targets amyloid‐beta protein‐like protein and arachidonate 5‐lipoxygenase of SAA .
Figure 8.
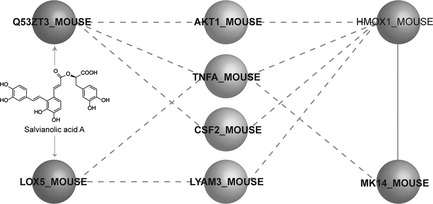
Identification of of salvianolic acid A‐inhibited lipopolysaccharide‐stimulated inflammatory pathways.
Discussion
Salvianolic acid A, a phenylpropanoid isolated from S. miltiorrhiza Bge., effectively exhibited anti‐inflammatory activity by reducing production of NO in LPS‐treated RAW264.7 macrophages. As there have been no previous reports on its anti‐inflammatory effects, we investigated underlying mechanisms in further studies.
Macrophages are an important component of the human immune defence mechanism. During progress of inflammation, macrophages participate in inflammatory responses by releasing pro‐inflammatory cytokines such as TNFα and IL‐6, as well as other inflammatory factors, such as iNOS, COX‐2 to protect the body from infection or tissue injury 36. These pro‐inflammatory mediators play an important role in pathogenesis of many acute and chronic inflammatory conditions. Thus, we have investigated the influence of SAA on production of pro‐inflammatory mediators. The results provide evidence that SAA inhibited development of inflammation by reducing levels of iNOS, COX‐2, TNF α and IL‐6 in LPS‐stimulated RAW 264.7 macrophages.
NF‐κB is a dimeric transcription factor formed by hetero‐ or homo‐dimerization of proteins of the Rel family, including p50 and p65; it coordinates expression of pro‐inflammatory enzymes and cytokines 37. It is present in cytoplasm in an inactive form through association with IκB family members. On stimulation by LPS, IκB kinase is activated by IKK α and IKK β, resulting in translocation of NF‐κB to nuclei 38, 39. In our study, SAA inhibited transfer of NF‐κB to nuclei and phosphorylation of IKKα/β, in a dose‐dependent manner, indicating that SAA reduced NF‐κB transcription activation due to inhibition of IKKα/β activation.
The MAPK family includes ERKs‐1 and ‐2, c‐Jun, N‐terminal kinase (JNK) and p38. Activation of a distinct MAPK subtype cascade depends on types of cells and the stimuli; functional role of each MAPK subtype might be different according to cell type 40. The ERK pathway is predominantly activated by mitogens through a Ras‐dependent mechanism and is required for cell proliferation and differentiation; however, JNK and p38 are activated by pro‐inflammatory cytokines and various environmental stresses 41, 42. It has been reported that MAPK participates in synthesis and release of pro‐inflammatory mediators by activated macrophages in the process of inflammatory development 34; thus, roles of MAPK were examined by use of ERK inhibitor PD98059, JNK inhibitor SP600125 and p38 inhibitor SB203580 in our study. Results indicat that only p38 inhibitors SB203580 markedly increased NO production compared to SAA and LPS‐treated group, indicating that p38 was activated and involved in the anti‐inflammatory process of SAA. Moreover, western blot analysis further confirmed this result as SAA markedly triggered phosphorylation of p38 in contrast to the LPS alone‐treated group.
