

Fibrocytes, easily isolated from peripheral blood, hold potential as a cell source for regenerative medicine.
Abstract
Fibrocytes (FCs) are hematopoietic lineage cells that migrate to sites of injury, transition to a mesenchymal phenotype, and help to mediate wound repair. Despite their relevance to human fibrotic disorders, there are few data characterizing basic FC biology. Herein, using proteomic, bioenergetic, and bioengineering techniques, we conducted deep phenotypic characterization of differentiating and mature FCs. Differentiation was associated with metabolic reprogramming that favored oxidative phosphorylation. Mature FCs had distinct proteomes compared to classic mesenchymal cells, formed functional stromae that supported epithelial maturation during in vitro organotypic culture, and exhibited in vivo survival and self-tolerance as connective tissue isografts. In an in vitro scratch assay, FCs promoted fibroblast migration and wound closure by paracrine signaling via the chemokine CXCL8 (interleukin-8). These findings characterize important aspects of FC differentiation and show that, in addition to their role in wound healing, FCs hold potential as an easily isolated autologous cell source for regenerative medicine.
INTRODUCTION
Cell-based therapies hold substantial promise in regenerative medicine. Some therapies, such as blood transfusion and hematopoietic stem cell transplantation, are widely available and accepted as clinical standard of care (1); others remain in the preclinical and clinical trial pipelines or have received regulatory approval for specific indications (2, 3). Irrespective of a given candidate therapy’s biological target and approach, successful translation requires a robust cell source. Pluripotent and multipotent stem cells and tissue-specific somatic cells are all under investigation: Each population confers a distinct set of advantages and challenges (2, 3), meaning that appropriately targeted clinical therapies will likely depend on different cell sources. Moreover, as experimental advances spur the arrival of sophisticated, multicellular engineered tissue and organ grafts (4–6), there is a need to expand the toolbox of source cells available for the myriad applications of regenerative medicine. Desirable features of translationally relevant cells are ease of access with little harvest-related morbidity, plasticity, hypoimmunogenicity or potential for autologous use, and suitability for bespoke applications in isolation or in concert with other cells, scaffolds, and biologics.
Fibrocytes (FCs), first identified 25 years ago (7), are a subpopulation of leukocytes that are recruited to sites of injury and inflammation, differentiate toward a mesenchymal phenotype, and play a role in both adaptive and maladaptive wound healing (8, 9). FC accumulation and its sequelae have been linked to pathologic severity in human inflammatory and fibrotic disorders (10–20); however, FCs are also multipotent (21, 22), hold immunomodulatory capacity (23), and have shown therapeutic effect in multiple preclinical disease models (23–25). Despite their increased acceptance as effector cells in connective tissue remodeling, little is known about the cellular reprogramming that drives FC differentiation from monocyte precursors, the relationship between FCs and other (stromal or nonstromal) cells, and whether the mesenchymal phenotype assumed by mature FCs is fully functional.
Given their plasticity, capacity for mesenchymal function, and therapeutic potential under permissive conditions, we examined whether FCs might, in addition to their much-studied role within the wound milieu, hold value as a cell source for regenerative medicine. We interrogated proteomic and metabolic remodeling during human FC differentiation from adherent monocytes in vitro; tested the ability of FCs to assemble into functional bioengineered stromae, support epithelial maturation in organotypic culture, and survive as connective tissue isografts in vivo; and examined the influence of FCs on fibroblast behavior in an in vitro wound model. This deep phenotypic and functional characterization provides a foundation for future work in FC-based therapeutics.
RESULTS
FC isolation and differentiation in culture
FCs are classically identified by coexpression of markers associated with both mesenchymal [e.g., prolyl-4-hydroxylase β (P4HB), collagen type I (COL1)] and hematopoietic (e.g., CD11b, CD34, and CD45) lineages, as well as fibroblastic morphology in tissue and following in vitro culture (9). To date, most characterization research has focused on mature, differentiated FCs (26, 27); there are fewer data describing FCs (or their precursors) in circulating blood, particularly in healthy donors (11, 28). One reason for this is the challenge of separating FCs from other leukocyte subpopulations, which share many surface markers and can also synthesize collagen under certain conditions (29). We previously validated an FC detection panel in a rat vocal fold (VF) mucosal injury model, defined by positive expression of the collagen synthesis enzyme P4HB and leukocyte-specific integrin CD11b and negative expression of the leukocyte-specific Fcγ receptor CD16 (30). As FC precursors arise from a subset of CD11b+ monocytes (31) but are CD16− (9), we reasoned that negative selection of CD16+ cells would eliminate contaminating monocytes, granulocytes, and natural killer cells, yielding an ultrapure FC population for downstream analysis. We performed flow cytometry of human peripheral blood mononuclear cells (PBMCs) and found that 5.8 to 8.7% of circulating CD16− cells were P4HB+CD11b+ FCs (Fig. 1, A and B). CD16−P4HB+CD11b+ FCs comprised 0.7 to 1.5% of the PBMC population and exhibited volume and granularity properties associated with the monocyte subpopulation (Fig. 1C), consistent with their reported lineage (31, 32). They also expressed the hematopoietic (and FC) markers CD34 and CD105, as well as the β-1 integrin CD29 (Fig. 1D) (9), further supporting the specificity of our detection panel.
Fig. 1. Flow cytometric characterization of circulating human FCs.
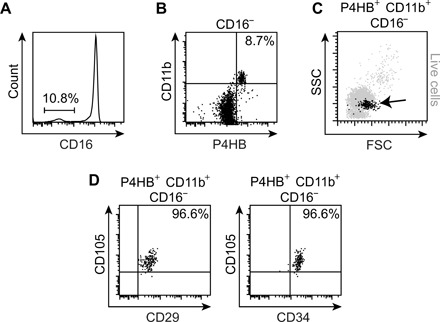
(A) CD16 expression in freshly isolated PBMCs. The gate represents the CD16− subpopulation. (B) P4HB and CD11b expression in the CD16− subpopulation. (C) Forward scatter (FSC) and side scatter (SSC) features of P4HB+CD11b+CD16− FCs (black) compared to all live cells (gray). (D) CD29, CD34, and CD105 expression in P4HB+CD11b+CD16− FCs.
The reliance of FC identification upon intracellular staining of P4HB (or COL1) via fixation and membrane permeabilization has rendered FC precursor cell sorting and in vitro investigation impossible with current technology. Previous in vitro studies have therefore relied on culture-induced differentiation of FCs from adherent monocytes, following plating of PBMCs or splenocytes. Using a primary readout of FC yield, these studies have refined cell isolation and culture parameters (33, 34) and identified an array of signaling cells and molecules that either promote or inhibit FC differentiation (31, 35–38). There remains, however, little understanding of the cell biology of FC differentiation itself. We used a standard culture technique [suspension of freshly isolated PBMCs in serum-supplemented Dulbecco’s modified Eagle’s medium (DMEM); plating at 1 × 105 to 5 × 105 cells/cm2 density on culture substrate with medium change every 48 hours] (39) and assayed differentiating FCs at 1, 7, and 14 days. A subset of adherent monocytes showed early somatic protrusions at day 1 (Fig. 2A); the majority developed bipolar (or multipolar) fibroblastic morphology over 14 days, steadily increasing in both volume and granularity (Fig. 2, A and B). P4HB expression increased with culture duration, whereas CD11b expression remained static (Fig. 2C and fig. S1A). Differentiated FCs retained positive expression of the previously reported monocyte lineage marker CD14, as well as the progenitor cell homing receptor CXCR4 (also known as CD184) (fig. S1B).
Fig. 2. Characterization of human FC differentiation in vitro.
