

High-content functional screening of primary patient blood cells reveals repurposed psychiatric drug candidates.
Abstract
There is a paucity of efficacious new compounds to treat neuropsychiatric disorders. We present a novel approach to neuropsychiatric drug discovery based on high-content characterization of druggable signaling network responses at the single-cell level in patient-derived lymphocytes ex vivo. Primary T lymphocytes showed functional responses encompassing neuropsychiatric medications and central nervous system ligands at established (e.g., GSK-3β) and emerging (e.g., CrkL) drug targets. Clinical application of the platform to schizophrenia patients over the course of antipsychotic treatment revealed therapeutic targets within the phospholipase Cγ1–calcium signaling pathway. Compound library screening against the target phenotype identified subsets of L-type calcium channel blockers and corticosteroids as novel therapeutically relevant drug classes with corresponding activity in neuronal cells. The screening results were validated by predicting in vivo efficacy in an independent schizophrenia cohort. The approach has the potential to discern new drug targets and accelerate drug discovery and personalized medicine for neuropsychiatric conditions.
INTRODUCTION
In few areas of postgenomic drug discovery is the disconnect between improved scientific resources and the lack of novel drug entities as devastatingly apparent as in the case of neuropsychiatric disorders (1). Despite their enormous burden on worldwide health, accounting for 31% of years lived with disability and a lifetime prevalence of up to 20% of the global population (2), drugs with novel mechanisms of action are lacking (1, 2). Derivatives of drugs found serendipitously over four decades ago (3), and thought to target brain monoamine receptors, are effective in only 40 to 60% of affected individuals (4, 5). This is largely due to a lack of understanding of the pathophysiology of neuropsychiatric diseases, incomplete characterization of the molecular targets of existing drugs, a scarcity of relevant preclinical models, and an inability to accurately predict clinical treatment response as a result of disease heterogeneity (1, 6).
Primary neuropsychiatric drug targets, such as the dopamine 2 receptor (DRD2) or glutamate receptor subunit genes (GRM3, GRIN2A, and GRIA1) in schizophrenia (SCZ), have only recently been linked to genetic risk of disease at the population level through large-scale, genome-wide association studies (GWAS) (7). However, polygenic risk scores currently explain only a fraction of disease liability, e.g., 7% in SCZ (4) relative to up to 80% heritability derived from family and monozygotic twin studies (8). Putative individual GWAS risk alleles account only for a marginal increase in disease risk with odds ratios typically under 1.1 and differences in allele frequencies between cases and controls less than 2% (4). The concept that each neuropsychiatric patient has a different combination of multiple common risk alleles, each with a small incremental contribution, has led to the use of in silico pathway analysis to identify cell signaling pathways that represent convergent drug targets (1, 4, 7). Although recent expression of quantitative trait loci and transcriptome-wide association studies have provided a means for prioritizing neuropsychiatric GWAS risk loci (9), there remains a formidable challenge in summarizing the functional implications of these studies relative to the molecular and cellular complexity of the human nervous system (7).
Furthermore, because of the impracticalities of accessing live human brain tissue, there is a lack of relevant and scalable cellular models in which to empirically test cell signaling alterations and their interactions with novel pharmacological probes on the genomic background of neuropsychiatric disease. Neurons derived from induced pluripotent stem cells (iPSCs) of neuropsychiatric patients have partially addressed this issue (10). While the cost of the iPSC screening technologies has decreased relative to increased throughput, challenges in end fate specificity, technical variation, and attrition of donor phenotypes continue to hinder the differentiation process (11) and limit the application of these models to comprehensive drug screening. In contrast, peripheral blood mononuclear cells (PBMCs) are a physiologically relevant and readily accessible primary tissue amenable to high-content functional analysis (12, 13). Recent work suggests that neuropsychiatric diseases are systemic disorders resulting from disruption of cell signaling networks with parallel manifestations in both the brain and peripheral tissues (14, 15). PBMCs express many central nervous system (CNS) receptors and downstream signaling proteins that are implicated in neuropsychiatric disorders (16). Subsets of protein alterations common to both cell lineages have been associated with disease status, symptom severity, and therapeutic efficacy (17–19). Furthermore, recent GWAS data suggest the enrichment of single-nucleotide polymorphisms (SNPs) associated with neuropsychiatric (SCZ) risk loci within PBMC subtype–specific gene expression enhancers (7) and the coexpression of neuropsychiatric genetic risk phenotypes in both peripheral and CNS tissues (20, 21). Although the functional correspondence between shared signaling motifs in PBMCs and CNS cells remains to be fully explored, it is possible that subsets of pathways, or even individual protein-protein interactions, which do overlap, are clinically relevant and can be practically exploited for drug discovery. Moreover, evidence that peripheral alterations can induce functional changes in the CNS with corresponding behavioral symptomatology [e.g., psychotic symptoms linked to gut microbiome alterations (22) or depressive-like symptoms following proinflammatory cytokine release during microbial infection (23)] suggests that peripheral cellular models may not solely be confined to surrogate identification of CNS targets but may also represent underlying pathophysiological mechanisms.
Exploration of neuropsychiatric patient PBMCs, with regard to functional alterations that reflect the disease state, could have important implications for overcoming current obstacles in neuropsychiatric drug discovery. First, the use of primary patient cells in the early stages of the drug discovery pipeline is associated with a higher success rate for putative targets and compounds (24). Second, the use of functional testing in live cells enables the elucidation of relevant disease-specific alterations in cell signaling networks, which are not observable simply by quantification of protein levels in their basal state, including homeostatic and regulatory mechanisms (13). Third, the focus on cellular responses provides the opportunity to summarize complex genetic risk factors with heterogeneous manifestations as integrated phenotypes, which are more amenable to drug screening and clinical treatment response prediction (25). Last, targeting of key cellular functional transducers, such as G protein–coupled receptors, receptor tyrosine kinases, ion channels, and protein kinases and phosphatases, represents a heuristic method for screening of the most “druggable” part of the genome (26).
Here, we apply high-content single-cell screening of functional ligand-receptor interactions and downstream signaling mechanisms (27, 28) in PBMCs from neuropsychiatric patients and controls ex vivo (Fig. 1). Using fluorescent cell barcoding (27), automated sample preparation, and multiparameter phospho-specific flow cytometry (28), we measure the activation of up to 78 intracellular signaling epitopes in parallel [spanning key CNS and immune cell signaling pathways such as Akt/GSK-3β (glycogen synthase kinase 3β), PKA (protein kinase A), PKC, MAPK (mitogen-activated protein kinase), JAK (Janus kinase)/STAT (signal transducers and activators of transcription), IL-1R (interleukin-1 receptor)/TLR (Toll-like receptor), and TCR/BCR (T/B cell receptor) signaling] in response to 70 functional ligands (including CNS receptor agonists, neuropsychiatric medications, cytokines, hormones, growth factors, antigens, and intracellular signaling modulators) in primary PBMCs. We use a mixture of endogenous ligands and exogenous cellular stimulants to provide a balance of physiological relevance, receptor subtype specificity, potency, and heuristic targeting of the maximum number of downstream signaling alterations in limited clinical samples. This generates readouts for a total of 5460 ligand-epitope combinations (nodes), or parallel functional assays, conducted in an individual PBMC sample. We explore the utility of this functional array for neuropsychiatric drug discovery in four stages. First, we define the kinetic profile of cell signaling network responses to CNS ligands and known neuropsychiatric treatments in PBMCs from control donors [kinetic profiling (KP); Fig. 1B]. Second, we identify cell signaling responses, which are altered in PBMCs from drug-naïve patients with SCZ relative to controls and are subsequently normalized by efficacious clinical treatment in vivo [drug target identification (TI); Fig. 1C]. Third, we use the disease-associated cell signaling responses as the basis for phenotypic screening of U.S. Food and Drug Administration (FDA)–approved and experimental drugs capable of normalizing the aberrant cellular responses in PBMCs and neurons [drug repurposing (DR); Fig. 1D]. Last, we validate the clinical significance of the screening results by correlating the cellular activity of a subset of compounds with their in vivo efficacy in an independent cohort of drug-naïve patients with SCZ [clinical validation (CV); Fig. 1E]. Together, our platform provides a novel patient-centric paradigm for performing pharmacological studies and drug discovery for neuropsychiatric disorders.
Fig. 1. Ex vivo CNS drug discovery pipeline.
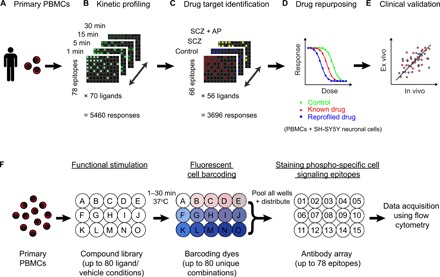
(A) Human primary PBMCs provide an accessible ex vivo model of physiological single-cell phenotypes in health and disease. (B) Time-course exploration of responses to 70 ligands (including CNS ligands and neuropsychiatric treatments) across 78 diverse cell signaling epitopes (5460 responses in total) in T cells from healthy control donors (n = 8) at 1, 5, 15, and 30 min ligand incubation times. (C) Identification of functional drug targets by comparing the T cell signaling response profiles of 56 ligands across 66 cell signaling epitopes (3696 responses) in PBMC samples from three clinical groups: healthy controls (n = 12), antipsychotic drug-naïve patients with SCZ (SCZ; n = 12), and the same patients following 6 weeks of clinical treatment with the atypical antipsychotic olanzapine (SCZ + AP; n = 10). (D) Modeling of disease-associated cellular responses and screening of U.S. Food and Drug Administration (FDA)–approved drugs (repurposing) and experimental neuropsychiatric compounds (n = 946 in total) in T cells from healthy control PBMC donors (n = 6 to 12) and human SH-SY5Y neuronal cells. (E) Validation of the ex vivo cellular model relative to in vivo clinical efficacy in an independent cohort of drug-naïve patients with SCZ (n = 30) treated with two of the study compounds. (F) Experimental design: PBMCs from each donor were distributed across a ligand library (A to O) and stimulated for 1 to 30 min at 37°C. Each ligand or vehicle well was stained with a unique combination of fluorescent barcoding dyes. Ligand wells were pooled and distributed across an array of antibodies (01 to 15) targeting specific phosphorylation or total protein sites on different intracellular signaling proteins. Changes in the activation status of the cell signaling proteins were analyzed at the single-cell level using flow cytometry for each donor individually. Assay plate dimensions are restricted to 15 wells for representation; full dimensions are detailed in brackets.
RESULTS
T lymphocytes as a surrogate model for kinetic drug interactions at CNS drug targets
We evaluated the potential of multiplexed phospho-specific flow cytometry (27, 28) for investigating signaling responses relevant to neuropsychiatric disorders in PBMCs in three stages. We focused on T lymphocytes (CD3+) as the most abundant (40 to 60%) PBMC subtype. First, we set up and tested the platform for reproducibility and dynamic range. We used induction of Stat3 (pY705) phosphorylation by IL-6 at 15 min as a model response [based on previously reported findings in phospho-specific flow cytometry (27, 28) and IL-6 serum alterations in SCZ (14)] and assessed whether cell signaling responses could be measured with precision independently of the position of the ligand within the barcoding matrix (n = 80 barcodes; figs. S1 and S2) and the antibody detection fluorophore (mean CV, 6%; mean Z′ factor, 0.65; figs. S3 and S4). Next, we assessed the variation in signaling responses across a subset of 3696 nodes (readouts from 56 compounds at 66 epitopes; tables S1 and S2) in PBMCs from the same healthy donor measured across six different experimental runs (mean CV per node, 11%; fig. S5A). Last, we compared functional responses in the above nodes between two independent cohorts of healthy PBMC donors from different collection sites (n = 8 and 12, respectively), which showed 75% overall replication of active nodes (100% replication for the JAK/STAT pathway and 85% replication for the Akt/GSK-3β pathway) and correlated fold changes (FCs; Spearman’s rank correlation coefficient rs = 0.94, P < 1 × 10−15; fig. S5B). Antibody stain indices [ratio of the median fluorescence intensity (MFI) in stained versus unstained vehicle condition across PBMC donors] at each epitope (fig. S6) were also reproduced between cohorts (rs = 0.96, P < 1 × 10−15; fig. S5C).
Among the selected time points [based on published data for prototypical T cell and CNS cell signaling responses (12, 28–31)] measured for the initial set of 5460 nodes (responses at 78 epitopes to 70 compounds at concentrations relevant to in vitro efficacy and clinical serum levels; Fig. 2, A and B, and tables S1 and S2), we observed maximal induction of significant cell signaling responses (P < 0.05, Wilcoxon rank sum test) at 30 min, where 228 of 5460 nodes were active, compared to 97, 146, and 211 nodes at 1, 5, and 15 min, respectively (Fig. 2, C and D, figs. S7 and S8, and tables S3 and S4). Although no time points longer than 30 min were tested, this kinetic profile is consistent with published data, which indicates maximal induction of phosphorylation of cell signaling proteins using conventional PBMC ligands at 30 min (12). The distribution of significant nodes with respect to ligand or epitope class was largely preserved across time points, such that 30 min represented the maximal induction of nodes for most classes within the time points tested (Fig. 2, C and D). Moreover, we observed widespread changes in cell signaling epitope expression for positive controls calyculin, phorbol 12-myristate 13-acetate (PMA)/ionomycin, thapsigargin, and staurosporine (table S3), in addition to dynamic regulation of key JAK/STAT signaling epitopes in response to immunomodulatory cytokines (fig. S9) at 30 min, consistent with published data (12, 31). Thus, we selected 30 min as the optimal time point of the four tested for the exploration of cell signaling nodes in clinical samples. To optimize use of the assay for clinical samples, in which cell numbers are limited, we excluded 14 ligands and 12 epitopes, which showed minimal activity at 30 min (tables S1 to S4), from the subsequent clinical study. This represented a reduction of the size of the functional matrix by a third and ensured that only the nodes with greater functionality, and hence greater potential for revealing disease-related drug targets, were carried forward to the clinical application of the platform.