HO is a rate‐limiting enzyme in haem catabolism, leading to generation of carbon monoxide (CO). Three HO isoforms (HO‐1, HO‐2 and HO‐3) that catalyse this reaction have been identified. HO‐1 is inducible and HO‐2 is constitutively synthesized and exists primarily in cells of the brain and testis; however, HO‐3 is a recently cloned gene product and is less well characterized. Cytoprotective effects of HO‐1 can be attributed to its products CO, associated with moderate or severe cell stress, such as occurs in inflammation 43. Other studies have shown that HO‐1 and CO can suppress production of pro‐inflammatory mediators such as tumour necrosis factor alpha, interleukin IL‐1β, IL‐6, iNOS and COX‐2, through inactivation of nuclear factor NF‐κB, in activated macrophages 44, 45, 46, 47, 48, 49. It has also been reported that activation of p38 induces HO‐1 expression in various cell types including macrophages 50. In our study, SAA of different concentrations increased expression of HO‐1 and p38 inhibitor SB203580 or p38 siRNA effectively reversed increasing expression of HO‐1. Also, HO‐1 inhibitor ZnPP or HO‐1 siRNA markedly promoted level of NO (product of iNOS) and transfer of NF‐κB to nuclei, demonstrating that anti‐inflammatory effects of SAA resulted from high expression of HO‐1, induced by p38 activation, and upregulation of HO‐1 reduced translocation of NF‐κB and consecutive expression of pro‐inflammatory mediators in LPS‐stimulated RAW 264.7 macrophages.
In silico analysis demonstrated that SAA targeted amyloid‐beta protein‐like protein and arachidonate 5‐lipoxygenase that might indirectly regulate p38 and HO‐1 by four other proteins AKT‐1, TNFA, CSF2 and LYAM3, which would shed new light on providing more novel direct targets and signalling pathways for SAA in future studies.
In conclusion, we first provided evidence that SAA has marked anti‐inflammatory effects on LPS‐stimulated RAW 264.7 cells. We further demonstrated levels of NO, TNF α and IL‐6, and reduced production of iNOS and COX‐2 by inhibition of transfer of NF‐κB to nuclei, by activation of the p38/HO‐1 pathway, which might be regulated by amyloid‐beta protein‐like protein and arachidonate 5‐lipoxygenase.
Acknowledgements
We are grateful to F. T. Wang and X. Wen for their valuable suggestions on this work. The authors acknowledge the funding for this study from National Natural Science Foundation of China (No. 20762008 and No. 81260628) and Technologies R & D Program of Shihezi University (No. gxji2007‐zdgg01 and RCZX200765).
References
- 1. Lundberg IE (2000) The role of cytokines, chemokines, and adhesion molecules in the pathogenesis of idiopathic inflammatory myopathies. Curr. Rheumatol. Rep. 2, 216–224. [DOI] [PubMed] [Google Scholar]
- 2. Adams DO, Hamilton TA (1984) The cell biology of macrophage activation. Annu. Rev. Immunol. 2, 283–318. [DOI] [PubMed] [Google Scholar]
- 3. Walsh LJ (2003) Mast cells and oral inflammation. Crit. Rev. Oral Biol. Med. 14, 188–198. [DOI] [PubMed] [Google Scholar]
- 4. Tilg H, Wilmer A, Vogel W, Herold M, Nolchen B, Judmaier G et al (1992) Serum levels of cytokines in chronic liver diseases. Gastroenterology 103, 264–274. [DOI] [PubMed] [Google Scholar]
- 5. Coker RK, Laurent GJ (1998) Pulmonary fibrosis: cytokines in the balance. Eur. Respir. J. 11, 1218–1221. [DOI] [PubMed] [Google Scholar]