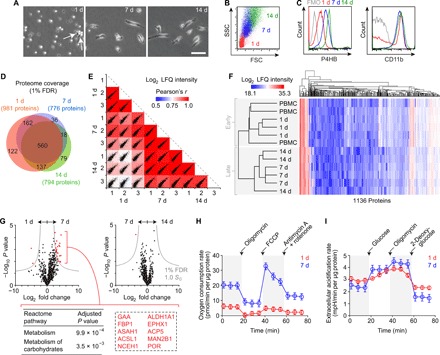
(A) Phase-contrast images showing cell morphology after 1, 7, and 14 days in monolayer culture. White arrows indicate early somatic protrusions. Scale bar, 50 μm. (B) FSC and SSC features on flow cytometry at 1, 7, and 14 days. (C) P4HB and CD11b expression at 1, 7, and 14 days. Fluorescence minus one (FMO) controls are shown in gray. (D) Venn diagram summarizing proteome coverage at 1, 7, and 14 days (n = 3 per condition). (E) Scatterplot matrix summarizing correlation analysis of log2 LFQ intensities (relative protein abundances) at 1, 7, and 14 days (n = 3 per condition). Correlation coefficients corresponding to each scatterplot (calculated using Pearson’s r) are represented by the heatmap overlay. (F) Hierarchical clustering analysis of log2 LFQ intensities in PBMCs and differentiating FCs at 1, 7, and 14 days (n = 3 per condition). (G) Volcano plots summarizing differential protein abundance at 1 day versus 7 days and 7 days versus 14 days (n = 3 per condition). Gray curves denote cutoff criteria, generated in Perseus; P values were calculated using Student’s t test. The table lists Reactome pathway terms significantly enriched in the protein set overrepresented at 7 days compared to 1 day; the dashed rectangle highlights the specific proteins associated with enrichment of both terms. The term list was generated using Enrichr; P values were calculated using Fisher’s exact test with Benjamini-Hochberg adjustment. Additional enrichment data are presented in table S1. (H) Real-time OCRs of differentiating FCs at 1 and 7 days (n = 10 per condition). Black arrows indicate timing of injection of the specified compounds. Data are shown as means ± SEM. Additional analyses are presented in fig. S2A. (I) Real-time extracellular acidification rates of differentiating FCs at 1 and 7 days (n = 4 per condition). Black arrows indicate timing of injection of the specified compounds. Data are shown as means ± SEM. Additional analyses are presented in fig. S2B.
Having obtained robust FC differentiation in vitro, we conducted liquid chromatography–tandem mass spectrometry to assess cellular proteome changes associated with the transition of adherent monocytes to differentiated FCs. We implemented a 1% false discovery rate (FDR) and identified 981, 776, and 794 proteins at 1, 7, and 14 days, respectively; the greatest overlap in protein identification was between the 7- and 14-day conditions (Fig. 2D). We further measured relative protein abundances using label-free quantification (LFQ) of spectral intensity (40) and evaluated group relationships via correlation and hierarchical clustering analyses. Log2 LFQ intensity correlations were strongest between the 7- and 14-day samples (r = 0.84 to 0.92) and weakest between the 1- and 14-day samples (r = 0.70 to 0.78; Fig. 2E). Hierarchical clustering, performed with the inclusion of an autologous PBMC condition, organized the samples into early (PBMCs and 1 day) and late (7 and 14 days) FC differentiation phases (Fig. 2F). As the greatest shift in proteome composition (as well as the most marked change in cell morphology; Fig. 2A) occurred during the initial 7-day period, we further interrogated the set of significantly overabundant proteins at 7 days compared to 1 day (Fig. 2G and table S1). Enrichment analysis using the Reactome pathway database (41) revealed significant up-regulation of cellular metabolism at 7 days, driven by a core set of 10 enzymes that are associated with lipid [e.g., long-chain fatty acid–coenzyme A ligase 1 (ACSL1)] and glucose [e.g., lysosomal α-glucosidase (GAA), fructose-1,6-bisphosphatase 1 (FBP1)] metabolism.
We sought to validate these proteomic data by characterizing cellular bioenergetics during the first 7 days of FC differentiation. Given the identification of enzymes associated with utilization of both lipids and glucose as cellular energy sources, we assayed mitochondrial respiration and glycolysis. FCs exhibited a significantly greater basal oxygen consumption rate (OCR) at 7 days compared to 1 day, consistent with increased oxidative phosphorylation within mitochondria (Fig. 2H). We further probed mitochondrial function by measuring real-time OCR during sequential treatment with the adenosine triphosphate inhibitor oligomycin, the protonophore and uncoupling agent carbonyl cyanide p-trifluoromethoxyphenyl-hydrazone (FCCP), and the electron-transport chain inhibitors antimycin A and rotenone. FCs at 7 days had greater maximum and reserve respiratory capacities; FCs at 1 day were operating near maximum capacity with no available reserve (fig. S2A). In contrast with this increase in mitochondrial fitness, differentiating FCs exhibited comparable basal extracellular acidification rates at 1 and 7 days, suggesting no difference in baseline glycolytic activity (Fig. 2I). Sequential treatment with glucose, oligomycin, and the glycolysis inhibitor 2-deoxyglucose revealed greater glycolytic capacity in the 7-day cells (fig. S2B).
These metabolic profiles indicate that differentiating FCs maintain a bivalent energy production phenotype while undergoing a substantial increase in metabolic capacity between 1 and 7 days. This increased capacity favors oxidative phosphorylation. To examine whether FC metabolic reprogramming is solely associated with differentiation or also supports proliferation, we additionally examined in vitro colony formation and population growth. During the initial 7-day differentiation period, most FC colonies were limited to 10 to 20 cells that exhibited greater maturity (larger cell size and longer somatic projections) at the periphery (fig. S1C). We rarely observed large, well-developed colonies; we never observed colony formation following subculture. Cell count data confirmed limited FC proliferation (fig. S1D), consistent with previous reports (13), alongside no evidence of cell membrane damage or death (fig. S1E). These data suggest that metabolic reprogramming during early FC differentiation does not support cell proliferation as well.
Assessment of mesenchymal function and potential for stromal replacement
A definitive feature of FCs is their ability to extravasate from the vasculature and assume a mesenchymal phenotype (32). To evaluate how closely differentiated FCs resemble classic mesenchymal lineage cells in humans, we compared the 14-day FC proteome to those of primary bone marrow–derived mesenchymal stromal cells (MSCs) and fibroblasts isolated from skin [dermal fibroblasts (DFs)] and upper airway mucosa [VF fibroblasts (VFFs)]. FCs exhibited a distinct proteomic signature compared to the other cell populations (Fig. 3, A and B), suggesting a unique (i.e., non-mesenchymal or non-classic mesenchymal) phenotype. Enrichment analysis of the differentially abundant proteins that defined the two largest hierarchical clusters (labeled according to mesenchymal or hematopoietic origin in Fig. 3B) identified the mesenchymal cells as more engaged in matrix synthesis and organization, as well as more proliferative; in contrast, FCs were more engaged in cellular metabolism, reactive oxygen and nitrogen species production, and immunologic signaling (Fig. 3C and tables S2 and S3).
Fig. 3. Mesenchymal features of differentiated human FCs.