Fig. 2. Kinetic distribution of T cell signaling responses across ligand and epitope categories.
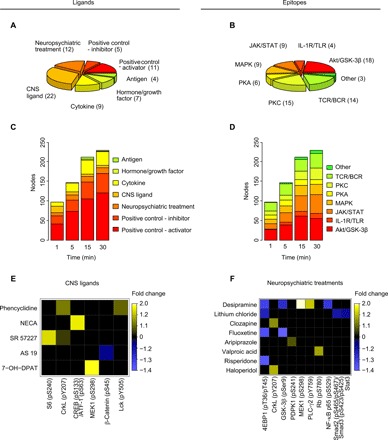
(A) Composition of the ligand array by class and (B) the epitope array by pathway. Number of ligands and epitopes per class is given in brackets. (C) Composition of the total number of significant responses (“nodes”; permuted P < 0.05, Wilcoxon rank sum test; minimum FC 10%) for each time point by ligand class and (D) by epitope pathway. (E) Significant kinetic responses (permuted P < 0.05, Wilcoxon rank sum test; sustained in the same direction for minimum two consecutive time points) to CNS ligands and (F) neuropsychiatric treatments. Legend shows the mean FC in epitope expression across significant time points, calculated per time point as median MFI of the ligand treatment/median MFI of the vehicle treatment across PBMC donors, with labels distributed evenly across the quantile range for negative and positive FCs separately. For down-regulated epitopes, legend shows −1/FC. Compounds and epitopes in the heat maps are ordered by the number of significant nodes across the time points. Targets of the CNS ligands include phencyclidine (an N-methyl-d-aspartate receptor antagonist/D2 receptor agonist/σ receptor agonist), NECA (5’-N-ethylcarboxamidoadenosine; an adenosine A1/2A/3 receptor agonist), SR 57227 (a 5-HT3 receptor agonist), AS 19 (a 5-HT7 receptor agonist), and 7-OH-DPAT (a D3 receptor agonist). Clinical indications of the neuropsychiatric treatments include desipramine and fluoxetine (antidepressants), lithium and valproic acid (mood stabilizers), and clozapine, risperidone, aripiprazole, and haloperidol (antipsychotics). All data represent eight healthy control PBMC donors. CREB, cyclic adenosine monophosphate response element–binding protein; PDPK1, 3-phosphoinositide-dependent protein kinase-1; MEK1, MAPK kinase 1; PLC-γ2, phospholipase Cγ2; Rb, retinoblastoma protein; NF-κB, nuclear factor κB.
Second, we evaluated the kinetic profiles of compounds known to interact with CNS pathways and compared them to the effects of established neuropsychiatric drugs and novel drug candidates. We focused on the Akt/GSK-3β pathway as one of the most widely implicated cell signaling pathways in neuropsychiatric drug discovery (32). The reference pathway activation range (i.e., maximum positive or negative FC in epitope expression in response to ligand stimulation) was defined using positive controls. Calyculin (a phosphatase inhibitor) and the PMA/ionomycin cocktail (a PKC activator/calcium flux inducer) caused widespread induction of the epitopes within the canonical signaling pathway, with incremental responses at later time points, while staurosporine (a nonselective protein kinase inhibitor) showed the opposite effect (Fig. 3A). We also examined the effects of specific inhibitors. GSK 690693 (a specific Akt inhibitor) inhibited phosphorylation downstream of Akt1 at GSK-3β (pS9), rapamycin [a specific mTORC1 (mammalian target of rapamycin complex 1) complex inhibitor] inhibited phosphorylation downstream of mTORC1 at 4EBP1 (pT36/pT45), and CHIR-99021 (a highly selective GSK-3β inhibitor) displayed inhibition of phosphorylation directly at the pS9 residue on GSK-3β (Fig. 3A).
Fig. 3. Kinetic exploration of neuropsychiatric treatments and novel inhibitors of the Akt/GSK-3β pathway in T cells.
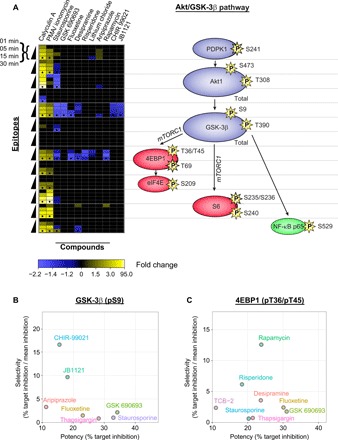
(A) Kinetic induction of responses (left) at key Akt/GSK-3β pathway epitopes (right) for positive controls (calyculin A, PMA/ionomycin, staurosporine, and GSK 690693) as compared to neuropsychiatric treatments (fluoxetine, desipramine, risperidone, lithium, and aripiprazole) and specific novel inhibitors (rapamycin, CHIR-99021, and JB1121). Only significant responses (permuted P < 0.05, Wilcoxon rank sum test; n = 8 healthy control PBMC donors) are shown. Black dots at 30-min time points represent replication in an independent PBMC cohort (n = 12). Legend shows FC in epitope expression (calculated as median MFI of the ligand treatment/median MFI of the vehicle treatment across PBMC donors), with labels distributed evenly across the quantile range for negative and positive FCs separately. FC is converted to −1/FC for down-regulated epitopes. Proteins are colored with respect to their cellular function: blue (kinase), red (translation), and green (transcription). The position of mTORC1 is shown for mechanistic interpretation, although no epitopes were measured on this protein. (B and C) Inhibition potency and selectivity across all 70 ligands used in the time course for targets (B) GSK-3β (pS9) and (C) 4EBP1 (pT36/pT45) at 30 min. Potency reflects the percentage of inhibition of phosphorylation at target site calculated as (1 − MFI of the ligand treatment/mean MFI of the vehicle treatment) × 100%, averaged across PBMC donors (n = 8). Selectivity reflects the ratio of the percentage of inhibition of phosphorylation at target site to mean percentage of inhibition across Akt (pS473), Akt (pT308), GSK-3β (pS9), and 4EBP1 (pT36/pT45) sites, averaged across eight donors. Only ligands with >10% potency are shown.
Several compounds used clinically to treat neuropsychiatric disorders displayed kinetic inhibition profiles, which overlapped with those described for GSK 690693, rapamycin, and CHIR-99021 (Fig. 3A). Antidepressant medications fluoxetine and desipramine showed significant inhibition of phosphorylation at both GSK-3β (pS9; maximum FCs, −1.48 and −1.35, respectively) and 4EBP1 (pT36/pT45; maximum FCs, −1.54 and −1.35, respectively). Antipsychotic medications aripiprazole and risperidone inhibited phosphorylation at either GSK-3β (pS9; maximum FC, −1.20) or 4EBP1 (pT36/pT45; maximum FC, −1.53), respectively. The mood stabilizer lithium exhibited phosphorylation inhibition at 4EBP1 (pT36/pT45; maximum FC, −1.25) but not at either of the GSK-3β phospho-epitopes. However, several of these psychoactive drugs also displayed inhibitory effects on phosphorylation at key sites of other Akt/GSK-3β signaling proteins. For example, lithium showed inhibition at PDPK1 (pS241; maximum FC, −1.05), aripiprazole at Akt (pT308; maximum FC, −1.09), and desipramine at nuclear factor κB p65 (pS529; maximum FC, −1.27). In contrast, JB1121, a member of the recently found series of highly selective tricyclic pyrazolo-tetrahydroquinolinone GSK-3 inhibitors, developed as candidates for treatment-resistant bipolar disorder and other neuropsychiatric disorders with dysregulated GSK-3 signaling (e.g., fragile X syndrome) (33), showed highly selective (−1.27- to −1.41-fold) phosphorylation inhibition of GSK-3β (pS9) across all the time points with no inhibition at any of the other selected pathway sites. This profile closely matched that of CHIR-99021, one of the most potent and selective GSK-3β inhibitors available (34). These data demonstrate multitarget modulation of known neuropsychiatric treatments, a property that is considered key to their therapeutic profile (35), in relevant CNS pathways ex vivo. Furthermore, it is possible to identify novel drug leads (CHIR-99021 and JB1121) or repurposing candidates currently indicated for other diseases (rapamycin) with enhanced specificity for putative neuropsychiatric target epitopes (Fig. 3, B and C).
Third, we examined the activities of CNS ligands and neuropsychiatric treatments across the full array of cell signaling epitopes. Kinetic screening of the cell signaling repertoire revealed 59 significant responses (FC, −1.28 to 1.35) for CNS receptor ligands at different time points. We prioritized the most robust responses as those with activity at a minimum of two consecutive time points in the same direction. Seven nodes fulfilled these criteria (Fig. 2E), including adenosine receptor agonist NECA/CREB [cyclic adenosine monophosphate (cAMP) response element–binding protein] (pS133)–ATF-1 (activating transcription factor 1) (pS63; mean FC, 1.25 for 5 to 30 min), D3 receptor agonist 7-OH-DPAT/MAPK kinase 1 (MEK1) (pS298; mean FC, 1.26 for 5 to 15 min), N-methyl-d-aspartate receptor antagonist phencyclidine/CrkL (pY207; mean FC, 1.10 for 1 to 5 min), and 5-HT3 receptor agonist SR 57227/S6 (pS240; mean FC, 1.15 for 15 to 30 min).
Similar prioritization of the cell signaling interactions outside of the Akt/GSK-3β pathway for drugs used in different neuropsychiatric indications revealed multiple kinetic responses in T cells, which reflect potentially novel or emerging neuropsychiatric drug targets (Fig. 2F). Both typical (haloperidol) and atypical (clozapine) antipsychotics increased the phosphorylation of CrkL (pY207; mean FCs, 1.12 and 1.11 for 1 to 5 min, respectively), a protein encoded by a gene within a SCZ risk locus (36) and previously unknown as a target of antipsychotic medication. In the case of the mood stabilizers, the specific induction of Rb (pS780) phosphorylation by valproic acid (mean FC, 1.11 for 15 to 30 min) is consistent with its role as a histone deacetylase inhibitor, a feature that distinguishes its putative mechanism of action from that of lithium (37). In contrast, the effect of lithium in decreasing Smad2 (pS465/pS467)/Smad3 (pS423/pS425) phosphorylation (mean FC, −1.16 for 15 to 30 min) is noteworthy in the context of the implication of Smad3 polymorphisms in antidepressant response latency (38). Similarly, the induction of MEK1 (pS298) phosphorylation by the antidepressant desipramine (mean FC, 1.90 for 5 to 30 min) is consistent with previous reports (39). The current data suggest that the above compound-epitope interactions reflect emerging mechanisms of action of these compounds in the CNS, which can be tracked in peripheral blood cells.
Drug target discovery using an in vivo antipsychotic intervention study design
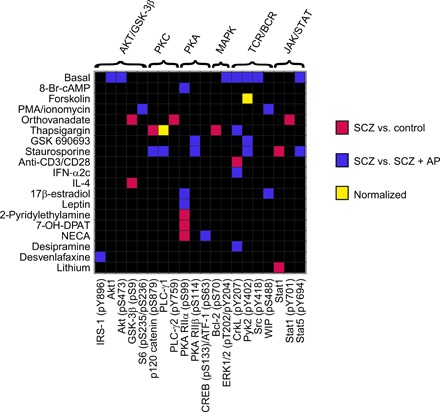
To test the suitability of the platform for neuropsychiatric drug target discovery, we used SCZ as a model disease and compared responses to 56 compounds at 66 epitopes at 30 min, in addition to 66 basal epitope levels (n = 3762 nodes in total; figs. S10 and S11 and tables S1 and S2), in T cells across three clinical groups—healthy controls (n = 12), antipsychotic drug-naïve patients with SCZ before treatment (n = 12), and the same patients with SCZ after 6 weeks of clinical treatment with the atypical antipsychotic olanzapine (n = 10; fig. S12 and table S5). Three hundred thirty-five nodes, including 48 basal epitopes and 287 ligand responses, satisfied the activity criteria of a minimum epitope stain index of two and, additionally, for ligand responses, a significant FC (P < 0.05, Wilcoxon rank sum test) of minimum 10% in at least one of the comparison groups. Of these, across both group comparisons (pretreatment SCZ versus healthy controls and pretreatment SCZ versus posttreatment SCZ), a total of 32 ligand responses and seven basal epitope levels were found to be significantly altered [P < 0.05, analysis of covariance (ANCOVA); Fig. 4, fig. S13, and tables S6 and S7], representing 1.0% of the conditions screened and 11.6% of active nodes. Notably, all the nodes significantly associated with disease in the pretreatment SCZ versus healthy control comparison and most of the nodes altered in the pretreatment versus posttreatment SCZ comparison (20 of 27 nodes) were functional responses to ligands, as opposed to changes in basal epitope expression. Six of the involved epitopes [CrkL (pY207), Pyk2 (pY402), Src (pY418), PKA RIIα (pS99), Akt1, and PLC-γ1 (phospholipase Cγ1)] have been previously shown to display altered functional connectivity in CD4+ T cells in SCZ (13). These results imply that there is a clinical representation of SCZ pathology and antipsychotic treatment effects within the T cell signaling repertoire, which is more readily observable in the presence of functional ligand stimulation.
