

Abstract
We propose a stochastic model of aging to explain deviations from exponential growth in mortality rates commonly observed in empirical studies. Mortality rate plateaus are explained as a generic consequence of considering death in terms of first passage times for processes undergoing a random walk with drift. Simulations of populations with age-dependent distributions of viabilities agree with a wide array of experimental results. The influence of cohort size is well accounted for by the stochastic nature of the model.
Fundamental studies of the aging process have of late attracted the interest of researchers in a variety of disciplines, linking ideas and theories from biochemistry to mathematics (1–3). Much of this recent activity is due to the possibility that one of the supposedly fundamental tenets of aging, namely the exponential growth of mortality rates proposed by Gompertz (4), may fail to describe the behavior of observed populations adequately. More specifically, studies using populations or “cohorts” of Saccharomyces cerevisiae, Caenorhabditis elegans, Drosophila, and humans demonstrate that mortality rates tend to level off and even decrease at later stages of life (5–9). Attempts have been made to explain these plateaus via parabolic hazard functions (10), age-dependent demographics (11, 12), and phenomenological bifurcation models (8). In this paper we propose a simple model that incorporates heterogeneous dynamics to explain the generic plateau in mortality rates commonly observed in large cohorts of organisms.
Consider a population of N organisms with a distribution of viabilities, vi ≥ 0, where vi = 0 signifies death. The dynamics of an individual viability will be modeled as follows:
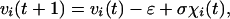 |
1 |
where ɛ > 0 is a constant drift, σ > 0 is the standard deviation of the fluctuations, and χi(t) is an uncorrelated Gaussian random variable with zero mean and unit standard deviation. The linear decline of viability is justified by the observation of a linear decline of physiological functions noted by Strehler and Mildvan (13) as well as similar results in more recent surveys (14). The stochasticity in the system may be related to the competition for resources, phenotypic differences, local environmental changes, or even stochastic gene expression (15) but does not necessarily depend on heterogeneity in the initial genotypic distribution. The inclusion of stochasticity at the individual level implies that Eq. 1 may be considered a changing frailty model as opposed to a fixed frailty model. A fixed frailty model preserves any initial heterogeneity in v throughout each individual lifespan (16).
Biologically, this model states that each organism drifts toward death but with low probability may occasionally increase its long-term chances for survival. The probability of dying at time t is equivalent to the probability of first passage time P(t|v0) of a random walk that begins at v = v0 and reaches the origin, v = 0, at time t. The likelihood of death is controlled by the relative strength of drift and fluctuations. In this paper we explain the basic mechanism associated with first passage time problems and then proceed to show how such a model captures the essential features of late life mortality plateaus.
Theory of First Passage Time Problems
Eq. 1 may be better understood by considering the limits of vanishing noise and then vanishing drift. When σ → 0, the organisms move in lock step toward an inevitable death. The hazard rate, μ(t), may be solved at t > 0 for any initial distribution of viabilities, n0(v),
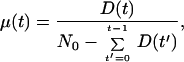 |
2 |
where N0 is the total number of initial organisms, and D(t) is the number of organisms that die at time t,
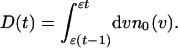 |
3 |
For a uniform distribution of initial viabilities, 0 < vi(0) < 1, the hazard rate reduces to
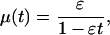 |
4 |
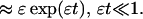 |
In the limit of slow drift the hazard rate grows exponentially for small t and continues to grow until the system is left desolate at t = 1/ɛ. At intermediate times the hazard rate is not increasing exponentially as one might expect from a Gompertz model. Regardless of its precise form, the monotonic increase of mortality rates as evidenced in this simple example leads to the natural question of what causes mortality rates to plateau in populations of fruit flies, yeast, and other organisms.
The first step in answering this question is to consider the other limit of Eq. 1, namely ɛ → 0, when fluctuations dominate the dynamics. In this regime an individual viability vi(t) follows a random walk that ends when v ≤ 0. Qualitatively the removal of individuals with v ≤ 0 is tantamount to increasing the average viability of the remaining cohort. With time the average viability should increase, and therefore the hazard rate should decrease. It is important to note that as the population dies off, it will become more susceptible to fluctuations and may exhibit an intermittent rise in hazard rate near complete elimination. This caveat notwithstanding, we begin to see why the combination of these two effects, drift and fluctuation, might give rise to just the sort of behavior observed in large-scale mortality studies.