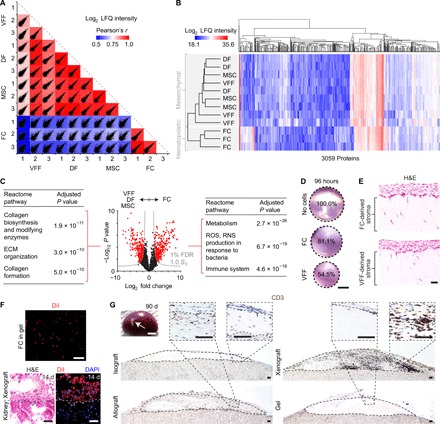
(A) Scatterplot matrix summarizing correlation analysis of log2 LFQ intensities (relative protein abundances) in FCs compared to MSCs, DFs, and VFFs (n = 3 per condition). Correlation coefficients corresponding to each scatterplot (calculated using Pearson’s r) are represented by the heatmap overlay. (B) Hierarchical clustering analysis of log2 LFQ intensities in all cell types (n = 3 per condition). (C) Volcano plot summarizing differential protein abundance across the two largest hierarchical clusters [FCs (hematopoietic origin) compared to MSCs, DFs, and VFFs (mesenchymal origin); n = 3 per condition]. Gray curves denote cutoff criteria, generated in Perseus; P values were calculated using Student’s t test. The tables list the top three Reactome pathway terms significantly enriched in the protein set overrepresented in MSCs, DFs, and VFFs (left) or FCs (right). Term lists were generated using Enrichr; P values were calculated using Fisher’s exact test with Benjamini-Hochberg adjustment. Additional enrichment data are presented in tables S2 and S3. (D) FC contractility in collagen matrix over 96 hours, compared to VFF and no-cell controls. Scale bar, 5 mm. (E) H&E-stained sections showing morphology of engineered mucosae generated using FCs or VFFs as the stromal cell source. Scale bar, 50 μm. (F) DiI-labeled, FC-derived engineered stromae in organotypic culture and 14 days after subrenal capsule xenoimplantation in rats. Scale bar, 50 μm. (G) CD3+ T cell infiltration of FC-derived engineered iso-, allo-, and xenografts, 90 days after implantation in rats. Dashed black contour lines indicate the boundary of each graft. Scale bars, 2 mm (gross specimen) and 50 μm (sections). ROS, reactive oxygen species; RNS, reactive nitrogen species.
Despite their distinct proteomic signature, we pursued additional experiments to assess the capacity of differentiated FCs to operate as functional stromal cells, with a view to their use for bioengineering autologous connective tissues. We reasoned that FCs hold potential as a tissue engineering cell source because of the following: (i) Despite phenotypic differences with MSCs and fibroblasts, FCs are collagen-producing cells (Fig. 2C and fig. S1A) (13). (ii) Similar to multipotent MSCs and fibroblasts (42, 43), FCs can be directed into chondrogenic, osteogenic, and adipogenic lineages (21, 22). (iii) Immunosignaling by FCs primarily involves antigen presentation via major histocompatibility complex (MHC) class II molecules (44) (which is of limited concern in an autologous therapy paradigm), and adoptive cell transfer of syngeneic FCs is well tolerated and, in certain scenarios, immunomodulatory (23, 24).
First, we assessed cytoskeletal contractility by seeding differentiated FCs in a three-dimensional collagen matrix. FCs exhibited moderate contractility over 96 hours compared to VFF-positive controls (Fig. 3D), consistent with previous work comparing FCs to DFs (32). In organotypic coculture (6), collagen-encapsulated FCs supported the proliferation and stratification of VF epithelial cells, yielding mucosae that were indistinguishable from those engineered using VFFs as the stromal cell source (Fig. 3E). Next, as CD14+ monocytes can assume an epithelial phenotype under permissive conditions (45, 46), we cultured adherent monocytes in epithelial cell–oriented medium in an attempt to derive functional epithelial cells that could be cocultured with FCs to engineer blood-sourced, multicellular, autologous tissues. At 14 days, the cultured cells expressed the epithelial progenitor markers ∆N-tumor protein 63 and keratin 5, as well as the junctional protein E-cadherin (CDH1, also known as CD324; fig. S3A). They exhibited a cuboidal morphology with no somatic protrusions (fig. S3B) and, in some cases, self-assembled into cellular chains (fig. S3C). Despite having these epithelial cell features, however, the cells were unable to mature beyond a monolayered epithelium in organotypic coculture (fig. S3D), irrespective of the stromal cell source and despite culture conditions favoring stratification.
Given the limited functionality of these monocyte-derived epithelial cells, we focused on the FC-derived stroma and conducted in vivo survival and immunotolerance assays. We used a rat subrenal capsule graft assay based on its precedence in immunobiology (47) and because rats have sufficient blood volumes and FC yields to allow stromal engineering. Brown Norway (BN/Crl; MHC haplotype RT1n) and Lewis (LEW/Crl; MHC haplotype RT1I) strain rats were each implanted with a subrenal capsule isograft (BN-to-BN; LEW-to-LEW), contralateral to an allograft (BN-to-LEW; LEW-to-BN) or xenograft (human-to-BN or human-to-LEW). We initially labeled mature FCs with the lipophilic dye DiIC18 (3) (DiI) and identified FC survival and localization within the grafts for 14 days after implantation (Fig. 3F), after which the dye signal faded to background autofluorescence levels, presumably due to either in vivo proliferation, cell-to-cell transfer, or leakage of DiI across the cell membrane (48). At 90 days after implantation, all grafts were intact (Fig. 3G). The allografts and xenografts exhibited minimal and moderate infiltration by CD3+ T cells, respectively, whereas the contralateral isografts showed no evidence of infiltration, consistent with self-tolerance by the adaptive immune system.
Together, these experiments support the use of differentiated FCs as an autologous cell source for bioengineering connective tissues. As FCs exhibit limited in vitro proliferation with standard isolation and culture techniques (fig. S1D), we explored additional strategies to maximize FC yield for translational applications. Previous work has shown enhanced differentiation efficiency when PBMCs are plated at ~8 × 104 cells/cm2 density on plastic and cultured in serum-free medium (33); however, withholding serum yields FCs with substantial phenotypic differences compared to those obtained using a standard approach (49). We therefore maintained serum supplementation and assessed an array of adhesion-promoting culture substrates, as well as the method used to isolate PBMCs from whole blood. FC yield at 14 days was highest on a commercial substrate derived from human plasma (MesenCult-SF; STEMCELL Technologies), followed by in-house preparations of human plasma and a collagen-fibronectin-serum albumin cocktail; yields were lowest (and FCs were morphologically less mature) on Matrigel, collagen, and plastic (Fig. 4A). Furthermore, FC yield at 14 days was 2.5- to 7.5-fold higher when PBMCs were isolated using ammonium-chloride-potassium (ACK) lysis of red blood cells, compared to density centrifugation with polyester gel separation (Vacutainer CPT; BD Biosciences) (Fig. 4B). Follow-up analyses confirmed that the PBMC isolation method had no effect on FC differentiation at the proteome level (Fig. 4, C to F).
Fig. 4. Optimization of human FC yield.
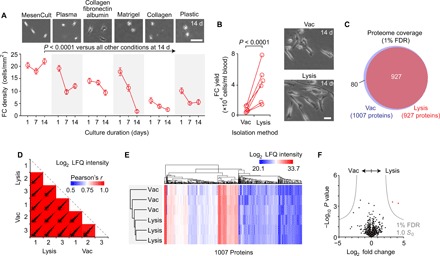
(A) Effect of culture substrate on FC yield. Data in the time-course plot are shown as means ± SEM. The P value was calculated using analysis of variance (ANOVA) (n = 4 to 12 per condition). The phase-contrast images show FC morphology at 14 days. Scale bar, 200 μm. (B) Effect of the PBMC isolation method [Vacutainer CPT (vac) versus ACK lysis (lysis) of red blood cells] on FC yield. The P value was calculated using Student’s t test (n = 6 per condition). The phase-contrast images show FC morphology at 14 days. Scale bar, 25 μm. (C) Venn diagram summarizing proteome coverage in FCs differentiated following vac or lysis (n = 3 per condition). (D) Scatterplot matrix summarizing correlation analysis of log2 LFQ intensities (relative protein abundances) in FCs differentiated following vac or lysis (n = 3 per condition). Correlation coefficients corresponding to each scatterplot (calculated using Pearson’s r) are represented by the heatmap overlay. (E) Hierarchical clustering analysis of log2 LFQ intensities in FCs differentiated following vac or lysis (n = 3 per condition). (F) Volcano plot summarizing differential protein abundance in FCs differentiated following vac or lysis (n = 3 per condition). Gray curves denote cutoff criteria, generated in Perseus; P values were calculated using Student’s t test.
