

Abstract
Water-mediated interactions play key roles in drug binding. In protein sites with sparse polar functionality, a small-molecule approach is often viewed as insufficient to achieve high affinity and specificity. Here we show that small molecules can enable potent inhibition by targeting key waters. The M2 proton channel of influenza A is the target of the antiviral drugs amantadine and rimantadine. Structural studies of drug binding to the channel using X-ray crystallography have been limited because of the challenging nature of the target, with the one previously solved crystal structure limited to 3.5 Å resolution. Here we describe crystal structures of amantadine bound to M2 in the Inwardclosed conformation (2.00 Å), rimantadine bound to M2 in both the Inwardclosed (2.00 Å) and Inwardopen (2.25 Å) conformations, and a spiro-adamantyl amine inhibitor bound to M2 in the Inwardclosed conformation (2.63 Å). These X-ray crystal structures of the M2 proton channel with bound inhibitors reveal that ammonium groups bind to water-lined sites that are hypothesized to stabilize transient hydronium ions formed in the proton-conduction mechanism. Furthermore, the ammonium and adamantyl groups of the adamantyl–amine class of drugs are free to rotate in the channel, minimizing the entropic cost of binding. These drug-bound complexes provide the first high-resolution structures of drugs that interact with and disrupt networks of hydrogen-bonded waters that are widely utilized throughout nature to facilitate proton diffusion within proteins.
Graphical abstract
INTRODUCTION
Protein channels and water-filled pores present particularly challenging targets for drug design. Typically, drugs bind their targets at extended allosteric or substrate-binding sites lined with multiple functional groups capable of forming numerous protein—small molecule interactions. Often structural and functional water molecules play critical roles in drug interactions.1,2 Water is also an important component in channels, but these proteins often have very constricted cavities with only sparse polar functionality available for drug design. The natural substrate in such cases can be as small as a single proton. Nevertheless, potent inhibitors of channels can still be achieved, possibly by targeting water molecules that serve to hydrate charged groups during ion conduction. Examples of this include channel blockers such as the chloride channel blocker picrotoxin3 and the adamantyl—amine class of influenza A virus matrix 2 (M2) proton channel inhibitors (Figure 1).4,5 These compounds achieve high affinity, ligand efficiency, and biologically useful specificity despite their relatively small sizes and low molecular weights (e.g., the MW of amantadine is 151 Da). Here we use X-ray crystallography to show the role of water in the binding and the mechanism of action of the adamantyl—amine class of M2 inhibitors. The hydrophobic groups of these molecules displace waters from the portion of the pore that faces the viral interior, while the drug’s positively charged ammonium group locks into water networks that normally hydrate and stabilize protons as they diffuse through the pore. Intriguingly, the α-helical pore-lining carbonyl groups are physicochemical chameleons that are easily dehydrated to hydrophobically stabilize the binding of apolar groups from M2 inhibitors in the drug-bound form and yet are also able to form stabilizing interactions with cations through water-mediated polar interactions in the drug-free form. We also elucidate several features of adamantane that explain its increasingly successful use in drug design.6
Figure 1.
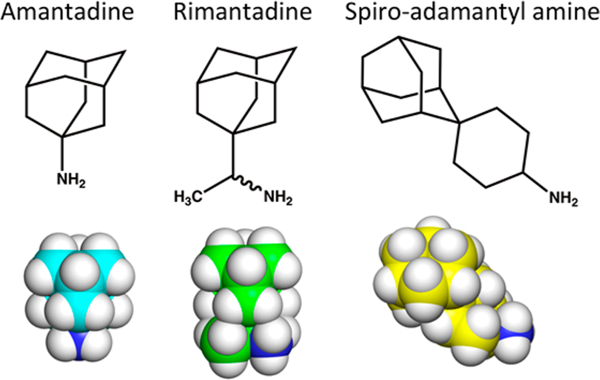
Chemical structures and space-filling models of amantadine (cyan), rimantadine (green), and spiro-adamantyl amine (yellow).
Influenza virus infections are perennial problems. The 2017–2018 influenza season is a timely reminder of the devastating impact of influenza: between October 1, 2017, and April 30, 2018, 30 451 laboratory-confirmed influenza-associated hospitalizations have been reported in the United States.7 Moreover, 2018 marks the 100-year anniversary of the 1918 Spanish Flu, which infected an estimated one-third of the human population and killed approximately 50 million people.8 In recent years, resistance to the adamantyl–amine class of drugs has become widespread, leaving the neuraminidase inhibitor oseltamivir (Tamiflu) as the sole orally bioavailable anti-influenza medication.9 Thus, elucidating the structural mechanism of inhibition of adamantyl–amines has specific relevance to the design of new compounds to target drug- resistant influenza infections as well as general relevance to the design of drugs that bind to the water-filled pores of channel proteins.
The M2 protein is a homotetrameric channel that serves several different functions during the life cycle of the virus,10–14 which enters the cell via receptor-mediated endocytosis. The transmembrane (TM) domain (residues 23–46) transports protons from the low-pH conditions of the endosome into the viral interior. The resulting drop in the intraviral pH triggers the dissociation of viral ribonucleoproteins (RNPs) from the matrix 1 protein.15 M2’s extracellular domain (residues 1–22) aids incorporation of M2 into the virion, but this domain is absent in influenza B viruses.16 An amphiphilic helix in the cytosolic tail of M2 (residues 46–60) assists viral budding and membrane scission, and a disordered domain at the C-terminus is involved in virus assembly through interactions with M1.15 The TM domain is the minimal construct needed for selective proton transport and amantadine binding.17–20 The rate of conductance of the M2 TM domain and its ability to be inhibited by amantadine are nearly identical to those of the full-length protein when the proteins are expressed in frog oocytes or reconstituted in phospholipid vesicles.18,21,22 In fact, the differences between the conductance rates of different-length constructs (when expressed or reconstituted under identical conditions) are smaller than those seen between point mutants found in different strains of the virus.23 Thus, much crystallographic work has focused on the TM domain because the extra-membrane domains appear to impede crystallization.