Fig. 4. Target identification by functional profiling of cell signaling abnormalities in SCZ T cells.
The drug target was selected on the basis of the normalization of aberrant cell signaling responses following in vivo pharmacological antipsychotic therapy. Shown are clinically associated T cell signaling nodes (n = 39 of 3762 total nodes screened), defined as individual ligand-epitope combinations, which displayed a significant effect (permuted P < 0.05, ANCOVA) between clinical group status and either ligand response (n = 32) or basal epitope expression (n = 7). All nodes were filtered for a minimum stain index of 2 and, for ligand responses, a significant (permuted P < 0.05, Wilcoxon rank sum test) FC of minimum 10% in at least one of the comparison groups. Nodes altered in the SCZ versus healthy control (n = 12 PBMC donors per group) or SCZ (n = 12) versus SCZ + antipsychotic (n = 10) group comparisons are shown in red and blue, respectively. Nodes altered in both comparisons in opposite directions (i.e., “reversed”) are shown in yellow. The yellow nodes represent putative normalization of SCZ cell signaling alterations following efficacious clinical treatment (fig. S12) and form the basis for drug target selection. Nodes are grouped by signaling pathway (top) and ligand class (left). Refer to tables S6 and S7 and fig. S13 for a detailed group comparison. IRS-1, insulin receptor substrate 1; WIP, Wiskott-Adrich syndrome protein (WASP)-Interacting Protein.
To identify nodes representing potential drug targets, we assessed which of the cell signaling alterations present in pretreatment SCZ compared to healthy control samples were reversed (“normalized”) over the course of long-term clinical olanzapine treatment and were supported by existing large-scale genetic screening evidence. Of 14 cell signaling nodes associated with disease in the SCZ versus control comparison, we found two nodes (14%) to be reversed after clinical treatment (Fig. 4). Considering that the number of clinically associated nodes was 4.2% (14 of 335 nodes) and 8.1% (27 of 335 nodes) of the total active nodes assayed for SCZ versus control and olanzapine treatment comparisons, respectively, this represents a significant enrichment of nodes for which the signaling profile was reversed (P = 1.7 × 10−3). The reversal nodes included ligand responses to calcium mobilizer thapsigargin at PLC-γ1 and cAMP inducer forskolin at Pyk2 (pY402) and also remained significant after controlling for variables (smoking and alcohol consumption), which differed between the groups (table S5).
Thapsigargin/PLC-γ1 was the most significant ligand response altered in pretreatment SCZ compared to the healthy control group (P = 0.0001, Q = 0.018, two-way ANCOVA; table S6). Furthermore, these alterations were not due to existing differences in the levels of PLC-γ1, as basal expression of PLC-γ1 was not altered in either of the clinical group comparisons. This is in line with putative calcium signaling dysfunction in SCZ as suggested by GWAS and exome sequencing studies, which implicate a range of calcium signaling genes, including sarcoplasmic/endoplasmic reticulum calcium adenosine triphosphatase 2 (ATP2A2) and voltage-gated calcium channel subunit genes (e.g., CACNA1C, CACNB2, and CACNA1I), among the foremost SCZ risk loci (7, 40). Subsets of these calcium channel subunits are expressed in T cells at levels comparable to excitable cells such as neurons and have been shown to modulate responses to thapsigargin (41). For the calcium pump ATP2A2, which is a direct target of thapsigargin, we observed a significant association between the genome-wide significant SCZ risk SNP rs4766428 (C > T) in the ATP2A2 gene (7) and the attenuated response to thapsigargin at PLC-γ1 in patients (P = 0.049, linear regression; fig. S14), suggesting a point of convergence between the genetic and functional screening results. Mutations in ATP2A2 also cause Darier disease, which cosegregates with SCZ, bipolar disorder, and affective psychosis in some cases and families, and are associated with neuropsychiatric disease severity (20). Concurrently, PLC isotypes (PLCB2, PLCL1, and PLCH2) have been implicated as primary SCZ risk loci (7) with altered expression in the brain of patients with SCZ postmortem (PLCB1) and SCZ-like behavioral abnormalities in knockout animal models (PLCG1 and PLCB1) (42, 43). The response elicited by thapsigargin, increased cytosolic calcium concentrations and changes in PLC-γ1 expression, is common to many endogenous ligands and signaling mechanisms implicated in SCZ (44–46), including dopamine-glutamate interactions in the neuronal postsynaptic density (44, 47) and receptor tyrosine kinase signaling in response to neurotrophins (48, 49). These results implicate the thapsigargin/PLC-γ1 response as the primary novel drug target to emerge from the functional screening of cells of patients with SCZ (Fig. 5A and fig. S15).
Fig. 5. Phenotypic drug repurposing based on cellular response.
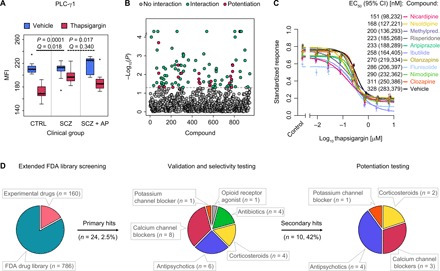
(A) Identification of functional cellular drug target. The attenuated response to thapsigargin at PLC-γ1 in T cells from drug-naïve patients with SCZ (permuted P = 0.0001, Q = 0.018, two-way ANCOVA), relative to healthy controls (CTRL), was reversed after 6 weeks of clinical treatment with the atypical antipsychotic drug olanzapine in vivo (permuted P = 0.017, Q = 0.340; n = 12 healthy controls, n = 12 drug-naïve patients with SCZ, and n = 10 patients with SCZ after antipsychotic treatment). Box plots show interquartile range with the median (horizontal line) and the minimum and maximum values (whiskers), excluding outliers (dots). (B) Results of primary drug screen. Permuted P values from thapsigargin drug interaction testing (two-way ANOVA; n = 6 to 12 healthy PBMC donors) are shown across the combined FDA-approved (n = 786) and experimental (n = 160) compound libraries. Dashed line represents threshold P value of 0.05. Significant hits are shown in green (n = 102), and compounds, which additionally showed selective potentiation of the thapsigargin/PLC-γ1 response (post hoc one-way ANOVA tests; fig. S18), are shown in magenta (n = 22; table S9). (C) Ranking of best selective potentiation candidates at 10 μM concentration in terms of half maximal effective concentration (EC50) shifts in the thapsigargin/PLC-γ1 dose-response curve. Shown are mean values from six healthy PBMC donors (points) with SEM (vertical bars) and fitted four-parameter logistic curves. The y axis represents the MFI standardized as a proportion of minimum and maximum values. Legend shows the EC50 values with 95% confidence intervals (CI). Methylpred., methylprednisolone. (D) Distribution of drug classes across repurposing stages. Extended FDA-approved library screening (left) refers to the primary compound screen at a single dose (20 μM unless otherwise specified in table S8) of compound (n = 946; B). Validation and selectivity testing (center) refer to dose-response titration (24 nM to 200 μM) and validation of selective potentiation candidates and structural class relatives (n = 24; fig. S20; n = 6 healthy PBMC donors). Potentiation testing (right) refers to the titration of thapsigargin (12.5 pM to 20 μM) in the presence of 10 μM concentration of validated compounds (n = 10; C). In vivo effect of olanzapine (A) was reproduced in vitro throughout.
Drug repurposing targeting disease-related functional cellular responses
On the basis of the functional response identified in SCZ patient T cells, we performed drug discovery and repurposing for SCZ in four stages (Fig. 5D). First, we modeled the attenuated T cell response to thapsigargin at PLC-γ1 ex vivo (figs. S16 and S17) and identified 102 compounds (of a total of 946; table S8) from the extended FDA-approved drug library that interacted with the thapsigargin/PLC-γ1 response [P < 0.05, two-way analysis of variance (ANOVA); Fig. 5B]. The top hits (P < 0.001, n = 18) were enriched for positive controls (calyculin, thapsigargin, and staurosporine) and calcium channel blockers (NNC 55-0396, penfluridol, loperamide, and nisoldipine), consistent with the established functions of the positive controls as broad-spectrum cell signaling modulators and the calcium channel blockers as mechanistic probes of calcium flux alterations. We subsequently identified a subset of 22 compounds, which displayed the selective potentiation profile (i.e., were inactive alone but potentiated the PLC-γ1 response selectively in the presence of thapsigargin; Fig. 5B, fig. S18, and table S9). These included nisoldipine as the top hit (P = 0.0002; 3.1-fold potentiation) and olanzapine (P = 0.009, ranked seventh; 2.9-fold potentiation), consistent with the clinical results in patients. The hits were also enriched for cephalosporin antibiotics (23%), corticosteroids (18%), and retinoids (9%). Eight of the initial 22 hits were validated in preliminary dose-response testing and were carried forward to the next stage, alongside 16 other members of the same therapeutic classes (calcium channel blockers, antipsychotic medications, corticosteroids, and antibiotics), which were included to explore potential structure-activity relationships (fig. S19).
In the second stage, using an extended dose-response range, we identified 10 compounds that showed selective potentiation of the PLC-γ1 response in the presence of thapsigargin, at concentrations below those that were active in the vehicle condition alone, indicating a potential therapeutic window (2.8 to 156.7 μM difference in 20% maximal inhibitory concentration, depending on the compound; fig. S20). The selective thapsigargin/PLC-γ1 potentiation profile of olanzapine was also shared by other atypical antipsychotic drugs (risperidone, aripiprazole, and clozapine), but not typical antipsychotics (haloperidol and penfluridol), consistent with differential effects of typical versus atypical antipsychotics on thapsigargin-induced calcium mobilization from the endoplasmic reticulum in vitro (46). Selective potentiation was also observed for 1,4-dihydropyridine (DHP) L-type calcium channel blockers with extended ester substitutions at the 3-position of the pyridine ring (nimodipine, nisoldipine, and nicardipine) but not for the other members of the DHP class (no effect for nilvadipine; potentiation without therapeutic window for nifedipine; opposing effects for amlodipine), the phenylalkylamine L-type calcium channel blocker (verapamil), or highly specific T-type calcium channel blockers (NNC 55-0396 and penfluridol). The last three compounds, which showed the selective potentiation profile, were the corticosteroids, methylprednisolone and flunisolide, and the potassium channel blocker, ibutilide. Three compounds showed a broader selectivity window than any of the measured antipsychotic drugs (maximum of 20.8 μM for olanzapine), namely, methylprednisolone (156.7 μM), nisoldipine (43.4 μM), and ibutilide (37.1 μM).
In the third stage, we ranked the compounds that showed the selective potentiation profile in terms of their potency to shift the half maximal effective concentration (EC50) of thapsigargin/PLC-γ1 dose-response curves at 10 μM concentration, on average 10 times higher than therapeutic plasma levels (50), to ensure adequate dosing in the 30-min time window and account for serum protein drug binding. Consistent with the previous results, all 10 drugs decreased the EC50 of the thapsigargin/PLC-γ1 response compared to the vehicle condition (Fig. 5C). The second-generation antipsychotics displayed a rank order of potency ranging from risperidone (EC50, 223 nM) to aripiprazole (233 nM), olanzapine (270 nM), and clozapine (311 nM). Furthermore, three drugs, namely, nicardipine (151 nM), nisoldipine (168 nM), and methylprednisolone (200 nM), showed stronger potentiation of the response than any of the measured antipsychotic medications. The effect of ibutilide (258 nM), flunisolide (286 nM), and nimodipine (290 nM) was within the range of the atypical antipsychotic medications.
Last, we validated the activity of the top two novel drug candidates (nisoldipine and nicardipine), in addition to the original clinical compound (olanzapine) as a positive control, at 10 μM concentration in βIII-tubulin–positive human SH-SY5Y neuronal cells (fig. S21). We observed potentiation of the thapsigargin/PLC-γ1 response (circa EC50; fig. S21) for all three compounds (Fig. 6) with similar (nicardipine, 2.1-fold; nisoldipine, 2.4-fold) or greater (olanzapine, 4.3-fold) FCs relative to those observed in PBMCs. Together, these results demonstrate the potential of the ex vivo platform to functionally stratify current SCZ medications, in terms of their ability to ameliorate disease-associated cellular responses, and, moreover, identify structurally diverse novel drug indications with activity in neuronal cells.
Fig. 6. Efficacy of top SCZ drug candidates in human SH-SY5Y neuronal cells.

(A) Response to 30-min sub-EC50 (40 nM; fig. S21B) thapsigargin stimulation at PLC-γ1 (yellow) in SH-SY5Y neuronal cells after a 45-min preincubation with the vehicle, top drug candidates (nisoldipine and nicardipine), or olanzapine (positive control) at 10 μM. Blue, 4′,6-diamidino-2-phenylindole (DAPI) (nuclei); magenta, βIII-tubulin (neuronal lineage marker). Scale bars, 100 μm; 60× magnification. Representative of three replicate experiments imaged using confocal microscopy at identical laser power and gain settings. (B) High-throughput wide-field microscopy analysis of (A) in SH-SY5Y cells positive for βIII-tubulin expression (fig. S21A). The y axis shows PLC-γ1 MFI standardized to the median control level in the vehicle condition. Boxes show median and interquartile range, whiskers extend to extreme values within 1.5 × interquartile range, and dots mark data points outside this range. Each box plot represents data from an average of 8126 cells from three replicate experiments. Top 0.5% values lie outside of the plot area and were included in statistical analysis. P values from two-way ANOVA for interaction between the drug condition and the thapsigargin/PLC-γ1 response; FC represents potentiation of the thapsigargin/PLC-γ1 response in drug relative to control conditions and is calculated as (1 − response ratio in drug condition)/(1 − response ratio in control condition). Wide-field images were acquired at the same light source and detector settings at 20× magnification.