FCs promote fibroblast migration and wound closure via paracrine CXCL8 signaling
FCs are associated with normal and aberrant wound healing; however, their role in the complex, multicellular wound milieu is controversial. Although FCs mobilize and migrate to wound sites (24, 32) and are detectable in circulation and fibrotic lesions at concentrations that correlate with pathologic severity (11, 12, 14), robust in vivo knockout data indicate that FCs synthesize minimal COL1 during matrix remodeling and are nonessential for fibrosis (50). Rather, FCs may primarily influence wound healing via paracrine signaling of tissue-resident cells, including fibroblasts (51, 52). To investigate this putative function, we assessed the influence of cocultured FCs on fibroblast behavior in an in vitro scratch assay. Monolayered VFFs and DFs received uniform scratch wounds and were immediately exposed to FCs via a Transwell insert. In this system, a 0.4-μm-pore–sized membrane allows transmission of secreted molecules but prevents direct-contact cellular signaling. Coculture with FCs resulted in greater fibroblast repopulation of the wound region at 20 hours compared to scratch-only controls, consistent with a paracrine effect (Fig. 5A). To determine whether this positive FC effect was associated with fibroblast proliferation, migration, or both, we measured cell density adjacent to the wound edge and performed immunohistochemistry for the proliferation marker Ki-67 (KI67), the microfilament protein F-actin (F-ACT), and the focal adhesion and spreading enzyme protein tyrosine kinase 2 (PTK2, also known as focal adhesion kinase). FCs had no impact on fibroblast density, KI67 expression, or overall F-ACT abundance adjacent to the wound edge but were associated with significantly increased PTK2 abundance within F-ACT+ fibroblast clusters (Fig. 5B). This suggests that FCs promote in vitro wound closure by up-regulating fibroblast motility, potentially via PTK2 (53).
Fig. 5. FCs promote fibroblast migration and wound closure via paracrine CXCL8 signaling.
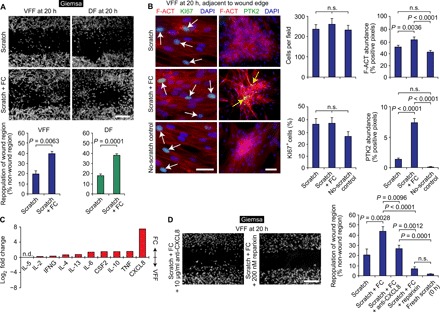
(A) In vitro repopulation of linear scratch wounds in fibroblast monolayers, with and without FC coculture. Scale bar, 500 μm. Data in the bar graphs are shown as means ± SEM. P values were calculated using Student’s t test (n = 3 to 6 per condition). (B) Cell density, as well as F-ACT, KI67, and PTK2 immunostaining patterns at 20 hours. White arrows indicate KI67+ nuclei; yellow arrows indicate F-ACT+PTK2+ cytoskeletal structures. Scale bars, 50 μm. Data in the bar graphs are shown as means ± SEM. P values were calculated using ANOVA (n = 6 to 18 per condition); n.s., not significant. (C) Differential cytokine production by FCs compared to VFFs, cultured under basal conditions for 24 hours. Data are shown as a single value per cytokine, assayed using pooled culture supernatant (n = 6 per condition); n.d., not detected. Raw concentration data are presented in table S4. TNF, tumor necrosis factor; IL, interleukin; IFNG, interferon-g; CSF2, colony-stimulating factor 2. (D) Effect of CXCL8 signaling inhibition on FC promotion of wound repopulation. Scale bar, 500 μm. Data in the bar graph are shown as means ± SEM. P values were calculated using ANOVA (n = 4 to 10).
To identify candidate signaling molecule(s) that might account for the FC effect on fibroblast motility, we assayed cytokine secretion using a multiplexed protein array. Under basal culture conditions and compared to VFFs, FCs secreted higher amounts of all cytokines detected in our panel (Fig. 5C and table S4). The greatest difference (a ~200-fold higher concentration in FC culture supernatant) was observed for the chemokine CXCL8 (also known as interleukin-8). Therefore, following a dose-response screen (Materials and Methods), we repeated the VFF scratch assay with the addition of an anti-CXCL8 neutralizing antibody or the drug reparixin, which acts as a CXCL8 inhibitor by allosterically binding and preventing activation of its primary receptors, CXCR1 and CXCR2 (54). In the presence of FCs, anti-CXCL8 (10 μg/ml) suppressed and 200 nM reparixin arrested VFF migration and repopulation of the wound region (Fig. 5D). These data indicate that FCs promote fibroblast migration and in vitro wound closure via paracrine CXCL8 signaling.
DISCUSSION
Our data show that FCs hold potential as an autologous cell source for regenerative medicine applications. This conclusion is notable, given that FCs have a reputation as key effector cells in chronic inflammation and fibrosis (8); however, FCs share the multipotent capacity of other mesenchymal progenitors (21, 22) and have shown therapeutic effect following adoptive transfer (or exosome delivery) in multiple preclinical disease models (23–25). Our results align with, and expand upon, these previous reports by showing that in vitro–differentiated FCs can be used to bioengineer functional connective tissues and by demonstrating one signaling mechanism by which FCs influence fibroblast behavior in a wound context. This research provides a foundation for future investigation of FC-based therapeutics; moreover, it provides a rationale for ongoing mechanistic work examining how FCs, like other monocyte lineage cells (55), exhibit starkly different phenotypes in response to their differentiation state and milieu.
FCs share a number of phenotypic features with MSCs, making it important to clarify whether these cell populations are distinct or overlapping. FCs and MSCs have similar morphologic and mesenchymal characteristics, are plastic adherent and multipotent in vitro, can synthesize cytokines including CXCL8 (56), and have regenerative potential. In addition, although classically isolated from bone marrow, adipose tissue, and other stromal sources, MSCs have been identified in peripheral blood (57, 58). Despite these shared characteristics, FCs and MSCs have substantially different in vitro proliferation capabilities, proteomes, and cell-surface immunophenotypes: FCs express cell surface markers consistent with their hematopoietic lineage (e.g., CD11b, CD14, CD34, and CD45) (9); in contrast, MSCs lack expression of these markers by definition (59), and peripheral blood–derived MSCs are isolated from the CD45− subset of circulating cells (58). This clear separation by lineage supports the notion that FCs and MSCs are phenotypically distinct.
Last, although defined by core characteristics, fibroblasts and MSCs are heterogeneous cell populations marked by phenotypic variation across donors, tissue sites (e.g., bone marrow versus adipose tissue), tissue subsites (e.g., upper versus lower dermis), individual clones, and individual cells (60–62). To our knowledge, there are no published data showing such biological heterogeneity among FCs; however, it is highly likely and therefore an important area of future investigation. Our findings help to define the human FC phenotype at the population level but, in doing so, might also mask biologically meaningful differences across and within FC subpopulations. These differences could be interrogated using single-clone and single-cell profiling of transcript and protein abundance, as well as cell behavior.
MATERIALS AND METHODS
Blood cell isolation and primary culture
Normal human peripheral blood samples were procured from leftover clinical specimens with the approval of the University of Wisconsin Health Sciences Institutional Review Board. The deidentified human donors were restricted to adult males and females with no known hematopoietic or immunologic disorders; all blood was processed within 4 hours of clinical venipuncture. Normal rat peripheral blood samples were obtained from experimentally naïve animals via jugular venipuncture with approval of the University of Wisconsin School of Medicine and Public Health Animal Care and Use Committee. PBMCs were isolated using either Vacutainer CPT tubes (BD Biosciences) or following ACK lysis of red blood cells (RBC lysis buffer, 5 to 6 min of incubation; eBioscience), according to the manufacturers’ protocols.