The structure of M2 has been studied using solution NMR, solid-state NMR (SSNMR), and X-ray crystallography. Two conformational states of the conductance domain have been observed, in which the C-terminus is either largely closed or open to the viral interior (Inwardclosed and Inwardopen, respectively). In both states, His37 is directed toward the center of the channel, where it serves as a selectivity filter and proton shuttle that imbues M2 with its strong proton selectivity.34,35 The Inwardclosed state has been characterized through solution NMR,24,25 SSNMR,26,27 and X-ray crystallog- raphy.28 The Inwardopen state has been characterized primarily through X-ray crystallography,20,29–31 as conformational equilibria between the multiple states at low pH result in peak broadening in NMR studies.24,25,32 The various crystallo-graphic structures of the Inwardopen state are in good agreement with molecular dynamics (MD) simulations that quantitatively explain experimental conductance curves.33 The conductance mechanism involves transporter-like cycling between different protonation states and the two conformations.36 Although the degree of protonation of His37 near the C-terminal region of the pore defines the stability of the Inwardopen and Inwardclosed states, the energetic difference between the two is not large, which assures dynamic switching between states during the transport mechanism. Thus, it is possible to isolate both conformational states in a variety of pH ranges using crystallography.
Previously, structure-based approaches have been used to inform the design of M2 inhibitors that are effective against wild-type (WT) and amantadine-resistant strains of the virus.37–39 However, high-resolution structural studies of M2 bound to inhibitors, particularly those with high enough resolution to examine the critical role of water in drug binding, have been lacking. The only previous drug-bound structures were obtained at relatively low resolution by solution NMR or SSNMR,40,41 and the sole crystal structure was solved at a diffraction limit of 3.5 Å.20 These studies have been sufficient to determine the drug’s general location but not its engagement of the pore-lining residues and water molecules in the channel. Here we report crystallographic structures of rimantadine bound to both the Inwardopen and Inwardclosed states of the WT M2 channel. We also determined structures of amantadine and a novel spiro-adamantyl amine inhibitor (Figure 1) bound to the Inwardclosed state of the WT M2 channel. These drug-bound structures describe the mechanism of binding and inhibition and inform the future design of drugs that interact with and disrupt the networks of hydrogen-bonded waters that are widely utilized throughout nature to facilitate proton diffusion within proteins.42
MATERIALS AND METHODS
M2TM peptide was synthesized and purified as previously described29,30 and reconstituted into the lipid cubic phase (LCP) with some modifications to the protocol described by Caffrey and Cherezov.43 Amantadine, rimantadine, and spiro-adamantyl amine44 were cocrystallized with M2TM. Crystallization conditions were tested in plastic 96-well LCP trays (Laminex) using an LCP crystallization robot (TTP Labtech). Trays were screened using visible-light and UV images (Figure S10). Crystals were harvested from the 96-well sandwich trays and then frozen in liquid nitrogen. Data were collected at the Advanced Light Source (ALS) beamline 8.3.1 at 100 K. Data collection, data processing (Table S2), model building, refinement (Table S3), and MD simulations were carried out as described in the Supporting Information (SI).
Indexing and integration were carried out in MOSFLM,45 and the data were scaled and merged using Aimless46 in the CCP4 suite.47,48 Molecular replacement was done using Phaser49 with previously solved structures as search models (3LBW28 for the Inwardclosed state and 4QK729 for the Inwardopen state). Zanuda50 was used to aid in space group determination. Refinement was carried out in PHENIX51 with model building in Coot52 and PyMOL.53
RESULTS AND DISCUSSION
The Adamantane Groups of Rimantadine and Amantadine Disrupt Networks of Hydrogen-Bonded Waters While the Ammonium Group Engages the Remaining Water Networks in the Inwardclosed State.
Using lipid cubic phase (LCP) crystallization techniques,43 we have solved X-ray crystal structures of the M2 TM domain (residues 22–46) (M2TM) in the Inwardclosed state bound to rimantadine (PDB code 6BKL) and amantadine (PDB code 6BKK) at a resolution of 2.00 A (Figure 2a,b). In both structures, the electron density (Figure S1) unambiguously defines the positions of the drug and surrounding water molecules. Crystals containing drug-bound M2 complexes formed at acidic pH (pH 5.6 for the amantadine-bound structure 6BKK and pH 4.5 for the rimantadine-bound structure 6BKL). A MNG detergent additive was used in crystallization trials to stabilize the Inwardclosed state. The binding of adamantane drugs also stabilizes this conformation and lowers the pKa of His37.56
Figure 2.
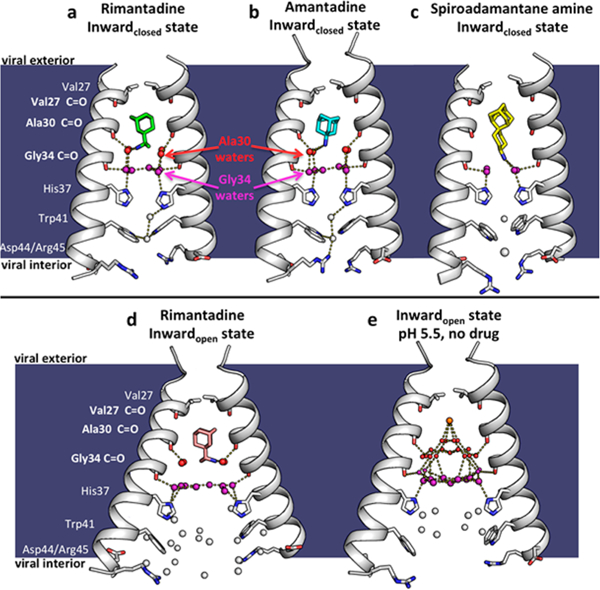
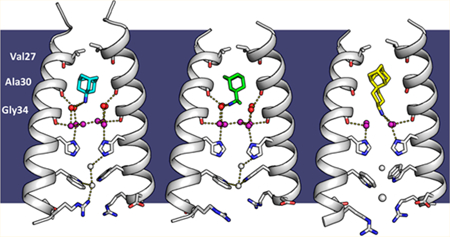
X-ray crystal structures of the M2 proton channel bound to drugs and inhibitors. The channel is a homotetramer, but here the front and back monomers have been removed to show the contents of the channel pore. Hydrogen bonds are shown as yellow dashes. The layer of waters forming hydrogen bonds to the Ala30 carbonyls (“Ala30 layer”) is shown as red spheres; the layer of waters forming hydrogen bonds to the Gly34 carbonyls (“Gly34 layer”) is shown as purple spheres. Top, left to right: (a) M2 bound to rimantadine in the Inwardclosed state (PDB code 6BKL, 2.00 A resolution, monomer subunits F and H); (b) M2 bound to amantadine in the Inwardclosed state (6BKK, 2.00 Å resolution, monomers B and D); (c) M2 bound to spiro-adamantyl amine in the Inwardclosed state (6BMZ, 2.63 Å resolution, monomers B and D). Bottom, left to right: (d) M2 bound to rimantadine in the Inwardopen state (6BOC, 2.25 Å resolution, monomers B and D); (e) Previously solved structure of M2 in the Inwardopen state at pH 5.0 in the absence of bound drug31 (5JOO, 1.41 Å resolution).