Clinical drug target validation using ex vivo functional cellular responses
To investigate the relevance of the functional drug target (thapsigargin/PLC-γ1) in relation to changes in psychopathology in patients and also explore its potential for personalized medicine applications, we correlated the ex vivo activity of two of the atypical antipsychotics identified in the DR stage (aripiprazole and risperidone) with their in vivo efficacy in a cohort of drug-naïve patients with SCZ who were longitudinally followed up over their treatment course (n = 30; table S10). Similar to the responses in PBMCs from healthy control donors, both compounds potentiated the thapsigargin/PLC-γ1 response at 10 μM in T cells from drug-naïve patients with SCZ ex vivo (Fig. 7). Furthermore, the shift in thapsigargin/PLC-γ1 EC50 evoked by each compound in T cells from patients before clinical treatment (0 weeks) was significantly associated with, and predictive of (P = 0.001 and 0.041, Q = 0.007 and 0.213, linear regression model; table S11), subsequent changes in positive symptomatology [Scale for the Assessment of Positive Symptoms (SAPS)] after 3 and 6 weeks of the respective drug administration in vivo (n = 12 aripiprazole, n = 18 risperidone; Fig. 7 and fig. S22A). The ex vivo readouts at 0 weeks for either drug were not associated with changes in negative symptomatology at either of these clinical time points (fig. S22, B to D), consistent with the clinical profile of atypical antipsychotics that are focused mainly on the amelioration of positive (“psychotic”) symptoms. This is also in line with the reported role of PLC-γ1 dysfunction in behavioral changes linked to positive symptoms in animal models of neuropsychiatric diseases (42, 43). Other compounds identified in the DR stage, including novel candidate compounds (nisoldipine, nicardipine, and methylprednisolone) and an antipsychotic compound (olanzapine), also shared the ex vivo activity profile of the clinical drugs (aripiprazole and risperidone) in PBMCs of drug-naïve patients with SCZ (fig. S23). Together, these findings suggest that the activity at the putative functional drug target identified in PBMCs is associated with clinical efficacy in treating characteristic subsets of SCZ symptoms in vivo. This supports the potential utility of the novel candidate compounds, which share the same mechanism of action at thapsigargin/PLC-γ1, and raises the possibility of treatment response prediction for drugs that are currently indicated for SCZ.
Fig. 7. Correlation of ex vivo drug-target activity with in vivo efficacy.

(A) Longitudinal clinical patient assessment of the in vivo efficacy of two antipsychotic medications (aripiprazole and risperidone), identified in the drug-screening phase (Fig. 5), on SCZ symptoms assessed using the SAPS in drug-naïve patients with SCZ (n = 12 and 18 for aripiprazole and risperidone treatments, respectively) between treatment time points at 0 weeks (w) and 12 months (m). Treatments were equally efficacious (P > 0.05, two-way ANOVA). Patients remained on the same treatment at least until the 3-month time point. (B) Correlation between the ex vivo efficacy of the two antipsychotic medications (aripiprazole and risperidone), measured as the shift in EC50 (ΔEC50) of the thapsigargin/PLC-γ1 response induced by the respective drug in PBMCs collected at baseline from individual patients with SCZ, and the in vivo efficacy in ameliorating positive symptoms (ΔSAPS) after 3 weeks of clinical treatment in the same patients. Linear regression model with 95% confidence intervals, adjusted for covariates selected in stepwise procedure using Bayesian information criterion.
DISCUSSION
Serendipitous clinical observations and structural improvement of existing treatments have been driving the field of neuropsychiatric drug discovery for decades and are currently approaching their limits (1–3). In this work, using an objective high-content screening approach for the functional characterization of signaling network responses in primary T cells ex vivo, we build on dynamic disease-related cellular phenotypes as a basis to explore novel therapeutic opportunities within the repertoire of approved drugs. The current findings have a number of potential implications for improved neuropsychiatric drug discovery and personalized medicine.
The elucidation ex vivo in primary patient samples of convergent abnormal cell signaling response phenotypes (“functional endophenotypes”) between neuropsychiatric patients with potentially varied genetic risk factors and environmental trajectories is the primary contribution of this work. It provides a tractable and accessible live cell model for de novo identification of drug candidates with novel mechanisms of action even when the pathological process is not fully understood. This model is amenable to DR such that therapeutic candidates with documented pharmacokinetic and safety profiles, for example, those presented for SCZ (Fig. 5C), can be readily validated in clinical trials. It also raises the possibility that the ex vivo cellular response phenotype could be used not only as a drug target but also as a companion biomarker predicting clinical efficacy in patient subpopulations (Fig. 7). The model helps to refine the generic indications suggested by GWAS and exome sequencing studies (such as T- and L-type calcium channel blockade for the treatment of SCZ) (7, 40, 51) by ranking which therapeutic class members best recapitulate the cellular hallmarks of in vivo efficacy. This information could prove useful for interpreting the heterogeneous clinical trial results obtained previously for calcium channel blockers in SCZ (51). While these clinical trials have shown potential therapeutic benefits of DHP L-type calcium channel blockers (51), the current results raise the possibility that the structural derivates with extended ester substitutions at the 3-position of the pyridine ring (nisoldipine and nicardipine), which have not yet been tested, could provide enhanced efficacy. This is supported by their in vitro activity in neurons (Fig. 6) and initial studies of brain penetrance and the reversal of aberrant behavior in the phencyclidine animal model of SCZ using these compounds (52, 53). Furthermore, the cellular response phenotype serves to identify compounds from structurally unrelated drug classes that show similar effects on cell signaling, for example, corticosteroids (methylprednisolone and flunisolide) or a potassium channel blocker (ibutilide), thus expanding the mechanistic diversity of neuropsychiatric drug candidates at early stages in the drug discovery pipeline. Last, it provides potential insight into the functional implications of interactions between multiple common but weak (7) and rare but penetrant (36, 40) genetic risk loci at a time when the majority of heritable genetic risk has yet to be determined (4).
The second contribution of this work is the identification in peripheral T cells of differential interactions of neuropsychiatric medications at multiple cell signaling targets possibly predictive of CNS efficacy. The results indicate mechanistic points of convergence between neuropsychiatric drugs with different indications [e.g., antidepressants, mood stabilizers, and antipsychotics at 4EBP1 (pT36/pT45); Fig. 2F] and divergent activities of neuropsychiatric drugs within the same indication [e.g., mood stabilizers such as lithium at 4EBP1 (pT36/pT45) and valproic acid at Rb (pS780) (Fig. 2F) or typical versus atypical antipsychotics at thapsigargin/PLC-γ1 (fig. S20)]. This, together with individual variation in drug sensitivity between patients (Fig. 7A), raises the possibility of treatment response prediction through ex vivo testing of the drugs that are administered clinically (Fig. 7B and fig. S22A). This has the potential to circumvent the challenges of therapeutic uncertainty and the identification of nonresponders early in the treatment course (3, 4). Moreover, the results emphasize the importance of associating putative drug target efficacy with changes in specific subsets of clinical symptomatology, both in terms of characterizing the drug target itself and determining its predictive value for personalized medicine.
While several of the reported neuropsychiatric drug-target interactions were already known, for example, at Akt/GSK-3β pathway epitopes (Fig. 3A) (32), others such as the induction of phosphorylation at CrkL (pY207) in response to antipsychotic administration ex vivo (Fig. 2F) and in vivo (Fig. 4) have not been previously reported, despite the emerging implication of the CRKL locus in genetic risk for SCZ (36). It is possible that these “off-target” effects may eventually be recognized as key components of therapeutic efficacy or alternatively toxicity. Therefore, their identification at early stages of the drug development pipeline could improve the efficiency of future drug discovery efforts. Moreover, the differential cell signaling signature of the approved medications may serve as a mechanistic scaffold to identify novel experimental drugs or repurposing candidates (e.g., JB1121 or rapamycin, respectively; Fig. 3). The ability to resolve pharmacological effects discretely at different sites in the cell signaling network (Fig. 3) also raises the possibility of exploiting synergistic interactions between highly specific ligands, such as CHIR-99021 (GSK-3β inhibitor) and rapamycin (mTORC1 inhibitor), to provide personalized combinatorial therapeutic options for neuropsychiatric disorders as a means to overcome treatment resistance (54).
This work aims to provide a proof-of-principle application of the novel drug discovery pipeline to neuropsychiatric disorders. However, there are several limitations relevant to future work. First, some of the cell signaling pathways interrogated in this screen are likely to respond differently in T cells versus CNS cells, consistent with cell lineage functional specificity. The aim of the study is therefore not to suggest that blood cells show an identical functional repertoire to neuronal or glial cells but to show that a subset of the pathways, or even individual protein-protein interactions, which do overlap, are clinically relevant and can be practically exploited for drug discovery and treatment response prediction. Given the high number of nodes afforded by high-content screening, even a small proportion of functional overlap could serve to identify a substantial number of novel targets that would, otherwise, not be available for functional exploration in primary neuropsychiatric patient cells. Nevertheless, as further ex vivo targets emerge, these will need to be carefully evaluated in CNS target tissues. Comparison of cell signaling responses between PBMCs and iPSC-derived CNS cells from the same patients offers a relevant opportunity in this respect. Moreover, although exogenous cellular stimulants have advantages in terms of receptor subtype specificity, potency, and heuristically targeting the maximum number of downstream signaling alterations in a limited clinical sample, disease-associated nodes identified using this approach require further characterization in the context of endogenous ligand responses. Second, the activity of second-generation antipsychotics at the target node, although not essential to the validity of the platform, provides a useful positive control to support the initial target discovery, the drug screening candidates, and the clinical potential of the platform in terms of in vivo efficacy. It also helps to refine the targets of existing drugs while providing therapeutic candidates with divergent mechanisms of action, increased potencies, or reduced side effects. However, future studies would benefit from focusing on pretreatment differences in normal versus disease samples to identify targets that do not overlap with existing treatments. In this respect, the application of the platform to subgroups of patients with refractory symptom subtypes (e.g., cognitive symptoms) is essential to identifying drug candidates with long-term clinical superiority. Third, diagnostic uncertainty, disease heterogeneity, and dimensional overlap between related neuropsychiatric disorders (1, 2, 55) are persistent limitations in the current diagnostic framework used to define clinical groups. Nevertheless, as biological markers that reflect the etiology of neuropsychiatric disease (such those suggested by the present findings) are validated clinically, these diagnostic tools can potentially be improved in future studies. In this respect, it is notable that aberrant cellular responses to thapsigargin (56), associated genetic mutations in ATP2A2 (20), and manic-like behavioral changes following PLC-γ1 knockout in animal models (43) have also been reported in the context of bipolar disorder, suggesting that the identified primary drug target might show transdiagnostic potential for the treatment of related psychotic disorders. Fourth, the modest sample sizes used in the target identification study reflect difficulties in obtaining viable PBMCs from strictly defined drug-naïve first-onset neuropsychiatric patients, controlled for treatment status, and combined with a longitudinal in vivo treatment follow-up. Despite validation of the principal drug target in an independent cohort, future studies will benefit from the application of the ex vivo methodology in more extensive patient groups. Last, despite indications of efficacy in animal models (52, 53), a more extensive validation of the DR candidates is necessary to determine their clinical utility.
The present study represents, to our knowledge, the most extensive (3696 responses) functional characterization of cell signaling abnormalities in live primary cells from neuropsychiatric patients. In addition to providing a conduit for novel drug discovery and clinical treatment response prediction, in the form of phenotypic screening, it allows the reevaluation of the concept that neuropsychiatric disorders involve composite dysregulation of whole canonical signaling pathways. Instead, current data suggest that cell signaling abnormalities are more complex and that aberrant responses appear sporadically, even in the case of broad spectrum signaling activators, at individual proteins within a tightly regulated network. The identification of specific stress points within these networks, which escape regulatory control (e.g., PLC-γ1), might ultimately prove more relevant for drug target discovery. In this respect, the incorporation of technologies such as mass cytometry (12, 31), high-dimensional computation [e.g., spanning-tree progression analysis of density-normalized events (57) and visualization by t-distributed stochastic neighbor embedding (58)], and combinatorial compound libraries (59) within the present workflow represents an unprecedented opportunity to explore causal relationships between cell signaling proteins and enhanced drug targets.