For FC purification and differentiation, freshly isolated PBMCs were rinsed and resuspended in complete medium [DMEM containing 10% fetal bovine serum (FBS) and antibiotic-antimycotic solution (100 U/ml; Sigma-Aldrich)] and seeded at a density of 1 × 105 to 5 × 105 cells/cm2 on a culture substrate [MesenCult-SF (40 μg/ml; STEMCELL Technologies) or, where indicated, one of the following: Vacutainer-isolated human plasma, diluted 1:1 (v/v) in DMEM; Earle’s Balanced Salt Solution (EBSS) containing COL1 (30 μg/ml), fibronectin (10 μg/ml), and bovine serum albumin (BSA; 10 μg/ml) (63); growth-factor–reduced Matrigel (200 μg/ml; Corning); COL1 (100 μg/ml); or plastic]. Cells were cultured at 37°C in 5% CO2 with medium change every 48 hours.
For monocyte-derived epithelial cell differentiation (beginning 3 days after PBMC seeding and culture), cells were transitioned to epithelial medium [DMEM/Ham’s F-12 supplemented with bovine pituitary extract (15 μg/ml), epidermal growth factor (10 ng/ml), epinephrine (0.5 μg/ml), insulin (5 μg/ml), transferrin (10 μg/ml), triiodo-l-thyronine (10 ng/ml), hydrocortisone (0.5 μg/ml), retinoic acid (0.1 ng/ml), BSA (1.5 μg/ml), antibiotic-antimycotic solution (100 U/ml), and 1% FBS (all reagents from Sigma-Aldrich)] (63). Cells were cultured at 37°C in 5% CO2 with medium change every 48 hours.
Phase-contrast images of live cells were captured using an Olympus CKX41 inverted microscope and a SPOT RT color digital camera (Diagnostic Instruments). To eliminate artifacts caused by incomplete FC detachment during trypsinization, we avoided hematocytometry and performed cell counts using low-magnification (4×) images of each well. We randomly selected four image fields per well, counted every cell in each field, and normalized the total to either cells per square millimeter (comparison of FC yield on various culture substrates) or cells per milliliter of whole blood (comparison of FC yield following Vacutainer CPT or ACK lysis isolation of PBMCs). Cell morphology was imaged at 10× and 20× magnification.
Nonblood cell isolation and primary culture
All primary human tissues and nonblood cells were procured or purchased commercially with approval of the University of Wisconsin Health Sciences Institutional Review Board. Normal VFFs and epithelial cells were isolated from three donors (58-year-old Caucasian female, 61-year-old Caucasian male, and 66-year-old Caucasian male) undergoing total laryngectomy with no clinical evidence of VF pathology, as previously described (6). Normal DFs were purchased from the American Type Culture Collection (ATCC) (40-year-old Asian female) and Lonza (42-year-old African American female and 59-year-old Caucasian male).
Normal bone marrow–derived MSCs were purchased from ATCC or isolated from leftover clinical specimens as previously described (64). Briefly, cells were isolated from filters retained after bone marrow harvest from donors (with no known hematopoietic or immunologic disorders) to human leukocyte antigen-matched sibling recipients. Specimens were rinsed in phosphate-buffered saline (PBS), and mononuclear cells were isolated using Ficoll-Hypaque (1.073 g/ml; GE Healthcare) and Leucosep tubes (Greiner Bio-One), according to manufacturer protocols. Following ACK lysis of red blood cells (3-min incubation), mononuclear cells were suspended in α-MEM containing 10% FBS (HyClone), 1× nonessential amino acids, and 4 mM l-glutamine (Sigma-Aldrich). Cells were cultured on plastic at 37°C in 5% CO2 with medium change every 72 hours. Nonadherent cells were removed at the first medium change; adherent cells were passaged before confluence and characterized as MSCs according to criteria of the International Society for Cellular Therapy (59).
Before experimental use, primary VFFs, DFs, and MSCs were seeded at a density of 0.5 × 104 to 1.0 × 104 cells/cm2 on plastic and cultured in complete medium at 37°C in 5% CO2. Medium was changed every 48 to 72 hours. Primary VF epithelial cells were seeded at a density of 0.5 × 104 to 1.0 × 104 cells/cm2 on a matrix substrate [EBSS containing COL1 (30 μg/ml), fibronectin (10 μg/ml), and BSA (10 μg/ml)] (63) and cultured in epithelial medium at 37°C in 5% CO2. Medium was changed every 24 to 48 hours.
Organotypic culture
Purified rat tail COL1 (BD Biosciences) was prepared according to the manufacturer’s instructions and seeded with 2 × 105 stromal cells (VFFs or FCs)/ml, yielding a final COL1 concentration of 2.4 mg/ml. The cell-scaffold mixture was then added to the apical chamber of a 24-well culture insert and polymerized at 37°C for 40 min. Complete medium was added to both apical and basolateral chambers; the cells were maintained in organotypic culture for up to 14 days with medium change every 24 hours.
A subset of organotypic cultures were seeded with epithelial cells following 24 hours in complete medium. VF or monocyte-derived epithelial cells were seeded on the polymerized scaffold surface within the apical chamber at a density of 2 × 105 cells per well. Epithelial medium was added to the apical chamber; a 1:1 ratio of complete and epithelial medium was added to the basolateral chamber. After an additional 48 hours in culture, the epithelial medium was aspirated from the apical chamber, leaving the epithelial cells at the air-liquid interface. The cells were maintained in organotypic culture for a total of 14 days with basolateral chamber medium change every 24 hours.
Rat transplantation experiments
Brown Norway (BN/Crl; MHC haplotype RT1n) and Lewis (LEW/Crl; MHC haplotype RT1I) rats aged 12 weeks were used for all in vivo experiments (Charles River Laboratories). All protocols were approved by the University of Wisconsin School of Medicine and Public Health Animal Care and Use Committee. Peripheral blood (up to 2 ml) was drawn from a subset of animals via jugular venipuncture and processed for PBMC isolation (via ACK lysis), followed by FC differentiation (14-day culture duration) and organotypic culture (an additional 14-day culture duration). Donor rats did not serve as graft recipients. Recipient rats were implanted with a subrenal capsule isograft (BN-to-BN; LEW-to-LEW), contralateral to an allograft (BN-to-LEW; LEW-to-BN) or xenograft (human-to-BN or human-to-LEW) and monitored for up to 90 days. Acellular polymerized COL1 served as a negative control. Grafts were harvested at the experimental endpoint and processed for histology and immunohistochemistry.
A subset of organotypic cultures intended for in vivo transplantation were rinsed in PBS and incubated in Vybrant CM-DiI cell-labeling solution (5 μM; Life Technologies) at 37°C for 5 min, followed by 4°C for 5 min. The staining procedure was stopped by adding 2:1 v/v complete medium, rinsing in PBS, and returning the organotypic culture to complete medium.
Fibroblast scratch experiments
Fibroblasts were seeded in 24-well plates at a density of 2 × 104 cells per well and cultured in complete medium for 48 to 72 hours, until 75% confluent. The cells were then serum-starved for 4 hours. A linear scratch injury was created across the cell monolayer using a sterile 1000-μl pipette tip, the cells were rinsed in serum-free medium, and a culture insert containing either 3 × 104 differentiated FCs (seeded in the apical chamber ~24 hours prior and cultured) or no cells was placed in each well. The cells were cultured in serum-free medium for an additional 20 hours and then processed for histology and immunocytochemistry.
We conducted additional experiments with the addition of an anti-CXCL8 neutralizing antibody (MAB208; R&D Systems) or reparixin (HY-15251; MedChem Express) following the scratch injury. We based our dosing regimen on published work (65, 66) and the following pilot. A dose-response screen showed greater suppression of fibroblast migration following treatment with 10 μg/ml, compared to 5 μg/ml, anti-CXCL8, as well as with 200 nM, compared to 100 nM, reparixin. Treatment with 400 nM reparixin resulted in loss of fibroblast adhesion and widespread detachment from the culture plate. We therefore used anti-CXCL8 (10 μg/ml; 20 hours post-scratch incubation time, concurrent with FC coculture) or 200 nM reparixin [30 min post-scratch incubation time, concurrent with the initiation of FC coculture (extended incubation time also caused fibroblast detachment)] for all experiments.