In order to simplify the analytical calculation of hazard rates we rewrite Eq. 1 in the case of continuous time,
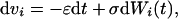 |
5 |
where Wi(t) is a stochastic Wiener process that satisfies dWi(t′)dWj(t) = δijδ(t′ − t)dt. The difference between Eqs. 1 and 5 is partly a matter of analytical convenience¶; however, the dynamics of aging may be related intimately to reproductive schedules, which would imply the bulk of aging takes place at discrete intervals (e.g. cell division in yeast) as opposed to taking place via a continuous process of senescence. The qualitative behavior of both models is essentially equivalent.
The closed form expression for the probability of crossing the origin at t given an initial viability v0, i.e., the probability of dying between t and t + dt, for systems behaving according to Eq. 5 is
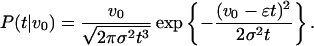 |
6 |
This probability distribution is known as the inverse Gaussian distribution (18–21). Researchers have considered its importance in the context of a broad range of lifetime problems (22); for example, it has proved useful in analyzing marriage durations (23), species extinctions (24), and even the shelf life of food (25). Other stochastic models of mortality have been proposed (26, 27), but to our knowledge none has recognized the importance of the inverse Gaussian distribution in the context of late-life mortality plateaus.
Given an initial distribution of viabilities, n0(v), the hazard rate in the continuous model (Eq. 5) can be written as
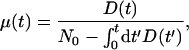 |
7 |
where
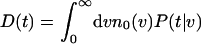 |
8 |
is the density of individuals dying as a function of age. For a completely homogeneous population, with vi(t) = v0 and N0 individuals, D(t) = N0P(t|v0). For arbitrary heterogeneous populations, D(t) may be calculated analytically or computed numerically. In the case of homogeneous populations, e.g. v0 = 1 for all individuals at t = 0, the hazard rate can be expressed analytically (28) as
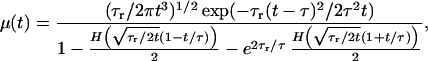 |
9 |
where τ ≡ v0/ɛ,
τr ≡
v /σ2, and H(x)
is the complementary error function (29).
/σ2, and H(x)
is the complementary error function (29).
Although the expression for hazard rate is complicated, the behavior is controlled by two time scales: τ, corresponding to the mean lifetime, and τr, corresponding to the mean time for an organism to change its viability by v0 via fluctuations alone. The mean lifetime does not depend on σ, the parameter responsible for changing frailty, although the variance of the lifetime (as well as higher moments) do depend on both ɛ and σ.
In Fig. 1 we show theoretical hazard rate curves when τ = 10 and τr = 2, 50, and 150. When τr ≫ τ, the system is dominated by drift, and hazard rates will increase rapidly after a brief delay at the initial stages before leveling off. When τr ≪ τ, the system is dominated by fluctuations, and hazard rates will actually decline with age after a brief increase. Finally, when τr > τ but still of the same order of magnitude, the system will exhibit an initial increase in hazard rate followed by a plateau. A range of behavior from essentially monotonic growth to plateaus to plateaus followed by declines is possible. Chhikara and Folks (30) have reached similar conclusions for the “failure rate” of any system, the probability of first passage times of which is modeled by Eq. 6. However, the rapid increase of hazard rate in all cases is slower than exponential. As it stands, the model is insufficient to explain mortality curves that have sustained Gompertz-type behavior at intermediate times. It is worth noting that any decrease in ɛ leads to an increase in the mean survival time, τ, as well as a postponement of the onset of the plateau, consistent with recent work on human populations (31).
Figure 1.

Mortality rates according to Eq. 9 with τ = 10 and τr = 2, 50, and 150 corresponding to the solid, dashed, and dot-dashed curves, respectively.
One important point is that the asymptotic hazard rate for the inverse Gaussian distribution is nonzero, i.e., limt→∞ μ(t) > 0. However, for finite size populations the fluctuations inevitably lead to the eventual decline of the entire population, and therefore μ(t) = 1 at some finite value of t. The results of numerical simulations of Eq. 1 can be found in Fig. 2. Notice that the generic shape of the curves are the same as those found in Fig. 1, although the presence of fluctuations becomes important as the cohort dwindles in number.
Figure 2.

Mortality rates obtained via numerical simulations of a homogeneous
population of N = 106 organisms, the
dynamics of which is that of Eq. 1, with
v0 = 1, τ = 10, and
τr = 2, 50, and 150 corresponding to the solid,
dashed, and dot-dashed curves, respectively. Note that ɛ = 1/τ
and σ = τ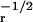 .
.