MATERIALS AND METHODS
Experimental design
The objective of this study was to identify functional drug targets in SCZ using patient primary blood cells and high-content phospho-specific flow cytometry and subsequently use the identified targets as a basis for DR and clinical treatment response prediction. This work was performed in four stages (Fig. 1). First, samples from eight healthy donors were analyzed in a time-course exploration of T cell responses to 70 ligands across 78 cell signaling epitopes (5460 responses) to select the most active nodes and time point (KP; Fig. 1B). Next, responses to a selection of 56 ligands across 66 cell signaling epitopes (3696 in total) were compared across three clinical groups [healthy controls (n = 12), antipsychotic drug-naïve patients with SCZ (n = 12), and the same patients following 6 weeks of clinical treatment with the atypical antipsychotic olanzapine (n = 10)] to identify functional SCZ drug targets (TI; Fig. 1C). The identified target, attenuated response to thapsigargin at PLC-γ1, was then modeled in blood cells from healthy control donors (n = 6 to 12; limited clinical sample availability did not allow the use of patient PBMCs at this stage) and used for screening of 946 FDA-approved drugs (repurposing) and experimental neuropsychiatric compounds (extended FDA-approved DR; Fig. 1D), in addition to hypothesis-driven genotype association analysis between selected SCZ risk genes and target response. Last, the candidate compounds were validated in the SH-SY5Y human neuronal cell line (n = 3 replicates) and blood cells from an independent cohort of drug-naïve patients with SCZ (n = 30) treated with two of the study compounds, aripiprazole and risperidone, to investigate the relationship between the ex vivo and in vivo drug efficacy (CV; Fig. 1E). Methods relevant to specific substudies (KP, TI, DR, and CV) are indicated by abbreviations. All samples were randomized across different plate positions and experimental days to minimize measurement-related batch effects. To allow randomization, investigators were not blinded to sample characteristics. Samples in which the lymphocyte gate contained less than 30% of events (e.g., because of low viability; one sample in the TI study) were excluded from statistical analysis. Data permutation procedures (n = 10,000 permutations) were applied to control for small sample sizes, non-normal distribution of data, presence of outliers, and simultaneous testing of multiple hypotheses. Sample sizes were based on the availability of viable PBMCs from strictly defined drug-naïve first-onset patients with SCZ, controlled for treatment status and with longitudinal in vivo treatment follow-up information available, as well as previous studies (12, 31). At 12 versus 12 and with a 20% coefficient of variation, the TI study was sufficiently powered (0.8) to detect a 23% difference between the groups at α = 0.05.
Clinical sample recruitment
For the KP and DR studies, healthy control PBMC donors were recruited at the Cambridge Centre for Neuropsychiatric Research (CCNR), University of Cambridge, UK. For the TI study, first-onset antipsychotic drug-naïve male patients with SCZ before (n = 12) and the same patients after (n = 10) 6 weeks of treatment with the atypical antipsychotic medication olanzapine (10 to 20 mg/day), in addition to matched controls (n = 12), were recruited at the Department of Psychiatry, Erasmus Medical Centre, Rotterdam, the Netherlands (table S5). For the CV study, first-onset antipsychotic drug-naïve patients with SCZ (n = 30) were recruited at the University Hospital Marqués de Valdecilla, Santander, Spain (table S10). These patients were subsequently treated with the antipsychotic medication aripiprazole (n = 12; 5 to 30 mg/day) or risperidone (n = 18; 2 to 4 mg/day). The medical faculty ethical committees responsible for the respective sample collection sites approved the study protocols. Informed consent was given in writing by all participants, and clinical investigations were conducted according to the Declaration of Helsinki and Standards for Reporting of Diagnostic Accuracy.
Diagnoses of neuropsychiatric pathology were conducted by experienced psychiatrists and were based on the Diagnostic and Statistical Manual of Mental Disorders–IV–Text Review. The severity of symptom subtypes in SCZ was measured using the Positive and Negative Syndrome Scale (TI) or the SAPS and the Scale for the Assessment of Negative Symptoms (CV). The exclusion criteria for patients and controls included as follows: additional neuropsychiatric diagnoses other than SCZ, other neurological disorders including epilepsy, mental retardation, multiple sclerosis, immune/autoimmune disorders, infectious disease, metabolic disorders including diabetes, obesity, cardiovascular disease, hepatic and renal insufficiency, gastrointestinal disorders, endocrine disorders including hypo-/hyperthyroidism and hypo-/hypercortisolism, respiratory diseases, cancer, severe trauma, substance abuse including psychotropic drugs and alcohol, somatic medication with brain side effects, and somatic medication affecting the immune system including glucocorticoids, anti-inflammatory/immunomodulating drugs, and antibiotics.
PBMC isolation and culture
PBMCs were prepared within 4 hours from blood collected into 7.5-ml sodium heparin tubes (BD Biosciences; KP, TI, and DR) or acid citrate dextrose solution A tubes (BD Biosciences; CV). Whole blood was pelleted at 500g for 10 min to remove platelet-rich plasma, diluted 1:1 with Dulbecco’s phosphate-buffered saline solution (PBS; Sigma-Aldrich), and centrifuged over Ficoll-Paque PLUS (GE Healthcare) at 750g for 20 min at 23°C. PBMCs were extracted from the interphase, washed three times with PBS at 300g for 10 min, and cryopreserved at 5 × 106 cells/ml in heat-inactivated fetal bovine serum (FBS; Sigma-Aldrich) containing 10% dimethyl sulfoxide (DMSO; Sigma-Aldrich). For cell culture, PBMCs were thawed at 37°C and resuspended in sterile conditions in complete RPMI media with deoxyribonuclease (DNAse) [RPMI-1640 with sodium bicarbonate (Sigma-Aldrich), 10% FBS (Life Technologies), penicillin (50 U/ml) and streptomycin (50 μg/ml; Life Technologies), 2 mM l-alanyl-l-glutamine dipeptide (Life Technologies), and DNAse (20 μg/ml; Sigma-Aldrich)]. The cells were counted using a Coulter counter (Beckman Coulter), pelleted, and resuspended at 1 × 106 cells/ml. The cells were rested for 24 hours at 37°C/5% CO2 either in cell culture flasks (Corning; KP and TI) or, via a 40-μm cell strainer (Corning), in 96-well polypropylene plates (StarLab; DR and CV). Samples from participants from different clinical groups, alongside quality control (QC) samples from healthy control donors, were randomized across different plate positions and experimental days to minimize measurement-related batch effects.
Preparation of functional ligands and compound libraries
Ligands used to stimulate PBMCs (including cell signaling activators/inhibitors and receptor agonists/antagonists) were purchased from Sigma-Aldrich, Tocris/Bio-Techne, eBioscience/Affymetrix, R&D Systems, Life Technologies, Abcam, Antibodies-online, and Enzo Life Sciences. CHIR-99021 and JB1121 were synthesized in-house following published protocols (33). Table S2 lists the ligands used with their primary mechanisms of action and final assay concentrations (selected on the basis of published in vitro efficacy data and maximal clinical serum concentrations in the presence of serum proteins where relevant). Stock solutions of ligands were prepared in sterile conditions. Initial solubilization was achieved using DMSO where possible and, alternatively, PBS, H2O, or H2O with equimolar NaOH as per the manufacturer’s instructions. Intermediate dilutions were made in PBS, and DMSO was added to equivalent amounts for each ligand and vehicle.
The FDA-approved compound library (v.2.0; Enzo Life Sciences) was extended to incorporate experimental compounds contributed by collaborators [L. Jones-Brando, R. Yolken, G. H. Posner, J. G. D’Angelo, and C. P. Hencken (Johns Hopkins University School of Medicine, Baltimore, MD, USA); J. McNulty (McMaster University, Hamilton, Ontario, Canada); F. F. Wagner and E. B. Holson (Broad Institute of MIT and Harvard, MA, USA); T. L. Petryshen and S. J. Haggarty (Massachusetts General Hospital, Boston, MA, USA); and R. S. Williams (Royal Holloway University of London, Egham, UK)] and also positive controls/neutraceuticals, related to putative drug targets, from the CCNR compound library (table S8). Initial solubilization was conducted using DMSO where possible and, alternatively, PBS or H2O as per the manufacturer’s instructions. Intermediate dilutions were prepared using complete RPMI media without penicillin-streptomycin [RPMI-1640 with sodium bicarbonate (Sigma-Aldrich), 10% FBS (Life Technologies) to account for physiological serum protein binding of drugs, and 2 mM l-alanyl-l-glutamine dipeptide (Life Technologies)]. DMSO was added to equivalent amounts across all compounds and the vehicle. The final assay concentration of most of the compounds in the extended FDA-approved library was 20 μM with some exceptions in the compounds from collaborators and the CCNR library (table S8). All stocks and dilutions of stimulants and compounds were stored at −80°C, and repeated freeze-thaw cycles were avoided.
Stimulation of PBMCs
Stimulation is defined broadly as the exposure of PBMC cells to a ligand that has the potential to perturb resting-state cell signaling dynamics by either increasing or decreasing the expression of cell signaling epitopes. PBMCs were pelleted and resuspended [via 30-μm cell strainer (Partec) for KP and TI studies] in 96-well polypropylene plates using complete RPMI media without penicillin-streptomycin at 2 × 106 cells/ml (KP), 0.75 × 106 cells/ml (TI), and 0.4 × 106 cells/ml (DR and CV). The cells were rested for 75 min (KP and TI) or 45 min (DR and CV) at 37°C before ligand exposure. For the DR and CV studies, this resting period was used to preincubate the cells with the extended FDA-approved library compounds and screening hits, respectively, before stimulation. A standardized sample amount of 5.25 × 106 PBMCs per donor was used in the TI study, and no significant differences were observed in T cell frequencies between the control (67.1 ± 12.9%), pretreatment (72.0 ± 11.9%), and posttreatment (74.0 ± 9.8%) SCZ groups (P > 0.27; Wilcoxon rank sum and signed-rank tests).
Stimulants and vehicle were reconstituted in complete RPMI media without penicillin-streptomycin and added to the cells using a Biomek NX liquid handler (Beckman Coulter) with an integrated compact shaker-heater-cooler system (Inheco). The final concentrations of DMSO in all conditions including vehicle were 0.1% (KP and TI) and 0.2% (DR and CV). Vehicle wells represented one-eighth of the total wells assayed (KP and TI), one-sixth of the total wells assayed (DR), or one-quarter of the total wells assayed (CV) and were spaced evenly across each 96-well plate. The cells were exposed to the stimulants at 37°C for 1, 5, 15, and 30 min (KP) or 30 min (TI, DR, and CV). Ligand exposure was halted by fixation for 10 min at 37°C using paraformaldehyde (Sigma-Aldrich) in PBS at a final concentration of 1.6%.
Fluorescent cell barcoding
Stock solutions of barcoding dyes CBD 450 (BD Biosciences), CBD 500 (BD Biosciences), and DL800 (Thermo Fisher Scientific) were prepared as per the manufacturer’s instructions in DMSO in polypropylene 96-well plates and stored at −80°C. Different combinations of these dyes were used to produce a different number of barcoded cell populations specific to each study as follows. For the KP study, 80 populations were resolved using final concentrations of CBD 450 (0.000, 0.015, 0.075, and 0.300 mg/ml) combined with CBD 500 (0.000, 0.038, 0.188, and 0.750 mg/ml) and DL800 (0.000, 0.011, 0.033, 0.100, and 0.300 μg/ml; figs. S1 and S2). For the TI study, 64 barcoded populations were resolved using final concentrations of CBD 450 (0.000, 0.015, 0.050, and 0.150 mg/ml) combined with CBD 500 (0.000, 0.038, 0.125, and 0.375 mg/ml) and DL800 (0.000, 0.017, 0.050, and 0.150 μg/ml; fig. S10). For the DR study, four populations were resolved using final concentrations of DL800 (0.000, 0.017, 0.050, and 0.150 μg/ml; fig. S16). Cells in the CV study were not barcoded and treated with the vehicle (PBS) instead.
Fixed cells were washed with PBS and permeabilized in 100 μl of ice-cold methanol (Fisher Chemical) for 20 min at 2°C using a Biomek NX liquid handler to allow penetration of antibodies against intracellular epitopes. The barcoding dyes were diluted in ice-cold PBS and 100 μl per well added to the suspension of cells in methanol. The final concentration of DMSO from the barcoding dyes at this stage was 3.5%. The barcoding reaction was incubated in the dark for 30 min at 2°C, and the cells were washed five (KP and TI) or four times (DR and CV) in ice-cold fluorescence-activated cell sorting (FACS) buffer [PBS with 0.5% bovine serum albumin (Sigma-Aldrich)]. The barcoding wells were pooled, washed, and resuspended at 1 × 106 cells/ml in FACS buffer for staining.
Intracellular staining of cell signaling epitopes in PBMC subsets
The suspension of fixed, permeabilized, barcoded cells was stained using either anti-human CD3 (UCHT1) phycoerythrin (PE)–cyanine 7 (Cy7) (0.5 μl/ml; eBioscience; KP and TI) or anti-human CD3 (UCHT1) allophycocyanin (APC) (1 μl/ml; eBioscience; DR and CV) and distributed across a 96-well polypropylene plate. The cells were stained using a Biomek NX liquid handler (Beckman Coulter) with fluorescently conjugated anti-human antibodies against intracellular signaling epitopes (table S1) for 45 min in the dark at room temperature, as per the manufacturer’s instructions. Antibodies were purchased from BD Biosciences, Cell Signaling Technology, Merck Millipore, and Bioss. Antibodies against intracellular epitopes were used either individually (KP, DR, and CV) or in groups of three antibodies per plex (TI). The cells were washed twice and resuspended in FACS buffer at 1 × 106 cells/ml for acquisition.
Immunophenotyping and cell viability
PBMCs were resuspended at 1 × 106 cells/ml in FACS buffer with 20% human Fc receptor–binding inhibitor (eBioscience) and incubated for 20 min at room temperature in 5-ml polystyrene FACS tubes (BD Biosciences). The PBMCs were stained in a total volume of 135 μl per tube with 0.5 μl of anti-human CD3 (UCHT1) PE-Cy7 (eBioscience), 0.5 μl of anti-human CD4 (SK3) PerCP-eFluor 710 (eBioscience), 0.5 μl of anti-human CD8 (SK1) APC-eFluor780 (eBioscience), and anti-human CD14 (MφP9) V500 (BD Biosciences). The cells were incubated in the dark for 30 min at room temperature. They were washed twice with 3 ml of FACS buffer and resuspended in 0.5 ml of FACS buffer with 1 μM 4′,6-diamidino-2-phenylindole (DAPI; Sigma-Aldrich) (fig. S11).