Cell metabolism assays
OCR and extracellular acidification rate (ECAR) were measured using the Seahorse XF96 extracellular flux instrument, the Mito and Glycolysis Stress Test kits, and manufacturer protocols (Agilent Technologies). Cells were seeded on Seahorse XF96 (96-well) plates at a density of 2 × 105 cells per well, in 200 μl of complete medium per well. After 24 hours of culture (37°C in 5% CO2), the cells were rinsed and the medium was replaced with 175 μl of assay-specific medium [for Mito stress test, Seahorse XF base medium was supplemented with 1 mM pyruvate, 2 mM glutamine, and 10 mM glucose (pH 7.4); for glycolysis stress test, Seahorse XF base medium was supplemented with 1 mM glutamine (pH 7.4)]. The cells were then incubated at 37°C with no CO2 for 1 hour.
Before starting the assays, each culture plate was placed on a sensor cartridge that had been hydrated in Seahorse XF calibrant (pH 7.4) at 37°C with no CO2 overnight. The assays were run with real-time collection of OCR and ECAR data over ~75 min. For the Mito stress test, 1 μM oligomycin, 0.3 μM FCCP, and 0.5 μM antimycin A plus 0.5 μM rotenone were sequentially injected as indicated in Fig. 2H; mix, wait, and measure durations were 90, 150, and 180 s, respectively. For the glycolysis stress test, 10 mM glucose, 1 μM oligomycin, and 50 mM 2-deoxyglucose were sequentially injected as indicated in Fig. 2I; mix, wait, and measure durations were 90, 90, and 180 s, respectively. All injected reagents were from Sigma-Aldrich; assay protocols and reagent doses were determined on the basis of the manufacturer’s guidance and a pilot experiment; all samples were run in technical quadruplicate.
Following data collection, cells were rinsed, incubated in 25 μl of radioimmunoprecipitation assay buffer per well (BioVision), and processed for measurement of protein concentration using a modified Lowry assay (DC Protein Assay; Bio-Rad), according to the manufacturer’s protocol. Absorbance was measured using a FlexStation 3 microplate reader (Molecular Devices); samples were run in technical triplicate. OCR and ECAR data were then normalized to total protein abundance per well, to adjust for the presumed effect of increased cell volume on metabolic function (Fig. 2, A and B). Metabolic data were analyzed using Wave 2.4 (Agilent Technologies).
Cell proliferation assay
Cells were seeded in 24-well plates at a density of 104 cells per well and cultured for 96 hours. As noted above for other cell count assays, we avoided hematocytometry and performed cell counts using low-magnification (4×) phase-contrast images of each well. We randomly selected four image fields per well, counted every cell in each field, and extrapolated to obtain an estimate of total cells per well.
Cell viability assay
Cell viability was measured via lactate dehydrogenase release using the CytoTox-ONE homogeneous membrane integrity assay (Promega), according to the manufacturer’s protocol. All samples were assayed in technical quadruplicate. Fluorescence was measured using a FlexStation 3 microplate reader (Molecular Devices).
Flow cytometry
Cells were washed and suspended in staining buffer (Hanks’ balanced salt solution containing 2% FBS and 10 mM Hepes). For cell surface staining, single-cell suspensions were incubated with fluorochrome-conjugated antibodies. For intracellular staining, unstained or surface-stained cells were washed and then fixed and permeabilized using Cytofix/Cytoperm (BD Biosciences) according to the manufacturer’s instructions. The permeabilized cells were then incubated with unconjugated or fluorochrome-conjugated antibodies against the intracellular targets, washed, and resuspended in staining buffer. Where necessary, an additional incubation step with the appropriate secondary antibody was performed. All antibodies are listed in table S5. Cells were also incubated with 4′,6-diamidino-2-phenylindole (DAPI): A change in DAPI fluorescence intensity was used to gate live versus dead cells. Samples were run on a four-laser, 14-color LSR II instrument (BD Biosciences), and data were analyzed using FlowJo 8.7.1 (Tree Star). We used compensation (OneComp eBeads; eBioscience), isotype (table S5), and fluorescence minus one (FMO) controls.
Histology, immunocytochemistry, and immunohistochemistry
Cultured cells (on various substrates in wells, dishes, or slide chambers) were rinsed with PBS and fixed using 2 to 4% paraformaldehyde. Native and engineered tissues were processed for paraffin (5-μm-thick) and frozen (8-μm-thick) sections. Routine hematoxylin and eosin (H&E) and Giemsa stains were used to assess tissue morphology and identify monolayered cells on cultureware, respectively.
Frozen sections used for immunostaining were incubated with 0.1% Triton X-100 for 15 min, Image iT-FX (Life Technologies) for 30 min, and Block ACE (AbD Serotec) and 5% donkey serum (Sigma-Aldrich) for 30 min. Fixed cells and sections were incubated with primary antibodies for 90 min followed by, where necessary, appropriate secondary antibodies for 90 min, with thorough wash steps between each incubation step. F-ACT was detected using rhodamine phalloidin (Invitrogen), according to the manufacturer’s instructions. A complete list of antibodies (and the phallotoxin) is provided in table S6. Cells and sections (including nonimmunostained sections containing DiI-labeled cells) were counterstained with DAPI, covered with VECTASHIELD antifade mounting medium (Vector Laboratories), and coverslipped.
Paraffin sections used for immunostaining were first processed for antigen retrieval in a decloaking chamber (Biocare Medical) using 10 mM citrate buffer (pH 6.0). Sections were permeabilized using 0.2% Triton X-100 for 10 min and incubated with 10% BSA in PBS for 60 min to block nonspecific binding before incubation with rabbit anti-rat CD3 primary antibody for 60 min (table S6). Thorough washing was performed between each incubation step. For horseradish peroxidase (HRP)–based detection, endogenous peroxidase was quenched using 3% hydrogen peroxide in PBS. ImmPRESS anti-rabbit immunoglobulin G (IgG) HRP polymer was used for secondary detection (30 min of incubation) and the ImmPACT DAB kit was used to develop the signal, according to the manufacturer’s instructions (all reagents from Vector Laboratories). Sections were counterstained with hematoxylin, dehydrated, cleared, and coverslipped.
Histological, immunocytochemical, and immunohistochemical images were captured using either a Nikon E600 upright microscope and an Olympus DP71 camera, or a Nikon Ti-S/L100 inverted microscope and a DS-Qi2 camera. Consistent exposure parameters were used for each immunostained protein of interest. Positive control cells and tissues (table S6) showed expected immunostaining patterns. Negative control sections, stained with an IgG isotype control or without the primary or secondary antibody, showed no immunoreactivity.
Proteomic analyses
Cells were lysed using 60 μl of 4% sodium dodecyl sulfate, 0.1 M tris-HCl (pH 7.5), and 0.1 M dithiothreitol; bath-sonicated for 3 min; and then heated to 95°C for 6 min. Next, 0.1% w/v deoxycholic acid in 8 M urea was added to a final volume of 600 μl. Cell lysates were then processed according to the enhanced filter-aided sample preparation protocol (67) using Amicon 0.5-ml 30,000 molecular weight cutoff (MWCO) centrifugal ultrafiltration units (EMD Millipore). Briefly, filters and collection tubes were passivated through overnight incubation with 1% CHAPS and thoroughly washed in Milli-Q water. Samples were loaded onto the filters, washed, reduced, alkylated with iodoacetamide, washed further, and then digested with trypsin [40:1 (w/w) protein/enzyme ratio] at 37°C overnight to generate peptides. These peptides, now small enough to pass through the MWCO filter, were collected in the filtrate following centrifugation, and 10% trifluoroacetic acid was added to a final concentration of 0.5%. Residual deoxycholic acid was removed via liquid-liquid extraction using ethyl acetate. Samples were evaporated to dryness in a SpeedVac, reconstituted in 180 μl of 0.1% trifluoroacetic acid, desalted using C18 Bond Elut OMIX 100-μl pipette tips (Agilent), and eluted with 0.1% trifluoroacetic acid in 70% acetonitrile. Last, peptides were dried in a SpeedVac and reconstituted in 5% acetonitrile and 0.2% formic acid.