Because the presence of plateaus is related in some sense to the
difference between the most- and least-fit individuals in the cohort,
it is useful also to calculate the ratio between the viability of those
individuals closest and farthest from death. If we denote
v̄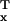 as the average viability of the
top x fraction of individuals and
v̄
as the average viability of the
top x fraction of individuals and
v̄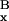 as the average viability of the
bottom x fraction of individuals, then the ratio of the most
fit to least fit can be expressed as rx =
v̄
as the average viability of the
bottom x fraction of individuals, then the ratio of the most
fit to least fit can be expressed as rx =
v̄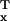 /v̄
/v̄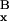 . In the
stochastic simulations of Fig. 2, the ratio between the top 10% to the
bottom 10% leveled off at approximately r0.1 =
15, 30, and 40 in the case of τ = 10 and
τr = 2, 50, and 150, respectively. Even if such
analysis does not constitute a formal proof, it does provide reassuring
evidence that the fitness gap between the most and least fit does not
grow without bound in this model.
. In the
stochastic simulations of Fig. 2, the ratio between the top 10% to the
bottom 10% leveled off at approximately r0.1 =
15, 30, and 40 in the case of τ = 10 and
τr = 2, 50, and 150, respectively. Even if such
analysis does not constitute a formal proof, it does provide reassuring
evidence that the fitness gap between the most and least fit does not
grow without bound in this model.
Application to Mortality Rate Data
The usefulness of this simple model is to see whether experimental data, specifically those exhibiting mortality plateaus, match with the predictions of theory. Given a set of times Xi specifying the lifespan of N organisms, the maximum likelihood estimates (MLEs) of the parameters ɛ and σ in Eq. 6 are
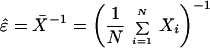 |
10 |
and
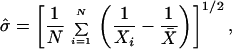 |
11 |
where v0 = 1 may be specified a priori. Using these estimates of parameters we now compare empirical observations to the analytical hazard rate assuming a homogeneous distribution as well as to the results of simulations. The empirical data on which we concentrate for the remainder of the paper is from an experiment where N0 = 1,203,646 genetically identical flies were observed for nearly 6 months, and the number of deaths was recorded on a daily basis (5).
The MLE estimates of the parameters are ɛ̂ = 0.0448 and σ̂ = 0.0975, which corresponds to τ̂ = 22.3 and τ̂r = 105. In Fig. 3 we compare theoretical predictions for the survival probability,
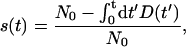 |
12 |
and the probability distribution P(t) in Eq. 6 to those calculated from the experimental data set. Notice the good agreement not only with the survival probability (which essentially is a cumulative measure) but with the actual probability distribution of dying at a given age.
Figure 3.

Survival probability at time t for experimental cohorts and theoretical predictions based on Eq. 6 where ɛ̂ = 0.0448 and σ̂ = 0.0975. (Inset) A comparison of the probability of dying at time t between experiment and theory. In both cases open circles signify data points from Carey et al. (5), and solid lines are predictions from theory.
By using these MLE estimates as a basis for further computational studies, we simulated Eq. 1 using v0 = 1 and calculated the mortality rates for various size cohorts. In the study by Carey et al. (5), mortality rate is plotted against time for cohorts of different sizes. Not surprisingly the mortality rate is ill defined for small cohorts and well defined for large cohorts until mortality rate levels off and then exhibits strong fluctuations about a mean. To compare against experimental data we generated random cohorts by sampling from the fly mortality data of Carey et al. (5). Fig. 4 demonstrates how stochasticity and drift lead to both plateau- and size-dependent fluctuations very much in agreement with real data.
Figure 4.

Age-specific mortality rates for cohorts of sizes ranging from n = 25 to 100,000. Going from top to bottom, rows 1 and 3 are randomly sampled from experimental data. Rows 2 and 4 are from simulations of Eq. 1 using MLE estimates as explained in Application to Mortality Rate Data.
The agreement should make it clear that small cohorts essentially veil the presence of mortality plateaus that would otherwise be evident in larger populations.
Discussion
This model of aging is in many ways an ideal construct. There is no upper limit on viability, no time-dependent drift to reflect developmental stages or the influence of natural selection, and no organism–organism interaction terms which might be important in large cohorts with limited resources. Modifications to the model perhaps could lead to better agreement with data but would not alter the main point we are trying to emphasize. The models we propose, Eqs. 1 and 5, and the behavior they exhibit demonstrate that mortality plateaus are a generic consequence of classifying lifespan as a first passage time problem for a random walk with drift. It is important also to note that although generic, mortality plateaus are not an inevitable outcome of this model for finite size cohorts.