Data acquisition using flow cytometry
PBMC suspensions were acquired using an eight-color FACSVerse flow cytometer (BD Biosciences) with 405-, 488-, and 640-nm laser excitations at an average flow rate of 2 μl/s and an average threshold event rate of 1000 to 2000 events/s. Multicolor Cytometer Setup and Tracking beads (BD Biosciences) were used for QC and standardization of photomultiplier tube detector voltages across multiple experimental runs. Fluorescence compensation for immunophenotyping was conducted using anti-mouse immunoglobulin G (IgG)κ antibody capture beads (Bangs Laboratories) labeled separately with each antibody. Fluorescence compensation for intracellular staining of cell signaling epitopes was conducted using anti-mouse IgGκ antibody capture beads labeled separately with anti-human CD3 (UCHT1) PE-Cy7 (eBioscience), anti-human Stat3 (pY705) (4/P-STAT3) Alexa Fluor 488 (AF 488; BD Biosciences), anti-human Stat3 (pY705) (4/P-STAT3) PE (BD Biosciences), and anti-human Stat3 (pY705) (4/P-STAT3) AF 647 (BD Biosciences; KP and TI) or anti-human CD3 (UCHT1) APC (eBioscience) and PLC-γ1 (10/PLCgamma) PE (BD Biosciences; DR and CV), alongside single-stain controls with maximum and minimum concentrations of each barcoding dye per PBMC sample.
Genotype analysis
Genotyping data from the CV samples (table S10) were obtained using the Illumina Infinium PsychArray Bead-Chip platform. SCZ risk SNPs related to calcium signaling were selected for analysis based on previous reports (7) and included variants in the ATP2A2 (rs4766428), CACNA1C (rs2159100, rs2007044, and rs758117), CACNB2 (rs7893279), CACNA1I (rs5757730), CALN1 (rs2944823), NLGN4X (rs12845396), RIMS1 (rs1339227), and TMEM110 (rs2535627) genes. For SNPs not included in the Illumina Infinium PsychArray Bead-Chip array (rs758117, rs7893279, rs12845396, and rs1339227), we used the LDproxy tool of LDlink (60) and the 1000 Genomes European population as a reference to identify measured SNPs that were in the highest linkage disequilibrium (R2 > 0.8) with the SNPs of interest (rs10491964, R2 = 0.93; rs4748466, R2 = 0.85; rs12835663, R2 = 0.83; and rs2789588, R2 = 0.82, respectively) and analyzed them as a proxy. Association between response to thapsigargin at PLC-γ1, expressed as the EC50, and the number of SCZ risk alleles per individual was assessed using fixed-effects linear regression (one-tailed test; H0, β ≤ 0; H1, β > 0) adjusted for age, gender, and body mass index (BMI). P values were obtained by permutation testing (n = 1000 permutations).
SH-SY5Y cell culture, stimulation, and microscopy
The SH-SY5Y human neuronal cell line (passage 5; European Collection of Authenticated Cell Cultures) was thawed at 37°C and seeded in cell culture flasks (Thermo Fisher Scientific) at approximately 0.1 M/ml in minimum essential medium (MEM)/F12 culture medium [1:1 MEM (Sigma-Aldrich) with 1% antibiotics (penicillin-streptomycin, Life Technologies) and Ham’s F12 nutrient mixture (Sigma-Aldrich) with 1% penicillin-streptomycin, 15% FBS (Life Technologies), 1% nonessential amino acids (Sigma-Aldrich), and 1% GlutaMAX (Life Technologies)]. Cells were rested overnight at 37°C/5% CO2, and the medium was changed the following day. The cells were cultured until reaching 70 to 75% confluence. For passaging, the cells were washed twice with MEM containing 1% penicillin-streptomycin and trypsinized using trypsin-EDTA (Life Technologies) for 5 min at 37°C, diluted with fresh culture medium, and centrifuged at 300g for 5 min. The cells were resuspended at approximately 0.3 M/ml in the culture medium and seeded into T75 flasks (Thermo Fisher Scientific). The medium was changed the next day after each passage. Cells were stored in liquid nitrogen in medium with 10% DMSO (Sigma-Aldrich).
Cells from passage 11 were used for DR microscopy experiments. Each experiment was performed in triplicate. The cells were seeded at 0.1 M/ml in 96-well imaging plates (Corning) and cultured for 48 hours at 37°C/5% CO2. Medium was changed 24 hours before stimulation experiment. Stimulation for 30 min was performed as in PBMCs using MEM/F12 culture medium without antibiotics in place of complete RPMI medium without antibiotics. Fixed cells were washed with PBS and permeabilized using 85 μl of ice-cold methanol for 20 min on ice, followed by addition of 85 μl of ice-cold PBS for another 30 min. The cells were washed three times with FACS buffer and stained with anti-β-tubulin class III AF 647 (50 μl/ml; BD Biosciences) and anti-human PLC-γ1 PE (300 μl/ml; BD Biosciences) in FACS buffer for 24 hours at 4°C. The cells were then washed three times with FACS buffer and stained with 3 μM DAPI (Sigma-Aldrich) in FACS buffer for 15 min at room temperature. The cells were washed once with FACS buffer and resuspended in FACS buffer for analysis.
Imaging was performed using confocal (LSM 710, Zeiss) and wide-field (DMI6000 with matrix HCS module, Leica) microscopy. For confocal imaging, laser excitation/fluorescence detection wavelengths were 405 nm/431 to 500 nm for DAPI, 543 nm/549 to 614 nm for PE, and 633 nm/644 to 708 nm for AF 647. For wide-field imaging, excitation/detection filters were DAPI/DAPI for DAPI, TRITC (tetramethyl rhodamine isothiocyanate)/TRITC for PE, and Cy5/Cy5 for AF 647. All replicate experiments were performed simultaneously and analyzed with identical laser/excitation power and gain settings.
Statistical analysis
Flow cytometry data were analyzed in flow cytometry standard 3.0 file format using FlowJo v.10.0.8 (TreeStar) and Kaluza Analysis v.1.3 (Beckman Coulter) software. Microscopy data were analyzed using CellProfiler Analyst v.2.0 (Broad Institute) and Fiji ImageJ 1.51n (National Institutes of Health). Statistical analysis was conducted using R software (R Core Team). PBMC samples in which the lymphocyte gate contained less than 30% of events, measured by forward scatter/side scatter, were excluded from further analysis. Individual treatment-epitope combinations (nodes) for which there were fewer than four data points (i.e., four PBMC samples), in the KP and DR studies, or fewer than eight data points (i.e., eight PBMC samples), in the TI study, were excluded from further analysis. Batch effects in MFIs caused by running samples on different days were removed for each epitope (TI) using the empirical Bayes algorithm—ComBat (R “sva” package). Experimental variables including positional effects within and across 96-well plates, barcoding dye fluorescence spillover, sample viability, cell counts, clinical group, and sample source were controlled for by principal components and Z factor analysis. Matching of clinical groups was conducted using the Wilcoxon rank sum test for continuous variables or the Fisher’s exact test for categorical variables.
For the determination of stimulant activity, the MFIs across PBMC samples, per stained epitope, were compared between each stimulant and the vehicle treatment using the conditional Wilcoxon rank sum test using the exact distribution for obtaining the P value (R “coin” package). The same test was also applied for each stimulant per functional fluorescence channel (AF 488, PE, and AF 647) in the unstained condition to determine whether the stimulant activity was an artifact of fluorescence spillover from adjacent channels or ligand autofluorescence (background fluorescence). In cases where the stimulant MFI was significantly altered with respect to vehicle MFI in the unstained condition, the epitopes labeled in the corresponding functional channel were only counted as active if the MFI response was in a different direction or had a 10% greater FC than the unstained condition. For stimulants with significant activity (permuted P < 0.05, Wilcoxon rank sum test) that superseded the background fluorescence, the response ratio was quantified as the median MFI of the stimulant divided by the median MFI of the vehicle across PBMC samples. For responses <1 (i.e., where the stimulant caused a decrease in MFI with respect to the vehicle), the response is reported as a negative FC (−1 divided by response ratio). The stain index of each antibody was calculated across PBMC samples, in the absence of stimulation, as the median MFI of the antibody stained sample divided by the median MFI of the corresponding unstained control. The two-tailed Spearman’s rank test was used to assess the correlation in FC responses for nodes replicated in independent experiments.
In the TI study, association of each active signaling node MFI to clinical group status was investigated using linear fixed-effects regression after adjusting for covariates. Active nodes were defined as those with a minimum epitope stain index of two and additionally, for ligand responses, a significant FC (P < 0.05, Wilcoxon rank sum test) of minimum 10% in at least one of the comparison groups, after adjusting for background fluorescence. Inactive nodes were excluded from the clinical group comparison analysis. The predictive variables of clinical group and ligand presence were set as interaction terms within the regression model to determine epitopes, which responded differently to individual ligand exposures across the clinical groups. Group status, without interaction, was set as the predictive variable to identify differences in basal epitope expression between clinical groups. Optional covariates, age and BMI (plus smoking and alcohol consumption in sensitivity analyses), were selected in the regression model in a stepwise procedure for each node separately using Bayesian information criterion. The mean of eight measurements in the vehicle condition and a single measurement in the stimulant condition per donor were used for the analysis. To account for the unknown distribution of the data, the small sample size, and the presence of outliers, the null distribution of the test statistic was estimated by randomly permuting sample labels 10,000 times for each node. Q values (P values corrected for multiple hypothesis testing) were calculated as the proportion of the permutation distribution for the maximal statistic that was greater than or equal to the originally obtained statistic.
In the DR study, association of PLC-γ1 MFI to compound and stimulant status was investigated using permutation-based linear fixed-effects regression (n = 10,000 permutations). The predictive variables of stimulant group (thapsigargin or vehicle) and compound exposure (compound or vehicle) were set as interaction terms within the regression model to determine compounds that modified the PLC-γ1 response to thapsigargin. For compounds that showed a significant interaction with the stimulant (permuted P < 0.05), post hoc one-way ANOVA tests and median values were used to determine the directionality of the effects. The same model (nonpermuted, due to large number of data points) was applied to neuronal DR validation data. In the validation experiments, dose-response data were fitted with four-parameter (thapsigargin dose-response experiments) or five-parameter (drug dose-response experiments) logistic regression models [R “nplr” package and GraphPad Prism 5 (GraphPad Software)].
In the CV study, ex vivo response to treatment, the shift in thapsigargin/PLC-γ1 EC50 (ΔEC50), was calculated as the difference in EC50 between the drug and vehicle conditions. In vivo response to treatment was calculated as the difference between basal and follow-up (3 weeks, 6 weeks, and 3 months) symptoms. Association between the ex vivo and in vivo responses to treatment was investigated using linear fixed-effects regression, after adjusting for covariates. Optional covariates, basal symptoms, treatment drug, age, gender, and BMI, were selected in the regression model in a stepwise procedure for each correlation separately using Bayesian information criterion. The null distribution of the test statistic was estimated by randomly permuting sample labels 10,000 times for each correlation. Q values were calculated as the proportion of the permutation distribution for the maximal statistic that was greater than or equal to the originally obtained statistic. Dose-response data were fitted with four-parameter logistic regression model. Data were visualized using FlowJo v.10.0.8, Kaluza Analysis v.1.3, R software, GraphPad Prism 5, Excel 2016 (Microsoft), PubChem (National Center for Biotechnology Information), Fiji, and Adobe Illustrator (Adobe Systems).
Supplementary Material
Acknowledgments
We would like to thank collaborators who contributed compounds to the screening library (L. Jones-Brando, R. Yolken, G. H. Posner, J. G. D’Angelo, C. P. Hencken, J. McNulty, F. F. Wagner, E. B. Holson, and R. S. Williams), J. D. Cooper for statistical discussion, N. Le Novère for bioinformatics advice, G. S. Kaminski-Schierle and K. Feeney for contribution of the neuronal cell line, P. D. Humphreys for microscopy acquisition and expertise, PBMC donors for provision of biological samples, and support staff at the affiliated institutions, especially those involved in sample collection, including the Department of Psychiatry, Erasmus Medical Centre, Rotterdam, the Netherlands and the HUMV-IFIMAV Biobank, Fundación Marques de Valdecilla, Santander, Spain. Funding: This work was supported by grants to S.B. from the Stanley Medical Research Institute (SMRI) and the Engineering and Physical Sciences Research Council UK (EPSRC CASE studentship and Impact Acceleration Award). S.G.L. was supported by EPSRC CASE studentship and Psynova Neurotech Ltd. J.T. was supported through grants awarded to Psynova Neurotech Ltd. by the European Union FP7 funding scheme: Marie Curie Actions Industry Academia Partnerships and Pathways(ref. 286334, PSYCH-AID project); by the Virgo consortium, funded by the Dutch Government (ref. FES0908); by the Netherlands Genomics Initiative (ref. 050-060-452); and by the Dutch Fund for Economic Structure Reinforcement, the NeuroBasic PharmaPhenomics project (ref. 0908). Additional compounds targeting GSK-3 were provided by S.J.H. who was funded by the SMRI (refs. R03MH08744 and R01MH095088). Author contributions: S.B., S.G.L., and J.T. conceived the study and designed experiments. N.J.v.B., B.C.-F., and P.S.-P. collected PBMC samples and were responsible for clinical diagnoses. S.J.H. and T.P. provided JB1121 (synthesized by J.A.B), CHIR-99021, and other compounds for the screening library. B.C.-F., J.V.-B., and S.P. provided the genotyping data. S.G.L., J.T., G.F.v.R., H.S., and N.R. conducted the experiments. S.G.L. and G.F.v.R. analyzed the raw data, and J.T. matched the study groups for demographic variables. J.T., J.M.R., and D.A.C. conducted the statistical analysis. S.G.L., J.T., and S.B. prepared the manuscript. Competing interests: S.G.L. was part funded by Psynova Neurotech Ltd. until October 2015. J.T. was a consultant for Psynova Neurotech Ltd. until April 2016. S.B. is a director of Psynova Neurotech Ltd. and Psyomics Ltd. S.J.H. is a consultant on the scientific advisory board for Rodin Therapeutics, Psy Therapeutics, and Frequency Therapeutics, a consultant for AstraZeneca, and a speaker for Amgen and Merck. All other authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. Materials provided under material transfer agreements can be requested pending availability, scientific review, and a completed material transfer agreement as follows: clinical samples from Erasmus University Medical Center, Rotterdam, the Netherlands (N.J.v.B.) and Department of Medicine and Psychiatry, University of Cantabria, Santander, Spain (B.C.-F.) and compounds in the extended compound library from the Broad Institute Cambridge, MA, USA (F. F. Wagner and E. B. Holson) and Royal Holloway University of London, Egham, UK (R. S. Williams).