Approximately 1 μg of reconstituted peptides, as estimated by a BCA assay (Pierce), was injected into a Waters nanoACQUITY HPLC coupled to an electrospray ionization ion-trap/Orbitrap mass spectrometer (LTQ Orbitrap Velos; Thermo Fisher Scientific). Peptides were separated at 60°C on a 100-μm-inner-diameter column packed with 20 cm of 1.7-μm ethylene-bridged hybrid C18 particles (Waters) and eluted at 0.3 μl/min in 0.2% formic acid with a gradient of increasing acetonitrile over 2.5 hours. A full mass scan [300 to 1500 mass/charge ratio (m/z)] was performed in the Orbitrap at 60,000 resolution. The 10 most intense MS1 peaks were selected for fragmentation by higher-energy collisional dissociation at 42% collision energy, and the resulting fragments were analyzed with a resolution of 7500 and an isolation width of 2.5 m/z. Dynamic exclusion was enabled with a repeat count of 2 over 30 s and an exclusion duration of 120 s.
The raw data files were analyzed using MaxQuant 1.5.3.30 (68) and the UniProt Homo sapiens database (20,278 reviewed canonical sequences; downloaded on 5 December 2013) supplemented with 262 common contaminant sequences. Precursor and fragment ion mass tolerances were set to 4.5 and 20.0 ppm, respectively; peptide and protein FDRs were set at 1%; fixed carbamidomethylation of cysteine, variable oxidation of methionine, and variable N-terminal acetylation were specified modifications; up to two missed tryptic cleavages were allowed; the minimum peptide length was set to seven amino acids; and a minimum of two unique peptides was required for protein identification. Protein groups containing matches to proteins from a reversed decoy database and to the contaminant sequences were discarded. The LFQ algorithm embedded in the MaxQuant software package was used (40). Quantification was limited to proteins identified in at least two independent biological replicates per condition; only unique and razor peptides were used; a minimum of two peptides per protein was required. Perseus 1.5.4.0 (69) was used for downstream data processing and analysis. LFQ intensities were log2-transformed, and missing values were inserted using data imputation based on an observed normal distribution, using an SD width of 0.5 and a mean distribution downshift of 1.8.
Protein array
Cells were seeded in 24-well plates at a density of 104 cells per well, cultured in complete medium for 72 hours, rinsed with PBS, and then serum-starved for 24 hours. Culture supernatants were harvested, pooled across six biological replicates per cell type, and processed for measurement of cytokine concentrations using the Quantibody Human T helper cell 1(TH1)/TH2 Array 1 platform (RayBiotech), according to the manufacturer’s instructions. Serum-free medium was used as a negative control and for serial dilution of cytokine standards. Each cytokine of interest was assayed in technical quadruplicate. The array was scanned and data were processed using Q-Analyzer software (RayBiotech).
Image analyses
In vitro wound repopulation analyses were performed using one low-magnification field (4×) per replicate; immunohistochemistry-based analyses of cell density and fluorescence intensity were performed using six non-overlapping, high-magnification fields (20×) per replicate.
For measurement of in vitro wound repopulation, 8-bit grayscale images of Giemsa-stained cells (encompassing the entire scratch) were analyzed using ImageJ (70). We implemented the following algorithm to control for any differences in basal cell density between replicates, as well as any change in background signal intensity in the wound region due to scratching the culture substrate. After setting thresholds to detect Giemsa-stained cells (applied consistently across all samples), mean signal intensity in the wound region (normalized to multiple cell-free background regions) was divided by mean signal intensity in a non-wound region of the same image (again, normalized to multiple cell-free background regions) and converted to a percentage.
For measurements of cell density and fluorescence intensity, 8-bit RGB (red-green-blue) images were analyzed using NIS-Elements Advanced Research 4.5 (Nikon). Fields were randomly selected from the region adjacent to the wound edge; negative (no-scratch) control fields were randomly selected from any image region. Cell density was measured by manual count of all DAPI+ nuclei; KI67+ cell density was measured by calculation of percent DAPI+ nuclei coexpressing KI67. Protein abundance was estimated by creating a binary layer, setting single-channel (red or green) thresholds to detect the target immunosignal (applied consistently across all samples), and measuring fluorescent signal intensity (percent positive pixels).
Statistical analyses
Quantitative proteomic data (log2 LFQ intensity) were analyzed in Perseus 1.5.4.0 (69) using a 1% FDR, generated using an artificial within-groups variance (S0) of 1.0 and permutation analysis based on a two-sample Student’s t test. Additional quantitative proteomic analyses were conducted in Perseus using hierarchical clustering and calculation of Pearson’s r. Enrichment analysis of differentially abundant proteins was performed using the Enrichr algorithm (71) (based on Fisher’s exact test with Benjamini-Hochberg adjustment) and Reactome database (41). Other statistical testing was performed using R (72). Metabolic, cell count, cell viability, and image analysis data were analyzed using a two-sample Student’s t test in cases of two experimental groups or one-way analysis of variance (ANOVA) in cases of more than two experimental groups. In all ANOVA models, if the F test revealed a significant difference, pairwise comparisons were performed using Tukey’s honest significant difference test. A preadjustment type I error rate of 0.01 was used; enrichment analyses were subject to an additional restriction of at least four protein identifications per pathway. All P values were two-sided.
Supplementary Material
Acknowledgments
We thank S. H. Dailey, G. K. Hartig, T. M. McCulloch, P. Hematti, J. A. Kink, and the staff of the UW Health Clinical Laboratory for providing tissue, blood, and cell isolates for primary cell culture; S. Kinoshita and D. A. Roenneburg for tissue processing and histology; Y. Yamaoka for image analysis; and B. M. Hanlon for statistical consultation. Funding: This work was supported by grants R01 DC004428 and R01 DC010777 from the National Institute on Deafness and Other Communication Disorders. Flow cytometry and cellular bioenergetic assays were performed in the Flow Cytometry Laboratory and Small Molecule Screening Facility of the University of Wisconsin (UW) Carbone Cancer Center, which was supported by grant P30 CA014520 from the National Cancer Institute. Author contributions: C.L. designed the experiments, performed all in vitro experiments, analyzed data, and helped write the manuscript. K.N. performed all in vivo experiments. Z.R. performed mass spectrometry and analyzed proteomic data. L.M.S. provided input on the proteomic data analysis and helped revise the manuscript. B.L.F. helped design the experiments, performed proteomic data analysis, and helped revise the manuscript. N.V.W. conceived the study, designed experiments, analyzed data, and helped write the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials; all raw MS data files are available from the MassIVE repository (http://massive.ucsd.edu) using the dataset identifier MSV000081656. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/5/eaav7384/DC1
Fig. S1. Additional characterization of human FC differentiation in vitro.
Fig. S2. Bioenergetic profiles of human FCs at 1 and 7 days.
Fig. S3. Characterization of monocytes following epithelial cell-oriented culture.
Table S1. Reactome pathways enriched in protein sets associated with FC differentiation.
Table S2. Reactome pathways enriched in the protein set overrepresented in VFFs, DFs, and MSCs compared to FCs.
Table S3. Reactome pathways enriched in the protein set overrepresented in FCs compared to VFFs, DFs, and MSCs.
Table S4. Cytokine production by FCs and VFFs, cultured under basal conditions for 24 hours.
Table S5. Antibodies and isotype controls used for flow cytometry.