Polder omit maps,54 which were calculated by removing the drug and solvent from the model, show good density for the drugs at a contour of 3σ (Figure S1, green meshes). For each complex, the hydrophobic adamantane moiety points upward toward the N-terminus on the exterior of the virus while the ammonium group is directed downward in the aqueous pore toward His37.The adamantane is bound in a predominantly hydrophobic pocket lined by the side chains and main chains of Val27, Ala30, and Ser31. The hydroxyl of Ser31 forms an internal hydrogen bond to a main-chain carbonyl of Val27, increasing the effective hydrophobicity of the environment. Rimantadine is a racemic drug, and the ligand electron density strongly suggests that the bound drug consists of an equimolar mixture of the R and S enantiomers, which were hence each modeled at half occupancy. Indeed, the two enantiomers have been shown to have equal potency in in vitro electrophysiology and cellular antiviral plaque assays.55
In previous high-resolution crystallographic and MD investigations of M2TM in the drug-free form, ordered waters form continuous hydrogen-bonding networks that span the pore from the exterior vestibule near Val27 to His37.29,31 The carbonyl groups of Val27, Ala30, and Gly34 form hydrogen bonds to successive layers of waters. Additional “bridging” waters bridge the carbonyl-associated waters to form continuous hydrogen-bonding networks from the Val27 to His37 (Figure 2e). In both the amantadine- and rimantadine- bound structures, the adamantane cage fits snugly into the top of the pore, displacing the layer of waters near Val27 (Figure 3a,d). Thus, the drug dehydrates the upper portion of the pore, snipping the continuous hydrogen-bonding network seen in the drug-free state.
Figure 3.
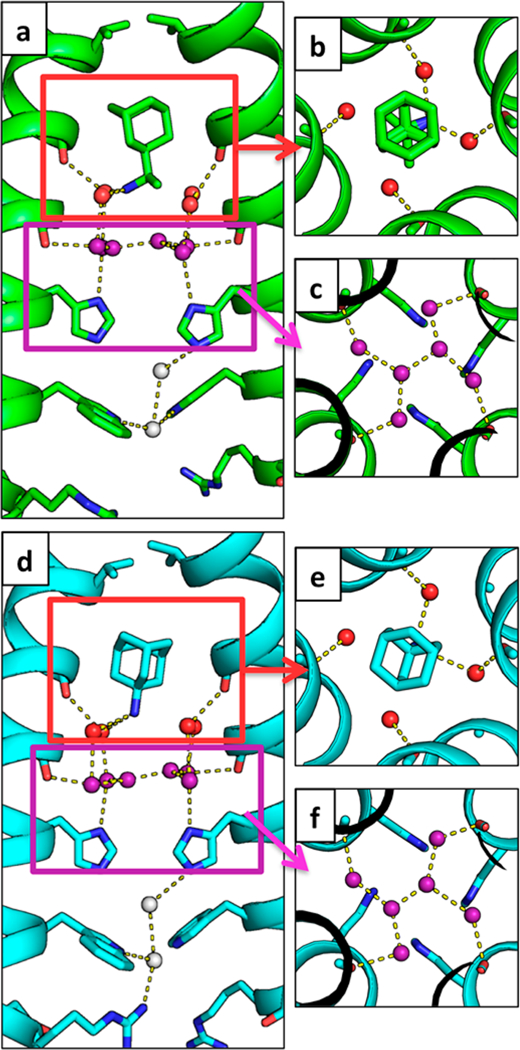
Water-mediated hydrogen bonds facilitate the binding of rimantadine and amantadine to the M2 pore. In both the rimantadine- bound (6BKL, green) and amantadine-bound (6BKK, cyan) structures of the Inwardclosed conformation, the drug ammonium group is positioned to hydrogen-bond with two of the four waters in the Ala30 water layer, shown as red spheres. The Gly34 water layer is shown as purple spheres. Hydrogen bonds are shown as yellow dashes. Amantadine and rimantadine bind asymmetrically and form hydrogen bonds with two of the four waters in the top solvent layer. (a, d) Side views of binding of rimantadine (a, monomer subunits F and H) and amantadine (d, monomers B and D). (b, e) Top-down views of binding of rimantadine (b, monomers E-H) and amantadine (e, monomers A-D) to the Ala30 water layer in the pore of the M2 channel. (c, f) Top-down views of the Gly34 water layer.
In contrast to the disruption of the waters adjacent to Val27, the lower two water layers remain similar to those in the drug- free state.28 The ammonium group is positioned proximal to the four waters comprising the Ala30 layer (Figure 3b,e), followed by the Gly34 water layer consisting of four carbonyl- associated and two bridging water molecules (Figure 3c,f). The B-factors of the waters bound directly to the Ala30 and Gly34 backbone carbonyls are generally similar to those of the backbone atoms, indicating that they are tightly associated, while the bridging waters show greater disorder. The overall water arrangement is strikingly similar to that observed in the crystal structure of the Inwardclosed conformation of M2TM in the absence of drug (PDB code 3LBW), indicating that the water structure that stabilizes the ammonium group is largely preorganized. This same pathway would appear to be ideally oriented for stabilizing and facilitating proton transfer and diffusion through this region of the channel in the absence of inhibitor. This arrangement is also in good agreement with earlier MD simulations of the water in a model of the amantadine complex based on the structure of drug-free M2TM (PDB code 3LBW).28
While the overall structure of the tetramer and pore waters is largely symmetrical, the placement of the drug within the pore deviates slightly from fourfold rotational symmetry. The rimantadine- and amantadine-bound M2 crystals presented here belong to space group P21, with two crystallographically distinct tetramers per asymmetric unit. The two drugs adopt similar asymmetric orientations in each of the two amantadine and rimantadine complexes. Their alkylammonium groups have three hydrogen-bond-donating protons, which cannot form symmetrical interactions with each of the four Ala30 waters without breaking symmetry. Thus, the drugs are slightly tilted within the site, displacing the ammonium group away from the central axis toward two of the Ala30 waters. The angle of tilt of the adamantyl threefold axis of symmetry relative to the channel’s fourfold axis of symmetry is 11.3 ± 0.7° for the amantadine-bound structure and 11 ± 3° for the rimantadine- bound structure, in good agreement with the value of 13° measured by SSNMR for amantadine.41
The Spiro-Adamantyl Amine Inhibitor Additionally Displaces a Second Layer of Water Molecules in the Inwardclosed State.