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/5/eaau9093/DC1
Fig. S1. Construction of a three-dimensional fluorescent cell barcoding matrix for multiplexing of 80 cellular treatments.
Fig. S2. Gating strategies for the functional analysis of 80 barcoded T cell populations.
Fig. S3. MFIs and CVs across 80 barcoded T cell populations for each functional fluorescence channel.
Fig. S4. Z factor analyses across 80 barcoded T cell populations for each functional fluorescence channel.
Fig. S5. Reproducibility across time and independent PBMC cohorts.
Fig. S6. Stain indices of antibody clones against T cell signaling epitopes used in the KP experiments.
Fig. S7. Kinetic induction of cell signaling responses across the ligand and epitope array (n = 5460 nodes; i.e., ligand-epitope combinations).
Fig. S8. Distribution of FCs for T cell signaling responses across time points.
Fig. S9. Dynamic regulation of JAK/STAT T cell signaling across time course.
Fig. S10. Gating strategies for the functional analysis of 64 barcoded T cell populations.
Fig. S11. Gating strategy for cell viability and immunophenotyping.
Fig. S12. Clinical response to antipsychotic treatment with olanzapine in patients with SCZ at 6 weeks.
Fig. S13. Altered T cell signaling nodes (ligand-epitope combinations) in pretreatment SCZ versus control and pretreatment versus posttreatment SCZ comparisons.
Fig. S14. Association between the drug target response to thapsigargin at PLC-γ1 in SCZ and the genome-wide significant SCZ risk SNP rs4766428 in the ATP2A2 gene.
Fig. S15. Normal regulatory response at PLC-γ1 to calcium release from the endoplasmic reticulum and hypothetical mechanism of action in SCZ, based on the altered response to thapsigargin at PLC-γ1 in T cells from patients with SCZ.
Fig. S16. Gating strategies for the functional analysis of PLC-γ1 expression in four barcoded T cell populations.
Fig. S17. Thapsigargin dose response at PLC-γ1.
Fig. S18. Selective potentiation of PLC-γ1 response in the presence of thapsigargin.
Fig. S19. Tanimoto structural similarity clustering of calcium channel blocker, antipsychotic, corticosteroid, and antibiotic compounds used in PLC-γ1 dose-response validation and selectivity testing.
Fig. S20. Validation and selectivity testing of calcium channel blocker, antipsychotic, corticosteroid, antibiotic, and other drug classes at PLC-γ1.
Fig. S21. Validation of top drug candidates in the SH-SY5Y neuronal cell line.
Fig. S22. Correlation of ex vivo drug-target activity with in vivo efficacy in the CV study.
Fig. S23. Potentiation of thapsigargin/PLC-γ1 dose response at 30 min by top drug candidates from the screening phase at 10 μM concentration in PBMCs from drug-naïve patients with SCZ.
Table S1. Antibodies used to detect intracellular cell signaling epitopes and PBMC subtypes.
Table S2. Ligands used to stimulate/alter cell signaling dynamics in PBMCs.
Table S3. Activity of ligands across the time course.
Table S4. Activity of epitopes across the time course.
Table S5. Demographic characteristics and matching of PBMC donors used in the TI study.
Table S6. Altered ligand responses at T cell signaling epitopes in healthy control versus pretreatment SCZ and pretreatment versus posttreatment SCZ comparisons.
Table S7. Altered basal expression of T cell signaling epitopes in pretreatment versus posttreatment SCZ comparison.
Table S8. Extended FDA-approved compound library.
Table S9. Extended FDA-approved library screening of compounds which selectively potentiate the PLC-γ1 response in the presence of 0.5 μM thapsigargin.
Table S10. Demographic characteristics and matching of PBMC donors used in the CV study.
Table S11. Prediction of in vivo response to treatment from ex vivo treatment activity.
REFERENCES AND NOTES
- 1.Agid Y., Buzsáki G., Diamond D. M., Frackowiak R., Giedd J., Girault J.-A., Grace A., Lambert J. J., Manji H., Mayberg H., Popoli M., Prochiantz A., Richter-Levin G., Somogyi P., Spedding M., Svenningsson P., Weinberger D., How can drug discovery for psychiatric disorders be improved? Nat. Rev. Drug Discov. 6, 189–201 (2007). [DOI] [PubMed] [Google Scholar]
- 2.Hyman S. E., A glimmer of light for neuropsychiatric disorders. Nature 455, 890–893 (2008). [DOI] [PubMed] [Google Scholar]
- 3.Wong M.-L., Licinio J., From monoamines to genomic targets: A paradigm shift for drug discovery in depression. Nat. Rev. Drug Discov. 3, 136–151 (2004). [DOI] [PubMed] [Google Scholar]
- 4.Kahn R. S., Sommer I. E., Murray R. M., Meyer-Lindenberg A., Weinberger D. R., Cannon T. D., O'Donovan M., Correll C. U., Kane J. M., van Os J., Insel T. R., Schizophrenia. Nat. Rev. Dis. Primers 1, 15067 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Huhn M., Tardy M., Spineli L. M., Kissling W., Förstl H., Pitschel-Walz G., Leucht C., Samara M., Dold M., Davis J. M., Leucht S., Efficacy of pharmacotherapy and psychotherapy for adult psychiatric disorders: A systematic overview of meta-analyses. JAMA Psychiat. 71, 706–715 (2014). [DOI] [PubMed] [Google Scholar]
- 6.Tomasik J., Schwarz E., Lago S. G., Rothermundt M., Leweke F. M., van Beveren N. J. M., Guest P. C., Rahmoune H., Steiner J., Bahn S., Pretreatment levels of the fatty acid handling proteins H-FABP and CD36 predict response to olanzapine in recent-onset schizophrenia patients. Brain Behav. Immun. 52, 178–186 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Schizophrenia Working Group of the Psychiatric Genomics Consortium , Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cross-Disorder Group of the Psychiatric Genomics Consortium , Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat. Genet. 45, 984–994 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gusev A., Mancuso N., Won H., Kousi M., Finucane H. K., Reshef Y., Song L., Safi A.; Schizophrenia Working Group of the Psychiatric Genomics Consortium, McCarroll S., Neale B. M., Ophoff R. A., O’Donovan M. C., Crawford G. E., Geschwind D. H., Katsanis N., Sullivan P. F., Pasaniuc B., Price A. L., Transcriptome-wide association study of schizophrenia and chromatin activity yields mechanistic disease insights. Nat. Genet. 50, 538–548 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brennand K. J., Simone A., Jou J., Gelboin-Burkhart C., Tran N., Sangar S., Li Y., Mu Y., Chen G., Yu D., McCarthy S., Sebat J., Gage F. H., Modelling schizophrenia using human induced pluripotent stem cells. Nature 473, 221–225 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schwartzentruber J., Foskolou S., Kilpinen H., Rodrigues J., Alasoo K., Knights A. J., Patel M., Goncalves A., Ferreira R., Benn C. L., Wilbrey A., Bictash M., Impey E., Cao L., Lainez S., Loucif A. J., Whiting P. J., Gutteridge A., Gaffney D. J.; HIPSCI Consortium , Molecular and functional variation in iPSC-derived sensory neurons. Nat. Genet. 50, 54–61 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bodenmiller B., Zunder E. R., Finck R., Chen T. J., Savig E. S., Bruggner R. V., Simonds E. F., Bendall S. C., Sachs K., Krutzik P. O., Nolan G. P., Multiplexed mass cytometry profiling of cellular states perturbed by small-molecule regulators. Nat. Biotechnol. 30, 858–867 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lago S. G., Tomasik J., van Rees G. F., Ramsey J. M., Haenisch F., Cooper J. D., Broek J. A., Suarez-Pinilla P., Ruland T., Auyeug B., Mikova O., Kabacs N., Arolt V., Baron-Cohen S., Crespo-Facorro B., Bahn S., Exploring the neuropsychiatric spectrum using high-content functional analysis of single-cell signaling networks. Mol. Psychiatry, 10.1038/s41380-018-0123-4 (2018). [DOI] [PubMed] [Google Scholar]
- 14.Miller B. J., Buckley P., Seabolt W., Mellor A., Kirkpatrick B., Meta-analysis of cytokine alterations in schizophrenia: Clinical status and antipsychotic effects. Biol. Psychiatry 70, 663–671 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Herberth M., Koethe D., Cheng T. M. K., Krzyszton N. D., Schoeffmann S., Guest P. C., Rahmoune H., Harris L. W., Kranaster L., Leweke F. M., Bahn S., Impaired glycolytic response in peripheral blood mononuclear cells of first-onset antipsychotic-naive schizophrenia patients. Mol. Psychiatry 16, 848–859 (2011). [DOI] [PubMed] [Google Scholar]
- 16.Gladkevich A., Kauffman H. F., Korf J., Lymphocytes as a neural probe: Potential for studying psychiatric disorders. Prog. Neuropsychopharmacol. Biol. Psychiatry 28, 559–576 (2004). [DOI] [PubMed] [Google Scholar]
- 17.Emamian E. S., Hall D., Birnbaum M. J., Karayiorgou M., Gogos J. A., Convergent evidence for impaired AKT1-GSK3β signaling in schizophrenia. Nat. Genet. 36, 131–137 (2004). [DOI] [PubMed] [Google Scholar]
- 18.Polter A., Beurel E., Yang S., Garner R., Song L., Miller C. A., Sweatt J. D., McMahon L., Bartolucci A. A., Li X., Jope R. S., Deficiency in the inhibitory serine-phosphorylation of glycogen synthase kinase-3 increases sensitivity to mood disturbances. Neuropsychopharmacology 35, 1761–1774 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rivera-Baltanas T., Olivares J. M., Martinez-Villamarin J. R., Fenton E. Y., Kalynchuk L. E., Caruncho H. J., Serotonin 2A receptor clustering in peripheral lymphocytes is altered in major depression and may be a biomarker of therapeutic efficacy. J. Affect. Disord. 163, 47–55 (2014). [DOI] [PubMed] [Google Scholar]
- 20.Gordon-Smith K., Green E., Grozeva D., Tavadia S., Craddock N., Jones L., Genotype-phenotype correlations in Darier disease: A focus on the neuropsychiatric phenotype. Am. J. Med. Genet. B Neuropsychiatr. Genet. 177, 717–726 (2018). [DOI] [PubMed] [Google Scholar]
- 21.O’Rourke L., Murphy K., Recent developments in understanding the relationship between 22q11.2 deletion syndrome and psychosis. Curr. Opin. Psychiatry 32, 67–72 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rogers G. B., Keating D. J., Young R. L., Wong M.-L., Licinio J., Wesselingh S., From gut dysbiosis to altered brain function and mental illness: Mechanisms and pathways. Mol. Psychiatry 21, 738–748 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dantzer R., O’Connor J. C., Freund G. G., Johnson R. W., Kelley K. W., From inflammation to sickness and depression: When the immune system subjugates the brain. Nat. Rev. Neurosci. 9, 46–56 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zheng W., Thorne N., McKew J. C., Phenotypic screens as a renewed approach for drug discovery. Drug Discov. Today 18, 1067–1073 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boyle E. A., Li Y. I., Pritchard J. K., An expanded view of complex traits: From polygenic to omnigenic. Cell 169, 1177–1186 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Santos R., Ursu O., Gaulton A., Bento A. P., Donadi R. S., Bologa C. G., Karlsson A., Al-Lazikani B., Hersey A., Oprea T. I., Overington J. P., A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 16, 19–34 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Krutzik P. O., Nolan G. P., Fluorescent cell barcoding in flow cytometry allows high-throughput drug screening and signaling profiling. Nat. Methods 3, 361–368 (2006). [DOI] [PubMed] [Google Scholar]
- 28.Krutzik P. O., Crane J. M., Clutter M. R., Nolan G. P., High-content single-cell drug screening with phosphospecific flow cytometry. Nat. Chem. Biol. 4, 132–142 (2008). [DOI] [PubMed] [Google Scholar]
- 29.Kalland M. E., Oberprieler N. G., Vang T., Taskén K., Torgersen K. M., T cell-signaling network analysis reveals distinct differences between CD28 and CD2 costimulation responses in various subsets and in the MAPK pathway between resting and activated regulatory T cells. J. Immunol. 187, 5233–5245 (2011). [DOI] [PubMed] [Google Scholar]