- 6. Lind L (2003) Circulating markers of inflammation and atherosclerosis. Atherosclerosis 169, 203–214. [DOI] [PubMed] [Google Scholar]
- 7. Bertolini A, Ottani A, Sandrini M (2001) Dual acting anti‐inflammatory drugs: a reappraisal. Pharmacol. Res. 44, 437–450. [DOI] [PubMed] [Google Scholar]
- 8. Cheng TO (2007) Cardiovascular effects of Danshen. Int. J. Cardiol. 121, 9–22. [DOI] [PubMed] [Google Scholar]
- 9. Yagi A, Fujimoto K, Tanonaka K, Hirai K, Takeo S (1989) Possible active components of tan‐shen (Salvia miltiorrhiza) for protection of the myocardium against ischemia‐induced derangements. Planta Med. 55, 51–54. [DOI] [PubMed] [Google Scholar]
- 10. Chang BB, Zhang L, Cao WW, Cao Y, Yang WL, Wang Y et al (2010) Pharmacokinetic interactions induced by content variation of major water‐soluble components of Danshen preparation in rats. Acta Pharmacol. Sin. 31, 638–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Han JY, Fan JY, Horie Y, Miura S, Cui DH, Ishii H et al (2008) Ameliorating effects of compounds derived from Salvia miltiorrhiza root extract on microcirculatory disturbance and target organ injury by ischemia and reperfusion. Pharmacol. Ther. 117, 280–295. [DOI] [PubMed] [Google Scholar]
- 12. Lee TY, Mai LM, Wang GJ, Chiu JH, Lin YL, Lin HC (2003) Protective mechanism of Salvia miltiorrhiza on carbon tetrachloride‐induced acute hepatotoxicity in rats. J. Pharmacol. Sci. 91, 202–210. [DOI] [PubMed] [Google Scholar]
- 13. Hu YY, Liu CH, Wang RP, Liu C, Liu P, Zhu DY (2000) Protective actions of salvianolic acid A on hepatocyte injured by peroxidation in vitro . World J. Gastroenterol. 6, 402–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tang MK, Ren DC, Zhang JT, Du GH (2002) Effect of salvianolic acids from Radix Salviae miltiorrhizae on regional cerebral blood flow and platelet aggregation in rats. Phytomedicine 9, 405–409. [DOI] [PubMed] [Google Scholar]
- 15. Fan HY, Fu FH, Yang MY, Xu H, Zhang AH, Liu K (2010) Antiplatelet and antithrombotic activities of salvianolic acid A. Thromb. Res. 126, e17–e22. [DOI] [PubMed] [Google Scholar]
- 16. Zhang YQ, Tang Y, Wu AL, Zhu HB (2010) Salvianolic acid A displays cardioprotective effects in in vitro models of heart hypoxia/reoxygenation injury. J. Asian Nat. Prod. Res. 12, 899–915. [DOI] [PubMed] [Google Scholar]
- 17. Titheradge MA (1998) The enzymatic measurement of nitrate and nitrite. Methods Mol. Biol. 100, 83–91. [DOI] [PubMed] [Google Scholar]
- 18. Kiefer F, Arnold K, Kunzli M, Bordoli L, Schwede T (2009) The SWISS‐MODEL Repository and associated resources. Nucleic Acids Res. 37, D387–D392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Keiser MJ, Roth BL, Armbruster BN, Ernsberger P, Irwin JJ, Shoichet BK (2007) Relating protein pharmacology by ligand chemistry. Nat. Biotechnol. 25, 197–206. [DOI] [PubMed] [Google Scholar]
- 20. Li X, Cai HY, Xu JB, Ying SC, Zhang YZ (2009) A mouse protein interactome through combined literature mining with multiple sources of interaction evidence. Amino Acids 38, 1237–1252. [DOI] [PubMed] [Google Scholar]
- 21. Guan Y, Myers CL, Lu R, Lemischka IR, Bult CJ, Troyanskaya OG (2008) A genomewide functional network for the laboratory mouse. PLoS Comput. Biol. 4, e1000165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang N, Xu HL, Zhao X, Wen X, Wang FT, Wang SY et al (2012) Network‐based identification of novel connections among apoptotic signaling pathways in cancer. Appl. Biochem. Biotechnol. 167, 621–631. [DOI] [PubMed] [Google Scholar]