Spiro-adamantyl amine derivatives are excellent inhibitors of the WT M2 channel and also the V27A drug-resistant mutants as assessed by antiviral plaque assay and electrophysiological assays.44,57 Moreover, these compounds have also been shown to rescue mice from lethal influenza infection.58 The more extended scaffold of the spiro-adamantyl amine (Figure 1) was designed to exclude more water from the channel and to bind with the ammonium group deeper in the WT channel relative to the ammonium position of amantadine.44 The X-ray crystal structure of spiro-adamantyl amine bound to the Inwardclosed conformation solved here at a resolution of 2.63 Å (PDB code 6BMZ; Figure 2c) shows that the drug indeed projects more deeply into the channel. The longer spiro-adamantyl amine compound displaces the waters of the Ala30 water layer (Figure 4a,b) previously observed in the amantadine- and rimantadine-bound structures, and the ammonium group of the spiro-adamantyl amine is positioned to form H-bonds with the four carbonyl-associated waters in the Gly34 solvent layer.
Figure 4.
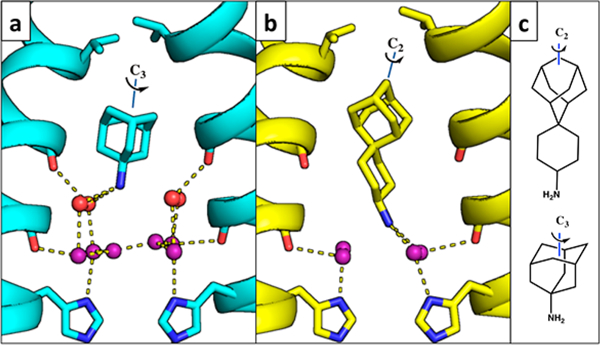
Binding of spiro-adamantyl amine to the Inwardclosed state. Hydrogen bonds are shown as yellow dashes. (a, b) Side views of the amantadine-bound Inwardclosed M2 structure (a, 6BKK, cyan, monomers B and D) and the spiro-adamantyl amine-bound Inwardclosed M2 structure (b, 6BMZ, yellow, monomers B and D). The spiro-adamantyl amine inhibitor binds with its ammonium group deeper in the channel. The Ala30 water layer (red) is completely displaced, and the inhibitor ammonium group is within hydrogen-bonding distance of the Gly34 water layer (purple). (c) Chemical structures of spiro-adamantyl amine (top) and amantadine (bottom) showing the adamantyl C2 and C3 symmetry axes.
Interestingly, the adamantane groups are oriented differently in the channel in the spiro-adamantyl amine complex versus the amantadine and rimantadine complexes (Figure 4). The adamantyl group projects one of its methylene groups along its own pseudo-twofold symmetry axis toward the top of the channel in the spiro-adamantyl amine complex. By contrast, a tertiary center along the threefold axis of adamantane projects upward in the amantadine and rimantadine complexes. This result highlights the near-spherical nature of the adamantane group and helps explain its versatility in drug design.6
Structure of Rimantadine Bound to M2 in the Inwardopen State.
Effective blockers of M2 inhibit proton conductance at high and low pH with the same or only slight reductions in potency (less than 2-fold) at lower pH.5,18 This versatility is likely an important property for drugs that need to remain bound as the pH of the endosome decreases prior to fusion of the viral and endosomal membranes. Nevertheless, this finding runs contrary to early biophysical studies, which appeared to show that amantadine was bound in the channel only under conditions in which the His residues are in the neutral state19,59 and that the amantadine dissociated at lower pH. However, these early studies focused on spectroscopic signatures of only the Inwardclosed state, suggesting that the drug must also bind to another conformational form at low pH. Thus, it was important to determine structures of drugs bound to the Inwardopen state in addition to the Inwardclosed conformational state.
We solved the structure of rimantadine bound to the Inwardopen state of the M2 channel (Figure 2d) to a resolution of 2.25 Å (PDB code 6BOC). The helices, which bend slightly at Gly34 in the Inwardclosed rimantadine complex (Figure 2a), straighten in the Inwardopen rimantadine complex. This conformational change increases the diameter of the bottom of the channel while slightly constricting the upper portion of the channel. The newly solved structure of rimantadine bound to the Inwardopen state is in good agreement with a previously solved low-resolution (3.5 Å) structure of the Inwardopen amantadine complex (PDB code 3C9J)20 (Cα RMSD = 0.98 Å). The adamantane drugs are observed to bind to the same position in both structures (Figure S2). However, the water molecules, which were not resolved in the low-resolution amantadine complex (3C9J), are well-defined in the current rimantadine complex (6BOC).
Hydrogen-bonded networks of water molecules play a similar role in stabilizing the Inwardopen and Inwardclosed rimantadine complexes (Figure 2a,d). In both complexes, the ammonium group is proximal to the four Ala30 carbonyl- associated water molecules, and the Gly34 waters are retained. However, the increased radius of the channel near the C- terminus of the Inwardopen complex leads to an increase in the number of bridging waters in the Gly34 layer, and a progressive opening of a channel from His37 downward. The structures of the Inwardopen and Inwardclosed complexes (Figure S3) are consistent with the known pH-independent binding of the drugs to the channel, which now can be understood in terms of the affinity of the individual conformational forms for drugs at a given pH. The Inwardclosed drug-bound conformation is stable at high pH, where the overall charge on His37 is 0 or at most +1. At lower pH, His37 residues are protonated, leading to repulsion between the imidazolium cations as well as the ammonium group of the drug in the highly restricted environment of the channel. However, at low pH, charge repulsions between protonated His37 groups as well as their interaction with the drug ammonium group would be more easily electrostatically screened in the solvent-exposed end of the Inwardclosed conformation. Also, water molecules are well- suited to stabilize charge changes by switching back and forth from donors to acceptors. Thus, the structures of the Inwardopen and Inwardclosed complexes explain how the drugs can inhibit over a wide range of pH with similar affinity.5,18
Structural Mechanism of Drug-Binding and Channel Inhibition.