- 30.Yan Z., Feng J., Fienberg A. A., Greengard P., D2 dopamine receptors induce mitogen-activated protein kinase and cAMP response element-binding protein phosphorylation in neurons. Proc. Natl. Acad. Sci. U.S.A. 96, 11607–11612 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bendall S. C., Simonds E. F., Qiu P., Amir E.-a. D., Krutzik P. O., Finck R., Bruggner R. V., Melamed R., Trejo A., Ornatsky O. I., Balderas R. S., Plevritis S. K., Sachs K., Pe'er D., Tanner S. D., Nolan G. P., Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science 332, 687–696 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beaulieu J.-M., A role for Akt and glycogen synthase kinase-3 as integrators of dopamine and serotonin neurotransmission in mental health. J. Psychiatry Neurosci. 37, 7–16 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wagner F. F., Bishop J. A., Gale J. P., Shi X., Walk M., Ketterman J., Patnaik D., Barker D., Walpita D., Campbell A. J., Nguyen S., Lewis M., Ross L., Weïwer M., An W. F., Germain A. R., Nag P. P., Metkar S., Kaya T., Dandapani S., Olson D. E., Barbe A.-L., Lazzaro F., Sacher J. R., Cheah J. H., Fei D., Perez J., Munoz B., Palmer M., Stegmaier K., Schreiber S. L., Scolnick E., Zhang Y.-L., Haggarty S. J., Holson E. B., Pan J. Q., Inhibitors of glycogen synthase kinase 3 with exquisite kinome-wide selectivity and their functional effects. ACS Chem. Biol. 11, 1952–1963 (2016). [DOI] [PubMed] [Google Scholar]
- 34.Pan J. Q., Lewis M. C., Ketterman J. K., Clore E. L., Riley M., Richards K. R., Berry-Scott E., Liu X., Wagner F. F., Holson E. B., Neve R. L., Biechele T. L., Moon R. T., Scolnick E. M., Petryshen T. L., Haggarty S. J., AKT kinase activity is required for lithium to modulate mood-related behaviors in mice. Neuropsychopharmacology 36, 1397–1411 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roth B. L., Sheffler D. J., Kroeze W. K., Magic shotguns versus magic bullets: Selectively non-selective drugs for mood disorders and schizophrenia. Nat. Rev. Drug Discov. 3, 353–359 (2004). [DOI] [PubMed] [Google Scholar]
- 36.Luo X., Huang L., Han L., Luo Z., Hu F., Tieu R., Gan L., Systematic prioritization and integrative analysis of copy number variations in schizophrenia reveal key schizophrenia susceptibility genes. Schizophr. Bull. 40, 1285–1299 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Machado-Vieira R., Ibrahim L., Zarate C. A. Jr., Histone deacetylases and mood disorders: Epigenetic programming in gene-environment interactions. CNS Neurosci. Ther. 17, 699–704 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ganea K., Menke A., Schmidt M. V., Lucae S., Rammes G., Liebl C., Harbich D., Sterlemann V., Storch C., Uhr M., Holsboer F., Binder E. B., Sillaber I., Müller M. B., Convergent animal and human evidence suggests the activin/inhibin pathway to be involved in antidepressant response. Transl. Psychiatry 2, e177 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Duman C. H., Schlesinger L., Kodama M., Russell D. S., Duman R. S., A role for MAP kinase signaling in behavioral models of depression and antidepressant treatment. Biol. Psychiatry 61, 661–670 (2007). [DOI] [PubMed] [Google Scholar]
- 40.Purcell S. M., Moran J. L., Fromer M., Ruderfer D., Solovieff N., Roussos P., O’Dushlaine C., Chambert K., Bergen S. E., Kähler A., Duncan L., Stahl E., Genovese G., Fernández E., Collins M. O., Komiyama N. H., Choudhary J. S., Magnusson P. K. E., Banks E., Shakir K., Garimella K., Fennell T., DePristo M., Grant S. G. N., Haggarty S. J., Gabriel S., Scolnick E. M., Lander E. S., Hultman C. M., Sullivan P. F., McCarroll S. A., Sklar P., A polygenic burden of rare disruptive mutations in schizophrenia. Nature 506, 185–190 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Badou A., Jha M. K., Matza D., Flavell R. A., Emerging roles of L-type voltage-gated and other calcium channels in T lymphocytes. Front. Immunol. 4, 243 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang Y. R., Kang D.-S., Lee C., Seok H., Follo M. Y., Cocco L., Suh P.-G., Primary phospholipase C and brain disorders. Adv. Biol. Regul. 61, 80–85 (2016). [DOI] [PubMed] [Google Scholar]
- 43.Yang Y. R., Jung J. H., Kim S.-J., Hamada K., Suzuki A., Kim H. J., Lee J. H., Kwon O.-B., Lee Y. K., Kim J., Kim E.-K., Jang H.-J., Kang D.-S., Choi J.-S., Lee C. J., Marshall J., Koh H.-Y., Kim C.-J., Seok H., Kim S. H., Choi J. H., Choi Y.-B., Cocco L., Ryu S. H., Kim J.-H., Suh P.-G., Forebrain-specific ablation of phospholipase Cγ1 causes manic-like behavior. Mol. Psychiatry 22, 1473–1482 (2017). [DOI] [PubMed] [Google Scholar]
- 44.Berridge M. J., Calcium signalling and psychiatric disease: Bipolar disorder and schizophrenia. Cell Tissue Res. 357, 477–492 (2014). [DOI] [PubMed] [Google Scholar]
- 45.Jang H.-J., Yang Y. R., Kim J. K., Choi J. H., Seo Y.-K., Lee Y. H., Lee J. E., Ryu S. H., Suh P.-G., Phospholipase C-γ1 involved in brain disorders. Adv. Biol. Regul. 53, 51–62 (2013). [DOI] [PubMed] [Google Scholar]
- 46.Mizoguchi Y., Kato T. A., Horikawa H., Monji A., Microglial intracellular Ca2+ signaling as a target of antipsychotic actions for the treatment of schizophrenia. Front. Cell. Neurosci. 8, 370 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.de Bartolomeis A., Tomasetti C., Calcium-dependent networks in dopamine-glutamate interaction: The role of postsynaptic scaffolding proteins. Mol. Neurobiol. 46, 275–296 (2012). [DOI] [PubMed] [Google Scholar]
- 48.Ferguson S. S. G., Receptor tyrosine kinase transactivation: Fine-tuning synaptic transmission. Trends Neurosci. 26, 119–122 (2003). [DOI] [PubMed] [Google Scholar]
- 49.Pitcher G. M., Kalia L. V., Ng D., Goodfellow N. M., Yee K. T., Lambe E. K., Salter M. W., Schizophrenia susceptibility pathway neuregulin 1-ErbB4 suppresses Src upregulation of NMDA receptors. Nat. Med. 17, 470–478 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Iglesias García C., Iglesias Alonso A., Bobes J., Concentrations in plasma clozapine levels in schizophrenic and schizoaffective patients. Rev. Psiquiatr. Salud Ment. 10, 192–196 (2017). [DOI] [PubMed] [Google Scholar]
- 51.Lencz T., Malhotra A. K., Targeting the schizophrenia genome: A fast track strategy from GWAS to clinic. Mol. Psychiatry 20, 820–826 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shah A. B., Poiletman R. M., Shah N. S., The influence of nisoldipine—A “calcium entry blocker” on drug induced stereotyped behavior in rats. Prog. Neuropsychopharmacol. Biol. Psychiatry 7, 165–173 (1983). [DOI] [PubMed] [Google Scholar]
- 53.Hori Y., Takeda H., Tsuji M., Matsumiya T., Differentiation of the inhibitory effects of calcium antagonists on abnormal behaviors induced by methamphetamine or phencyclidine. Pharmacology 56, 165–174 (1998). [DOI] [PubMed] [Google Scholar]
- 54.Thorne C. A., Wichaidit C., Coster A. D., Posner B. A., Wu L. F., Altschuler S. J., GSK-3 modulates cellular responses to a broad spectrum of kinase inhibitors. Nat. Chem. Biol. 11, 58–63 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bromet E. J., Kotov R., Fochtmann L. J., Carlson G. A., Tanenberg-Karant M., Ruggero C., Chang S.-w., Diagnostic shifts during the decade following first admission for psychosis. Am. J. Psychiatry 168, 1186–1194 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hayashi A., Kasahara T., Kametani M., Toyota T., Yoshikawa T., Kato T., Aberrant endoplasmic reticulum stress response in lymphoblastoid cells from patients with bipolar disorder. Int. J. Neuropsychopharmacol. 12, 33–43 (2009). [DOI] [PubMed] [Google Scholar]
- 57.Qiu P., Simonds E. F., Bendall S. C., Gibbs K. D. Jr., Bruggner R. V., Linderman M. D., Sachs K., Nolan G. P., Plevritis S. K., Extracting a cellular hierarchy from high-dimensional cytometry data with SPADE. Nat. Biotechnol. 29, 886–891 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Amir E.-a. D., Davis K. L., Tadmor M. D., Simonds E. F., Levine J. H., Bendall S. C., Shenfeld D. K., Krishnaswamy S., Nolan G. P., Pe'er D., viSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia. Nat. Biotechnol. 31, 545–552 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lehár J., Zimmermann G. R., Krueger A. S., Molnar R. A., Ledell J. T., Heilbut A. M., Short G. F., Giusti L. C., Nolan G. P., Magid O. A., Lee M. S., Borisy A. A., Stockwell B. R., Keith C. T., Chemical combination effects predict connectivity in biological systems. Mol. Syst. Biol. 3, 80 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Machiela M. J., Chanock S. J., LDlink: A web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics 31, 3555–3557 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/5/eaau9093/DC1
Fig. S1. Construction of a three-dimensional fluorescent cell barcoding matrix for multiplexing of 80 cellular treatments.
Fig. S2. Gating strategies for the functional analysis of 80 barcoded T cell populations.
Fig. S3. MFIs and CVs across 80 barcoded T cell populations for each functional fluorescence channel.
Fig. S4. Z factor analyses across 80 barcoded T cell populations for each functional fluorescence channel.
Fig. S5. Reproducibility across time and independent PBMC cohorts.
Fig. S6. Stain indices of antibody clones against T cell signaling epitopes used in the KP experiments.
Fig. S7. Kinetic induction of cell signaling responses across the ligand and epitope array (n = 5460 nodes; i.e., ligand-epitope combinations).
Fig. S8. Distribution of FCs for T cell signaling responses across time points.
Fig. S9. Dynamic regulation of JAK/STAT T cell signaling across time course.
Fig. S10. Gating strategies for the functional analysis of 64 barcoded T cell populations.
Fig. S11. Gating strategy for cell viability and immunophenotyping.
Fig. S12. Clinical response to antipsychotic treatment with olanzapine in patients with SCZ at 6 weeks.
Fig. S13. Altered T cell signaling nodes (ligand-epitope combinations) in pretreatment SCZ versus control and pretreatment versus posttreatment SCZ comparisons.
Fig. S14. Association between the drug target response to thapsigargin at PLC-γ1 in SCZ and the genome-wide significant SCZ risk SNP rs4766428 in the ATP2A2 gene.
Fig. S15. Normal regulatory response at PLC-γ1 to calcium release from the endoplasmic reticulum and hypothetical mechanism of action in SCZ, based on the altered response to thapsigargin at PLC-γ1 in T cells from patients with SCZ.
Fig. S16. Gating strategies for the functional analysis of PLC-γ1 expression in four barcoded T cell populations.
Fig. S17. Thapsigargin dose response at PLC-γ1.
Fig. S18. Selective potentiation of PLC-γ1 response in the presence of thapsigargin.
Fig. S19. Tanimoto structural similarity clustering of calcium channel blocker, antipsychotic, corticosteroid, and antibiotic compounds used in PLC-γ1 dose-response validation and selectivity testing.
Fig. S20. Validation and selectivity testing of calcium channel blocker, antipsychotic, corticosteroid, antibiotic, and other drug classes at PLC-γ1.
Fig. S21. Validation of top drug candidates in the SH-SY5Y neuronal cell line.
Fig. S22. Correlation of ex vivo drug-target activity with in vivo efficacy in the CV study.
Fig. S23. Potentiation of thapsigargin/PLC-γ1 dose response at 30 min by top drug candidates from the screening phase at 10 μM concentration in PBMCs from drug-naïve patients with SCZ.
Table S1. Antibodies used to detect intracellular cell signaling epitopes and PBMC subtypes.
Table S2. Ligands used to stimulate/alter cell signaling dynamics in PBMCs.
Table S3. Activity of ligands across the time course.
Table S4. Activity of epitopes across the time course.
Table S5. Demographic characteristics and matching of PBMC donors used in the TI study.
Table S6. Altered ligand responses at T cell signaling epitopes in healthy control versus pretreatment SCZ and pretreatment versus posttreatment SCZ comparisons.
Table S7. Altered basal expression of T cell signaling epitopes in pretreatment versus posttreatment SCZ comparison.
Table S8. Extended FDA-approved compound library.
Table S9. Extended FDA-approved library screening of compounds which selectively potentiate the PLC-γ1 response in the presence of 0.5 μM thapsigargin.
Table S10. Demographic characteristics and matching of PBMC donors used in the CV study.
Table S11. Prediction of in vivo response to treatment from ex vivo treatment activity.