There has been a long-standing interest on the effect of heterogeneity on mortality rates, although models have relied typically on notions of “fixed” frailty where individuals are fixed at the same relative risk compared to others in the same cohort (16). In the context of the present model, fixed frailty is analogous to having v0 differ among individuals but change by drift alone. However, a review of gerontological studies shows that the variability of a majority of biological and cognitive health indicators tends to increase with age (32). Likewise a longitudinal study of blood pressure among nearly 4,000 men over a 40-year period found increasing variability with increasing age (33). These empirical results on age-related variability are more compatible with a changing frailty model even if some combination of changing and fixed frailty models is ultimately necessary to explain gerontological data.
It is only in the past half-century that substantive theories have been proposed to explain the underlying causes as well as the definition of aging. Different aging mechanisms are thought to involve oxidative damage from free radicals (34, 35), senescence genes (36), shortening telomeres (37), programmed cell death (38), simple “clogging” of cellular machinery in the nucleolus from hyperreplication of rDNA plasmids (39), mitotic misregulation (40), and of course combinations of all these as well (3, 41). The implications of these and other explanations of aging for population level mortality rates have not been well established. Subsuming these various mechanisms into a single measure of viability leads to the natural question of how ɛ and σ are correlated with body size, environmental conditions, and even genetic differences, if at all.
In order to test the model one might attempt a series of experiments wherein a deleterious treatment (e.g. radiation, DNA damaging agents, etc.) could be used to artificially control the level of environmental fluctuations influencing viability. For example, one might consider exposing a cohort of Drosophila to normally distributed levels of treatment with a nonzero level of treatment as control. In this way it would be possible to test the effects of increasing fluctuations σ with a fixed ɛ on cohort mortality rates.
Although this model demonstrates why an aging process should exhibit mortality plateaus, it does not claim to uncover the fundamental causes of aging. Bridging the gap between the causes and consequences of aging would provide a natural framework to explain how different mechanisms of aging lead to different types of mortality curves.
Acknowledgments
We thank L. Demetrius, P. Dodds, D. Rothman, N. Schorghofer, D. Sinclair, and K. Wachter for helpful comments. We also thank the anonymous member editor and referees for their suggestions and for pointing out a number of useful references. J.S.W. acknowledges the support of a National Defense Science and Engineering Graduate Fellowship. This work was supported in part by National Science Foundation Grant DMS-9709607.
Abbreviation
- MLE
maximum likelihood estimate
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
The probability of first passage times for the discrete version of the model are referred to as ruin problems (17).
References
- 1.Finch C. Longevity, Senescence, and the Genome. Chicago: Univ. of Chicago Press; 1990. [Google Scholar]
- 2.Wachter K, Finch C, editors. Between Zeus and the Salmon: The Biodemography of Longevity. Washington, DC: Natl. Acad. Press; 1997. [PubMed] [Google Scholar]
- 3.Johnson F, Sinclair D, Guarente L. Cell. 1999;96:291–302. doi: 10.1016/s0092-8674(00)80567-x. [DOI] [PubMed] [Google Scholar]
- 4.Gompertz B. Philos Trans R Soc London A. 1825;115:513–525. [Google Scholar]
- 5.Carey J, Liedo P, Orozco D, Vaupel J. Science. 1992;258:457–461. doi: 10.1126/science.1411540. [DOI] [PubMed] [Google Scholar]
- 6.Brooks A, Lithgow G, Johnson T. Science. 1994;263:668–671. doi: 10.1126/science.8303273. [DOI] [PubMed] [Google Scholar]