The binding of the adamantane drugs to M2 presents particularly interesting problems for understanding the mechanism of drug action against channels in general as well as the design of drugs to address drug resistance in strains of M2 in particular. We hypothesize that amantadine acts as a mechanism-based inhibitor. The amantadine ammonium group exploits the channel’s essential capability to stabilize an excess hydrated proton. Thus, the ammonium groups do not form hydrogen bonds with pore-facing carbonyl groups; instead, they are hydronium mimics that interact with functional water networks connected directly to the proton-shuttling and gating residue His37. This hypothesis is consistent with the fact that replacing the ammonium group of amantadine with hydroxyl strongly decreases the antiviral activity.60 Moreover, the large adamantane displaces loosely bound waters near the top of the pore, thereby stabilizing the complex through hydrophobic van der Waals interactions, while also inhibiting conduction by occluding entry of protons into the pore. The more extended spiro-adamantyl amine places its ammonium group further down the channel into a lower site lined by four water molecules that simultaneously receive hydrogen bonds from both the carbonyl of Gly34 and the imidazole of His37.
Interestingly, our structures show that the pore-lining carbonyl groups act as physicochemical chameleons that stabilize hydrated cations through polar interactions in some complexes (e.g., the Ala30 carbonyls in structures 6BKL and 6BKK) but hydrophobically stabilize apolar aliphatic groups in others (e.g., the Ala30 carbonyls in structure 6BMZ). This behavior is consistent with the known tendency of the carbonyl groups of helices to form a single strong helical hydrogen bond with an amide at position i — 4 when in the hydrophobic core of a protein but to form an additional hydrogen bond to water molecules when exposed to water.61 The ability to switch between these two states offers intriguing and potentially generalizable possibilities for small-molecule drug design.
Explaining the Effectiveness of Adamantane as a Substituent for Drug Design.
Adamantane derivatives are commonly used in drugs that treat influenza (amantadine and rimantadine), Alzheimer’s disease (memantine), and diabetes (saxagliptin and vildagliptin), among other uses.6,62 The adamantyl group has been called a “liphophilic bullet”6 that can be added onto drugs to increase their hydrophobicity and structural stability. The adamantane drugs are hydrophobic, though less so than one would expect on the basis of the number of carbons they contain. Adamantane has 10 carbons contained within four interconnected cyclohexane rings that form a cage with a greatly reduced apolar surface area compared with the corresponding unbranched alkane. In fact, the hydrophobicity of adamantane (LogP = 3.98) is far smaller than that of n-decane (LogP = 5.98) and instead is similar to that of n-hexane (LogP = 3.87).63 The decreased hydro-phobicity of adamantane is a simple consequence of its highly compact, highly cross-linked structure. For example, the solvent-accessible surface area of adamantane is 262 Å2, while that of n-decane in an extended conformation 361 Å2.
A second attractive feature of adamantane is that it has no rotatable bonds and hence loses very little conformational entropy when it binds in a protein pocket. By comparison, an alkane of similar hydrophobicity such as n-pentane could lose up to 2.6 kcal/mol (ΔS = 4RT ln(3), associated with the three rotamers of each of its four rotatable bonds) when immobilized in a receptor-binding site. Moreover, adamantane has a very smooth surface (Figure 1), which allows it to rotate extremely rapidly about its C3 axis even in the crystalline state at room temperature.64 Because of this rapid rotational averaging, adamantane effectively has a smooth ellipsoidal shape with a round cross section that belies its jagged appearance when drawn as a stick figure in two dimensions. Thus, the apparent mismatch between the C3 structural symmetry of amantadine and the M2 tetramer is an artifact of considering the complex as a static object. The smooth cross section of the adamantane ring also assures a low torsional barrier for rotation of the amantadine C1—N bond, which again contributes to minimal loss in configurational entropy of binding. In summary, adamantane is a highly rigid group with a hydrophobicity similar to that of n-hexane that can contribute to binding with a greatly lower conformational entropy loss than for an unbranched alkane.
Molecular Dynamics Simulations Confirm the Structural Mechanism of Drug Binding and Inhibition.
To test and further refine these conclusions, we turned to all-atom MD simulations of the M2 channel in a POPC bilayer (200 lipid molecules) (see the SI). We examined the amantadine and spiro-adamantyl amine complexes in the neutral Inwardclosed conformation to allow a comparison of the two distinct drug classes in the same conformation of the channel. The simulated complexes were structurally stable and showed no large conformational changes over 200 ns (the Cα RMSD to the crystallographic structures was ≤1 Å; Table S1 and Figure S4). In accord with SSNMR measurements,41 amantadine undergoes rapid rotation about its major axis (correlation time τc = 40 ps) (Figure S5). Torsional averaging about the C1-NH2 bond of the drug also occurs with τc = 30 ps. Also in agreement with SSNMR, the main axis of the drug undergoes rapid motion within a cone (mean tilt angle = 9 ± 5°; Figure S6) rather than being fixed in a single orientation. Thus, the fixed asymmetric orientation seen in the cryo-cooled crystal structure represents a single snapshot of the time-averaged structure seen by SSNMR and MD simulations.
Although the water molecules undergo rapid motions in the channel, their average positions are in excellent (<1 Å) agreement with the positions seen in the crystal structure of the corresponding amantadine complex. As in the crystallo-graphic structure, the waters form tight clusters within hydrogen-bonding distance of the Ala30 and Gly34 carbonyls, with more diffuse density in the region bridging the four Gly34 waters (Figures 5a,b and S7a,b). Examination of the probability density function for the amine–water interaction (Figure 6a) shows that on average three to four waters are within hydrogen-bonding distance of the amine and that other features of the waters are in quantitative agreement with the crystallographic structure. In summary, the MD simulations of the amantadine Inwardclosed complex are in excellent agreement with the experimental results and confirm the expectation that the complex is entropically stabilized by rapid axial rotation and torsional motions of the bound amantadine.
Figure 5.
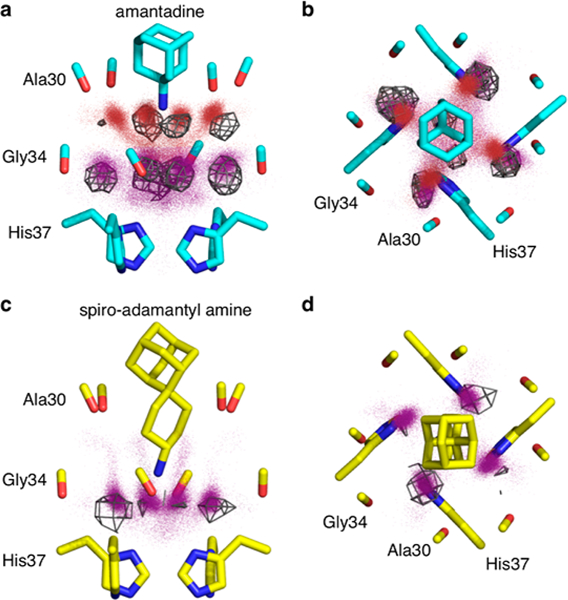
Molecular dynamics simulations reproduce the positions of the crystallographic waters. X-ray crystal structures are shown as sticks. Red and purple dots are water oxygen positions from MD snapshots of the largest amine clusters. Gray wireframe shows 1σ contours of water electron density from the X-ray crystal structure. (a, c) Side views. (b, d) Top views. Details of the simulations and treatment of the data are given in the SI.