- 23. Fu LL, Yang Y, Xu HL, Cheng Y, Wen X, Ouyang L et al (2013) Identification of novel caspase/autophagy‐related gene switch to cell fate decisions in breast cancers. Cell Prolif. 46, 67–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lang PT, Brozell SR, Mukherjee S, Pettersen EF, Meng EC, Thomas V et al (2009) DOCK 6: combining techniques to model RNA‐small molecule complexes. RNA 15, 1219–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dolinsky TJ, Czodrowski P, Li H, Nielsen JE, Jensen JH, Klebe G et al (2007) PDB2PQR: expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 35, W522–W525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Payne DN (2003) Nitric oxide in allergic airway inflammation. Curr. Opin. Allergy Clin. Immunol. 3, 133–137. [DOI] [PubMed] [Google Scholar]
- 27. Andreakos E, Foxwell B, Feldmann M (2004) Is targeting Toll‐like receptors and their signaling pathway a useful therapeutic approach to modulating cytokine‐driven inflammation? Immunol. Rev. 202, 250–265. [DOI] [PubMed] [Google Scholar]
- 28. Moffat FL Jr, Han T, Li ZM, Peck MD, Jy W, Ahn YS et al (1996) Supplemental L‐arginine HCl augments bacterial phagocytosis in human polymorphonuclear leukocytes. J. Cell. Physiol. 168, 26–33. [DOI] [PubMed] [Google Scholar]
- 29. Laskin DL, Pendino KJ (1995) Macrophages and inflammatory mediators in tissue injury. Annu. Rev. Pharmacol. Toxicol. 35, 655–677. [DOI] [PubMed] [Google Scholar]
- 30. Lee SH, Soyoola E, Chanmugam P, Hart S, Sun W, Zhong H et al (1992) Selective expression of mitogen‐inducible cyclooxygenase in macrophages stimulated with lipopolysaccharide. J. Biol. Chem. 267, 25934–25938. [PubMed] [Google Scholar]
- 31. Hanada T, Yoshimura A (2002) Regulation of cytokine signaling and inflammation. Cytokine Growth Factor Rev. 13, 413–421. [DOI] [PubMed] [Google Scholar]
- 32. Karin M (1999) The beginning of the end: IkappaB kinase (IKK) and NF‐kappaB activation. J. Biol. Chem. 274, 27339–27342. [DOI] [PubMed] [Google Scholar]
- 33. Mercurio F, Zhu H, Murray BW, Shevchenko A, Bennett BL, Li J et al (1997) IKK‐1 and IKK‐2: cytokine‐activated I kappaB kinases essential for NF‐kappaB activation. Science 278, 860–866. [DOI] [PubMed] [Google Scholar]
- 34. Bondeson J (1997) The mechanisms of action of disease‐modifying antirheumatic drugs: a review with emphasis on macrophage signal transduction and the induction of proinflammatory cytokines. Gen. Pharmacol. 29, 127–150. [DOI] [PubMed] [Google Scholar]
- 35. Otterbein LE (2002) Carbon monoxide: innovative anti‐inflammatory properties of an age‐old gas molecule. Antioxid. Redox Signal. 4, 309–319. [DOI] [PubMed] [Google Scholar]
- 36. Shapira L, Soskolne WA, Houri Y, Barak V, Halabi A, Stabholz A (1996) Protection against endotoxic shock and lipopolysaccharide‐induced local inflammation by tetracycline: correlation with inhibition of cytokine secretion. Infect. Immun. 64, 825–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jeong JB, Jeong HJ (2010) Rheosmin, a naturally occurring phenolic compound inhibits LPS‐induced iNOS and COX‐2 expression in RAW264.7 cells by blocking NF‐kappaB activation pathway. Food Chem. Toxicol. 48, 2148–2153. [DOI] [PubMed] [Google Scholar]