- 7.Vaupel J, Johnson T, Lithgow G. Science. 1994;266:826. doi: 10.1126/science.7973641. [DOI] [PubMed] [Google Scholar]
- 8.Jazwinski S, Kim S, Lai C, Benguria A. Exp Gerontol. 1998;33:571–580. doi: 10.1016/s0531-5565(98)00029-1. [DOI] [PubMed] [Google Scholar]
- 9.Vaupel J, Carey J, Christensen K, Johnson T, Yashin A, Holm N, Iachine I, Kannisto V, Khazaeli A, Liedo P, et al. Science. 1998;280:855–860. doi: 10.1126/science.280.5365.855. [DOI] [PubMed] [Google Scholar]
- 10.Yashin A, Manton K, Woodbury M, Stallard E. J Math Biol. 1995;34:1–16. doi: 10.1007/BF00180134. [DOI] [PubMed] [Google Scholar]
- 11.Mueller L, Rose M. Proc Natl Acad Sci USA. 1996;93:15249–15253. doi: 10.1073/pnas.93.26.15249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wachter K. Proc Natl Acad Sci USA. 1999;96:10544–10547. doi: 10.1073/pnas.96.18.10544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Strehler B, Mildvan A. Science. 1960;132:14–21. doi: 10.1126/science.132.3418.14. [DOI] [PubMed] [Google Scholar]
- 14.Sehl M, Yates F. J Gerontol Biol Sci. 2001;56:B198–B208. doi: 10.1093/gerona/56.5.b198. [DOI] [PubMed] [Google Scholar]
- 15.McAdams H, Arkin A. Proc Natl Acad Sci USA. 1997;94:814–819. doi: 10.1073/pnas.94.3.814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vaupel J, Mantan K, Stallard E. Demography. 1979;16:439–454. [PubMed] [Google Scholar]
- 17.Feller W. An Introduction to Probability Theory and Its Applications. 3rd Ed. Vol. 1. New York: Wiley; 1968. p. 342. [Google Scholar]
- 18.Schrodinger E. Phys Z. 1915;289:16. [Google Scholar]
- 19.Tweedie M. Ann Math Stat. 1957;28:362–377. [Google Scholar]
- 20.Tweedie M. Ann Math Stat. 1957;28:696–705. [Google Scholar]
- 21.Folks J, Chhikara R. J R Stat Soc B. 1978;40:263–289. [Google Scholar]
- 22.Sherif Y. Microelectronics Reliability. 1982;22:217–226. [Google Scholar]
- 23.Whitmore G. Statistician. 1986;35:207–219. [Google Scholar]
- 24.Dennis B, Munholland P, Scott J. Ecol Monogr. 1991;61:115–143. [Google Scholar]
- 25.Gacula M J, Kubala J. J Food Sci. 1975;40:404–409. [Google Scholar]
- 26.Sacher G, Trucco E. Ann NY Acad Sci. 1962;96:985–1007. doi: 10.1111/j.1749-6632.1962.tb54116.x. [DOI] [PubMed] [Google Scholar]
- 27.Yashin A, Manton K, Vaupel J. Theor Popul Biol. 1985;27:154–175. doi: 10.1016/0040-5809(85)90008-5. [DOI] [PubMed] [Google Scholar]
- 28.Shuster J. J Am Stat Assoc. 1968;63:1514–1516. [Google Scholar]
- 29.Abramowitz A. Handbook of Mathematical Functions with Formulas, Graphs, and Mathematical Tables, Applied Mathematics Series. 10th Ed. Washington, DC: U.S. Govt. Print. Office; 1972. p. 297. [Google Scholar]
- 30.Chhikara R, Folks J. Technometrics. 1977;19:461–468. [Google Scholar]
- 31.Horiuchi S, Wilmoth J. Demography. 1998;35:391–412. [PubMed] [Google Scholar]
- 32.Nelson E, Dannefer D. Gerontology. 1992;32:17–23. doi: 10.1093/geront/32.1.17. [DOI] [PubMed] [Google Scholar]
- 33.Tate R, Manfreda J, Krahn A, Cuddy T. Am J Epidemiol. 1995;142:946–954. doi: 10.1093/oxfordjournals.aje.a117742. [DOI] [PubMed] [Google Scholar]
- 34.Harman D. J Gerontol. 1956;11:298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- 35.Harman D. Free Radicals, Aging, and Degenerative Diseases. New York: Liss; 1986. pp. 3–49. [Google Scholar]
- 36.Lin Y, Seroude L, Benzer S. Science. 1998;282:943–946. doi: 10.1126/science.282.5390.943. [DOI] [PubMed] [Google Scholar]
- 37.Harley C, Vaziri H, Counter C, Allsopp R. Exp Gerontol. 1992;27:375–382. doi: 10.1016/0531-5565(92)90068-b. [DOI] [PubMed] [Google Scholar]
- 38.Warner H, Hodes R, Pocinki K. J Am Geriatr Soc. 1997;45:1140–1146. doi: 10.1111/j.1532-5415.1997.tb05981.x. [DOI] [PubMed] [Google Scholar]
- 39.Sinclair D, Guarente L. Cell. 1997;91:1033–1042. doi: 10.1016/s0092-8674(00)80493-6. [DOI] [PubMed] [Google Scholar]
- 40.Ly D, Lockhart D, Lerner R, Schultz P. Science. 2000;287:2486–2492. doi: 10.1126/science.287.5462.2486. [DOI] [PubMed] [Google Scholar]
- 41.Finch C, Schneider E, editors. Handbook of the Biology of Aging. 2nd Ed. New York: Van Nostrand Reinhold; 1985. [Google Scholar]