Figure 6.
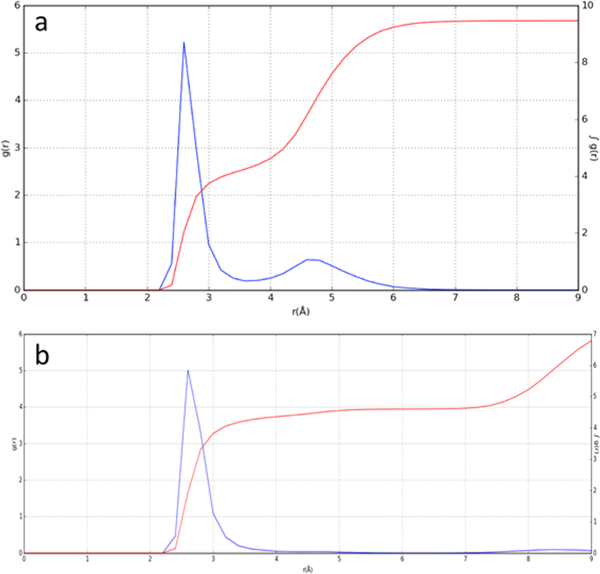
(a) Radial distribution function (RDF) between the amantadine amino group and water oxygen atoms, extracted from a 200 ns simulation. The RDF g(r) function (blue, left axis) shows a strong peak near 2.7 Å, associated with waters forming hydrogen bonds to the ammonium group. The cumulative integrated intensity ∫ g(r) (red, right axis) indicates that 3.5−4 waters are associated with the ammonium on average. The second broad peak near 4.8 Å represents the bridging and Gly34 waters and integrates to approximately 5−6 waters, in good agreement with the crystallographic structure. (b) RDF between the spiro-adamantyl amine amino group and water oxygens, extracted from a 200 ns simulation. Four waters associated with the ammonium group are observed, in agreement with the crystal structure.The second peak near 8 Å reflects more distant waters between His37 and Trp41 of the channel.
Simulations of the spiro-adamantyl amine complex were similarly in good agreement with SSNMR44 and the crystallo- graphic structures. The drug undergoes rapid rigid-body and torsional rotations about its pseudo-C2 axis (τc = 190 and 70 ps, respectively) (Figure S8), similar to the rotation of amantadine about its C3 axis. The water density obtained from the ensemble-averaged structures also matches the crystallographic structure (Figures 5c,d, S7c,d, and 6b), with dense clusters of water seen near the carbonyls of Gly34. Throughout the simulations of both M2 bound to amantadine and M2 bound to the spiro-adamantyl amine, the ammonium group remained near the center of the channel, within 1 Å of its position in the starting crystallographic structures (Figure S9).
CONCLUSIONS
Implications for the Design of Drugs That Target Influenza A Virus.
M2 undergoes changes in conformation and the protonation state of the gating His37 tetrad as the pH of the endosome gradually decreases. Thus, a successful drug must inhibit the channel over a wide range of pH and conformational states. If drugs such as amantadine and rimantadine bound exclusively to the Inwardclosed state, then one would expect that the drug would dissociate as the pH was lowered below the pKa of the His37 residues (Le Châtelier’s principle). However, the affinity of amantadine is relatively invariant with pH between neutral pH and acidic endosomal pH, as assessed by electrophysiological measurements at pH as low as 5.5.5,18 It was therefore important to define the mode of inhibition of drugs when bound to the Inwardopen form of the channel, which is the more stable form in native membranes at low pH.36 The crystal structures shown here define similar modes of drug binding in the two forms, with the ammonium group engaged in hydrogen bonds with ordered waters lower in the channel and the adamantane cage displacing water from the hydrophobic region near Val27. Thus, the ability of drugs to bind over a range of pH and conformational states appears to be important for viral inhibition. Along these lines, it is noteworthy that a series of inhibitors that appear to bind the S31N variant of M2’s TM domain exclusively in the Inwardopen state (with a straight helix lacking the kink that allows formation of the Inwardclosed state) are very weak inhibitors of this protein’s proton channel activity and are also inactive in antiviral assays.65
The structures presented here should also inform the design of compounds that inhibit M2 mutants from amantadine- and rimantadine-resistant forms of the virus. These mutants cluster in the upper portion of the channel between residues 26 and 31, disrupting the hydrophobic adamantane-binding site, while the pore-lining residues that are important for water-mediated conduction (Ala30, Gly34, His37) remain unchanged in more than 99% of the virus identified in humans, pigs, and animals to date.66 Here we have shown that inhibitors can be designed to inhibit either by hydrogen bonding to the Ala30-associated waters in the amantadine and rimantadine complexes or by displacing these same water molecules in the spiro-adamantyl amine complex. The structures presented here will provide critical information to aid in the design of inhibitors that also displace waters that associate with the absolutely conserved His37 imidazole and Gly34 carbonyls. Such compounds might be able to bind lower in the channel, avoiding the more resistance-prone upper region of the pore altogether.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Pil Seok Chae at Hanyang University (Seoul, South Korea) for providing MNG detergent and George Meigs and James Holton at ALS 8.3.1 for support during data collection. J.L.T., N.F.P., and W.F.D. were supported by NIH Grants GM122603 and GM117593. N.F.P. was supported by a T32 Grant from NIH (4 T32 HL 7731-25). J.W. was supported by NIH Grant AI119187. Use of the LCP crystallization robot was made possible by National Center for Research Resources Grant 1S10RR027234-01. This research used resources of the Advanced Light Source, a DOE Office of Science User Facility under Contract DE-AC02-05CH11231. Beamline 8.3.1 at the Advanced Light Source is operated by the University of California Office of the President, Multicampus Research Programs and Initiatives Grant MR-15-328599 and NIGMS Grants P30 GM124169 and R01 GM124149. A.K. thanks Chiesi Hellas, which supported this research (SARG 10354).