- 38. Bosca L, Zeini M, Traves PG, Hortelano S (2005) Nitric oxide and cell viability in inflammatory cells: a role for NO in macrophage function and fate. Toxicology 208, 249–258. [DOI] [PubMed] [Google Scholar]
- 39. Jeong SJ, Pise‐Masison CA, Radonovich MF, Park HU, Brady JN (2005) A novel NF‐kappaB pathway involving IKKbeta and p65/RelA Ser‐536 phosphorylation results in p53 Inhibition in the absence of NF‐kappaB transcriptional activity. J. Biol. Chem. 280, 10326–10332. [DOI] [PubMed] [Google Scholar]
- 40. Shinoura N, Yoshida Y, Asai A, Kirino T, Hamada H (1999) Relative level of expression of Bax and Bcl‐XL determines the cellular fate of apoptosis/necrosis induced by the overexpression of Bax. Oncogene 18, 5703–5713. [DOI] [PubMed] [Google Scholar]
- 41. Raingeaud J, Gupta S, Rogers JS, Dickens M, Han J, Ulevitch RJ et al (1995) Pro‐inflammatory cytokines and environmental stress cause p38 mitogen‐activated protein kinase activation by dual phosphorylation on tyrosine and threonine. J. Biol. Chem. 270, 7420–7426. [DOI] [PubMed] [Google Scholar]
- 42. Chen YR, Meyer CF, Tan TH (1996) Persistent activation of c‐Jun N‐terminal kinase 1 (JNK1) in gamma radiation‐induced apoptosis. J. Biol. Chem. 271, 631–634. [DOI] [PubMed] [Google Scholar]
- 43. Motterlini R, Foresti R, Bassi R, Green CJ (2000) Curcumin, an antioxidant and anti‐inflammatory agent, induces heme oxygenase‐1 and protects endothelial cells against oxidative stress. Free Radic. Biol. Med. 28, 1303–1312. [DOI] [PubMed] [Google Scholar]
- 44. Lee TS, Tsai HL, Chau LY (2003) Induction of heme oxygenase‐1 expression in murine macrophages is essential for the anti‐inflammatory effect of low dose 15‐deoxy‐Delta 12,14‐prostaglandin J2. J. Biol. Chem. 278, 19325–19330. [DOI] [PubMed] [Google Scholar]
- 45. Wiesel P, Foster LC, Pellacani A, Layne MD, Hsieh CM, Huggins GS et al (2000) Thioredoxin facilitates the induction of heme oxygenase‐1 in response to inflammatory mediators. J. Biol. Chem. 275, 24840–24846. [DOI] [PubMed] [Google Scholar]
- 46. Morse D, Pischke SE, Zhou Z, Davis RJ, Flavell RA, Loop T et al (2003) Suppression of inflammatory cytokine production by carbon monoxide involves the JNK pathway and AP‐1. J. Biol. Chem. 278, 36993–36998. [DOI] [PubMed] [Google Scholar]
- 47. Suh GY, Jin Y, Yi AK, Wang XM, Choi AM (2006) CCAAT/enhancer‐binding protein mediates carbon monoxide‐induced suppression of cyclooxygenase‐2. Am. J. Respir. Cell Mol. Biol. 35, 220–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Oh GS, Pae HO, Choi BM, Chae SC, Lee HS, Ryu DG et al (2004) 3‐Hydroxyanthranilic acid, one of metabolites of tryptophan via indoleamine 2,3‐dioxygenase pathway, suppresses inducible nitric oxide synthase expression by enhancing heme oxygenase‐1 expression. Biochem. Biophys. Res. Commun. 320, 1156–1162. [DOI] [PubMed] [Google Scholar]
- 49. Ryter SW, Alam J, Choi AM (2006) Heme oxygenase‐1/carbon monoxide: from basic science to therapeutic applications. Physiol. Rev. 86, 583–650. [DOI] [PubMed] [Google Scholar]
- 50. Ma W, Lim W, Gee K, Aucoin S, Nandan D, Kozlowski M et al (2001) The p38 mitogen‐activated kinase pathway regulates the human interleukin‐10 promoter via the activation of Sp1 transcription factor in lipopolysaccharide‐stimulated human macrophages. J. Biol. Chem. 276, 13664–13674. [DOI] [PubMed] [Google Scholar]