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b06741.
Crystallographic data for M2 bound to amantadine in the Inwardclosed conformation (PDB code 6BKK) (CIF) Crystallographic data for M2 bound to rimantadine in the Inwardclosed conformation (PDB code 6BKL) (CIF) Crystallographic data for M2 bound to spiro-adamantyl amine in the Inwardclosed conformation (PDB code 6BMZ) (CIF)
Crystallographic data for M2 bound to rimantadine in the Inwardopen conformation (PDB code 6BOC) (CIF) Supplemental figures and tables, detailed experimental methods (sample preparation, crystallization, and data collection), and detailed methods for MD simulations (PDF)
The authors declare no competing financial interest.
Data Availability. Atomic coordinates and structure factors have been deposited to the Protein Data Bank with the following accession codes: 6BKK, 6BKL, 6BOC, and 6BMZ.
REFERENCES
- (1).Leidner F; Kurt Yilmaz N; Paulsen J; Muller YA; Schiffer CA J. Chem. Theory Comput. 2018, 14 (5), 2784–2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Spyrakis F; Ahmed MH; Bayden AS; Cozzini P; Mozzarelli A; Kellogg GE J. Med. Chem. 2017, 60 (16), 6781–6827. [DOI] [PubMed] [Google Scholar]
- (3).Olsen RW Proc. Natl. Acad. Sci. U. S. A. 2006, 103 (16), 6081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Hay AJ; Wolstenholme AJ; Skehel JJ; Smith MH EMBO J. 1985, 4 (11), 3021–3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Wang C; Takeuchi K; Pinto LH; Lamb RA J. Virol. 1993, 67 (9), 5585–5594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Wanka L; Iqbal K; Schreiner PR Chem. Rev. 2013, 113 (5), 3516–3604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).World Health Organization. Influenza (Seasonal), 31 January 2018. http://www.who.int/en/news-room/fact-sheets/detail/influenza-(seasonal) (accessed June 1, 2018).
- (8).Taubenberger JK; Morens DM Emerging Infect. Dis. 2006, 12 (1), 15–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Shaw ML ACS Infect. Dis. 2017, 3 (10), 691–694. [DOI] [PubMed] [Google Scholar]
- (10).Chizhmakov IV; Geraghty FM; Ogden DC; Hayhurst A; Antoniou M; Hay AJ J. Physiol. 1996, 494 (2), 329–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Lin TI; Schroeder CJ Virol. 2001, 75 (8), 3647–3656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Mould JA; Drury JE; Frings SM; Kaupp UB; Pekosz A; Lamb RA; Pinto LH J. Biol. Chem. 2000, 275 (40), 31038–31050. [DOI] [PubMed] [Google Scholar]
- (13).Pinto LH; Holsinger LJ; Lamb RA Cell 1992, 69 (3), 517–528. [DOI] [PubMed] [Google Scholar]
- (14).Shimbo K; Brassard DL; Lamb RA; Pinto LH Biophys. J. 1996, 70 (3), 1335–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Martin K; Heleniust A Cell 1991, 67 (1), 117–130. [DOI] [PubMed] [Google Scholar]
- (16).Park EK; Castrucci MR; Portner A; Kawaoka YJ Virol. 1998, 72 (3), 2449–2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Duff KC; Ashley RH Virology 1992, 190 (1), 485–489. [DOI] [PubMed] [Google Scholar]
- (18).Ma CL; Polishchuk AL; Ohigashi Y; Stouffer AL; Schon A; Magavern E; Jing XH; Lear JD; Freire E; Lamb RA; DeGrado WF; Pinto LH Proc. Natl. Acad. Sci. U. S. A. 2009, 106 (30), 12283–12288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Salom D; Hill BR; Lear JD; DeGrado WF Biochemistry 2000, 39 (46), 14160–14170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Stouffer AL; Acharya R; Salom D; Levine AS; Di Costanzo L; Soto CS; Tereshko V; Nanda V; Stayrook S; DeGrado WF Nature 2008, 451 (7178), 596–U13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Liang R; Swanson JMJ; Madsen JJ; Hong M; DeGrado WF; Voth GA Proc. Natl. Acad. Sci. U. S. A. 2016, 113 (45), E6955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Tobler K; Kelly ML; Pinto LH; Lamb RA J. Virol. 1999, 73 (12), 9695–9701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Balannik V; Carnevale V; Fiorin G; Levine BG; Lamb RA; Klein ML; DeGrado WF; Pinto LH Biochemistry 2010, 49 (4), 696–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Schnell JR; Chou JJ Nature 2008, 451 (7178), 591–U12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Wang J; Wu Y; Ma C; Fiorin G; Wang J; Pinto LH; Lamb RA; Klein ML; Degrado WF Proc. Natl. Acad. Sci. U. S. A. 2013, 110 (4), 1315–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Hu FH; Schmidt-Rohr K; Hong MJ Am. Chem. Soc. 2012, 134 (8), 3703–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Sharma M; Yi M; Dong H; Qin H; Peterson E; Busath DD; Zhou HX; Cross TA Science 2010, 330 (6003), 509–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Acharya R; Carnevale V; Fiorin G; Levine BG; Polishchuk AL; Balannik V; Samish I; Lamb RA; Pinto L; DeGrado WF; Klein ML Proc. Natl. Acad. Sci. U. S. A. 2010, 107 (34), 15075–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Thomaston JL; Alfonso-Prieto M; Woldeyes RA; Fraser JS; Klein ML; Fiorin G; DeGrado WF Proc. Natl. Acad. Sci. U. S.A. 2015, 112 (46), 14260–14265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Thomaston JL; DeGrado WF Protein Sci. 2016, 25 (8), 1551–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Thomaston JL; Woldeyes RA; Nakane T; Yamashita A; Tanaka T; Koiwai K; Brewster AS; Barad BA; Chen Y; Lemmin T; Uervirojnangkoorn M; Arima T; Kobayashi J; Masuda T; Suzuki M; Sugahara M; Sauter NK; Tanaka R; Nureki O; Tono K; Joti Y; Nango E; Iwata S; Yumoto F; Fraser JS; DeGrado WF Proc. Natl. Acad. Sci. U. S. A. 2017, 114 (51) 13357–13362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Li C; Qin H; Gao FP; Cross TA Biochim. Biophys. Acta} Biomembr. 2007, 1768 (12), 3162–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Liang R; Li H; Swanson JMJ; Voth GA Proc. Natl. Acad. Sci. U. S. A. 2014, 111 (26), 9396–9401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Hu J; Fu R; Nishimura K; Zhang L; Zhou HX; Busath DD; Vijayvergiya V; Cross TA Proc. Natl. Acad. Sci. U. S. A. 2006, 103 (18), 6865–6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Venkataraman P; Lamb RA; Pinto LH J. Biol. Chem. 2005, 280 (22), 21463–21472. [DOI] [PubMed] [Google Scholar]
- (36).DiFrancesco ML; Hansen U-P; Thiel G; Moroni A; Schroeder I PLoS One 2014, 9 (9), e107406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Alhadeff R; Assa D; Astrahan P; Krugliak M; Arkin IT Biochim. Biophys. Acta, Biomembr. 2014, 1838 (4), 1068–1073. [DOI] [PubMed] [Google Scholar]
- (38).Leonov H; Astrahan P; Krugliak M; Arkin IT J. Am. Chem. Soc. 2011, 133 (25), 9903–9911. [DOI] [PubMed] [Google Scholar]
- (39).Wang J; Li F; Ma C Biopolymers 2015, 104 (4), 291–309. [DOI] [PubMed] [Google Scholar]
- (40).Cady SD; Mishanina TV; Hong MJ Mol Biol. 2009, 385 (4), 1127–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Cady SD; Schmidt-Rohr K; Wang J; Soto CS; DeGrado WF; Hong M Nature 2010, 463 (7281), 689–U127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Swanson JMJ; Maupin CM; Chen H; Petersen MK; Xu J; Wu Y; Voth GA J. Phys. Chem. B 2007, 111 (17), 4300–4314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Caffrey M; Cherezov V Nat. Protoc. 2009, 4 (5), 706–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Wang J; Ma CL; Fiorin G; Carnevale V; Wang T; Hu FH; Lamb RA; Pinto LH; Hong M; Klein ML; DeGrado WFJ Am. Chem. Soc. 2011, 133 (32), 12834–12841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Battye TGG; Kontogiannis L; Johnson O; Powell HR; Leslie AGW Acta Crystallogr. Sect. D: Biol. Crystallogr. 2011, 67, 271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Evans P Acta Crystallogr., Sect. D: Biol. Crystallogr. 2006, 62 (1), 72–82. [DOI] [PubMed] [Google Scholar]
- (47).Cowtan K; Emsley P; Wilson KS Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67 (4), 233–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Winn MD; Ballard CC; Cowtan KD; Dodson EJ; Emsley P; Evans PR; Keegan RM; Krissinel EB; Leslie AGW; McCoy A; McNicholas SJ; Murshudov GN; Pannu NS; Potterton EA; Powell HR; Read RJ; Vagin A; Wilson KS Acta Crystallogr. Sect. D: Biol. Crystallogr. 2011, 67 (4), 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).McCoy AJ; Grosse-Kunstleve RW; Adams PD; Winn MD; Storoni LC; Read RJ J. Appi. Crystallogr. 2007, 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Lebedev AA; Isupov MN Acta Crystallogr., Sect. D: Biol. Crystallogr. 2014, 70 (9), 2430–2443. [DOI] [PubMed] [Google Scholar]
- (51).Adams PD; Afonine PV; Bunkoczi G; Chen VB; Davis W; Echols N; Headd JJ; Hung LW; Kapral GJ; Grosse- Kunstleve RW; McCoy AJ; Moriarty NW; Oeffner R; Read RJ; Richardson DC; Richardson JS; Terwilliger TC; Zwart PH Acta Crystallogr. Sect. D: Biol. Crystallogr. 2010, 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Emsley P; Lohkamp B; Scott WG; Cowtan K Acta Crystallogr. Sect. D: Biol. Crystallogr. 2010, 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).The PyMOL Molecular Graphics System, version 1.3r1; Schrödinger, LLC: New York, 2010. [Google Scholar]
- (54).Liebschner D; Afonine PV; Moriarty NW; Poon BK; Sobolev OV; Terwilliger TC; Adams PD Acta Crystallogr. D 2017, 73 (2), 148–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Drakopoulos A; Tzitzoglaki C; Ma C; Freudenberger K; Hoffmann A; Hu Y; Gauglitz G; Schmidtke M; Wang J; Kolocouris A ACS Med. Chem. Lett. 2017, 8 (2), 145–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Hu J; Fu RQ; Cross TA Biophys. J. 2007, 93 (1), 276–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Balgi AD; Wang J; Cheng DYH; Ma C; Pfeifer TA; Shimizu Y; Anderson HJ; Pinto LH; Lamb RA; DeGrado WF; Roberge M PLoS One 2013, 8 (2), e55271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Hu Y; Musharrafieh R; Ma C; Zhang J; Smee DF; DeGrado WF; Wang J Antiviral Res. 2017, 140, 45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Hu J; Asbury T; Achuthan S; Li CG; Bertram R; Quine JR; Fu RQ; Cross TA Biophys. J. 2007, 92 (12), 4335–4343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Kolocouris A; Spearpoint P; Martin SR; Hay AJ; Lopez- Querol M; Sureda FX; Padalko E; Neyts J; De Clercq E Bioorg. Med. Chem. Lett. 2008, 18 (23), 6156–6160. [DOI] [PubMed] [Google Scholar]
- (61).Walsh STR; Cheng RP; Wright WW; Alonso DOV; Daggett V; Vanderkooi JM; DeGrado WF Protein Sci. 2003, 12 (3), 520–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Kim W; Zhu W; Hendricks GL; Van Tyne D; Steele AD; Keohane CE; Fricke N; Conery AL; Shen S; Pan W; Lee K; Rajamuthiah R; Fuchs BB; Vlahovska PM; Wuest WM; Gilmore MS; Gao H; Ausubel FM; Mylonakis E Nature 2018, 556, 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Raska I; Toropov A Eur. J. Med. Chem. 2006, 41 (11), 1271–1278. [DOI] [PubMed] [Google Scholar]
- (64).Vijayakumar V; Garg AB; Godwal BK; Sikka SKJ Phys.: Condens. Matter 2001, 13 (9), 1961–1972. [Google Scholar]
- (65).Tzitzoglaki C; Wright A; Freudenberger K; Hoffmann A; Tietjen I; Stylianakis I; Kolarov F; Fedida D; Schmidtke M; Gauglitz G; Cross TA; Kolocouris AJ Med. Chem. 2017, 60 (5), 1716–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Durrant MG; Eggett DL; Busath DD BMC Genet. 2015, 16 (2), S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.