Table S6. Antibodies and phallotoxin used for immunocytochemistry and immunohistochemistry.
REFERENCES AND NOTES
- 1.Weissman I. L., Shizuru J. A., The origins of the identification and isolation of hematopoietic stem cells, and their capability to induce donor-specific transplantation tolerance and treat autoimmune diseases. Blood 112, 3543–3553 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cossu G., Birchall M., Brown T., de Coppi P., Culme-Seymour E., Gibbon S., Hitchcock J., Mason C., Montgomery J., Morris S., Muntoni F., Napier D., Owji N., Prasad A., Round J., Saprai P., Stilgoe J., Thrasher A., Wilson J., Lancet commission: Stem cells and regenerative medicine. Lancet 391, 883–910 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Mount N. M., Ward S. J., Kefalas P., Hyllner J., Cell-based therapy technology classifications and translational challenges. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 370, 20150017 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nichols J. E., La Francesca S., Niles J. A., Vega S. P., Argueta L. B., Frank L., Christiani D. C., Pyles R. B., Himes B. E., Zhang R., Li S., Sakamoto J., Rhudy J., Hendricks G., Begarani F., Liu X., Patrikeev I., Pal R., Usheva E., Vargas G., Miller A., Woodson L., Wacher A., Grimaldo M., Weaver D., Mlcak R., Cortiella J., Production and transplantation of bioengineered lung into a large-animal model. Sci. Transl. Med. 10, eaao3926 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Marino D., Luginbühl J., Scola S., Meuli M., Reichmann E., Bioengineering dermo-epidermal skin grafts with blood and lymphatic capillaries. Sci. Transl. Med. 6, 221ra14 (2014). [DOI] [PubMed] [Google Scholar]
- 6.Ling C., Li Q., Brown M. E., Kishimoto Y., Toya Y., Devine E. E., Choi K.-O., Nishimoto K., Norman I. G., Tsegyal T., Jiang J. J., Burlingham W. J., Gunasekaran S., Smith L. M., Frey B. L., Welham N. V., Bioengineered vocal fold mucosa for voice restoration. Sci. Transl. Med. 7, 314ra187 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bucala R., Spiegel L. A., Chesney J., Hogan M., Cerami A., Circulating fibrocytes define a new leukocyte subpopulation that mediates tissue repair. Mol. Med. 1, 71–81 (1994). [PMC free article] [PubMed] [Google Scholar]
- 8.Reilkoff R. A., Bucala R., Herzog E. L., Fibrocytes: Emerging effector cells in chronic inflammation. Nat. Rev. Immunol. 11, 427–435 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bellini A., Mattoli S., The role of the fibrocyte, a bone marrow-derived mesenchymal progenitor, in reactive and reparative fibroses. Lab. Investig. 87, 858–870 (2007). [DOI] [PubMed] [Google Scholar]
- 10.Verstovsek S., Manshouri T., Pilling D., Bueso-Ramos C. E., Newberry K. J., Prijic S., Knez L., Bozinovic K., Harris D. M., Spaeth E. L., Post S. M., Multani A. S., Rampal R. K., Ahn J., Levine R. L., Creighton C. J., Kantarjian H. M., Estrov Z., Role of neoplastic monocyte-derived fibrocytes in primary myelofibrosis. J. Exp. Med. 213, 1723–1740 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shipe R., Burdick M. D., Strieter B. A., Liu L., Shim Y. M., Sung S.-s., Teague W. G., Mehrad B., Strieter R. M., Rose C. E. Jr., Number, activation, and differentiation of circulating fibrocytes correlate with asthma severity. J. Allergy Clin. Immunol. 137, 750–757.e3 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moeller A., Gilpin S. E., Ask K., Cox G., Cook D., Gauldie J., Margetts P. J., Farkas L., Dobranowski J., Boylan C., O'Byrne P. M., Strieter R. M., Kolb M., Circulating fibrocytes are an indicator of poor prognosis in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 179, 588–594 (2009). [DOI] [PubMed] [Google Scholar]
- 13.Schmidt M., Sun G., Stacey M. A., Mori L., Mattoli S., Identification of circulating fibrocytes as precursors of bronchial myofibroblasts in asthma. J. Immunol. 171, 380–389 (2003). [DOI] [PubMed] [Google Scholar]
- 14.Andersson-Sjöland A., de Alba C. G., Nihlberg K., Becerril C., Ramírez R., Pardo A., Westergren-Thorsson G., Selman M., Fibrocytes are a potential source of lung fibroblasts in idiopathic pulmonary fibrosis. Int. J. Biochem. Cell Biol. 40, 2129–2140 (2008). [DOI] [PubMed] [Google Scholar]
- 15.Isgrò M., Bianchetti L., Marini M. A., Bellini A., Schmidt M., Mattoli S., The C-C motif chemokine ligands CCL5, CCL11, and CCL24 induce the migration of circulating fibrocytes from patients with severe asthma. Mucosal Immunol. 6, 718–727 (2013). [DOI] [PubMed] [Google Scholar]
- 16.Medbury H. J., Tarran S. L., Guiffre A. K., Williams M. M., Lam T. H., Vicaretti M., Fletcher J. P., Monocytes contribute to the atherosclerotic cap by transformation into fibrocytes. Int. Angiol. 27, 114–123 (2008). [PubMed] [Google Scholar]
- 17.Yang L., Scott P. G., Giuffre J., Shankowsky H. A., Ghahary A., Tredget E. E., Peripheral blood fibrocytes from burn patients: Identification and quantification of fibrocytes in adherent cells cultured from peripheral blood mononuclear cells. Lab. Investig. 82, 1183–1192 (2002). [DOI] [PubMed] [Google Scholar]
- 18.Galligan C. L., Siminovitch K. A., Keystone E. C., Bykerk V., Perez O. D., Fish E. N., Fibrocyte activation in rheumatoid arthritis. Rheumatology 49, 640–651 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mathai S. K., Gulati M., Peng X., Russell T. R., Shaw A. C., Rubinowitz A. N., Murray L. A., Siner J. M., Antin-Ozerkis D. E., Montgomery R. R., Reilkoff R. A. S., Bucala R. J., Herzog E. L., Circulating monocytes from systemic sclerosis patients with interstitial lung disease show an enhanced profibrotic phenotype. Lab. Investig. 90, 812–823 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang C.-H., Huang C.-D., Lin H.-C., Lee K.-Y., Lin S.-M., Liu C.-Y., Huang K.-H., Ko Y.-S., Chung K. F., Kuo H.-P., Increased circulating fibrocytes in asthma with chronic airflow obstruction. Am. J. Respir. Crit. Care Med. 178, 583–591 (2008). [DOI] [PubMed] [Google Scholar]
- 21.Hong K. M., Burdick M. D., Phillips R. J., Heber D., Strieter R. M., Characterization of human fibrocytes as circulating adipocyte progenitors and the formation of human adipose tissue in SCID mice. FASEB J. 19, 2029–2031 (2005). [DOI] [PubMed] [Google Scholar]
- 22.Choi Y. H., Burdick M. D., Strieter R. M., Human circulating fibrocytes have the capacity to differentiate osteoblasts and chondrocytes. Int. J. Biochem. Cell Biol. 42, 662–671 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nemzek J. A., Fry C., Moore B. B., Adoptive transfer of fibrocytes enhances splenic T-cell numbers and survival in septic peritonitis. Shock 40, 106–114 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kao H.-K., Chen B., Murphy G. F., Li Q., Orgill D. P., Guo L., Peripheral blood fibrocytes: Enhancement of wound healing by cell proliferation, re-epithelialization, contraction, and angiogenesis. Ann. Surg. 254, 1066–1074 (2011). [DOI] [PubMed] [Google Scholar]
- 25.Geiger A., Walker A., Nissen E., Human fibrocyte-derived exosomes accelerate wound healing in genetically diabetic mice. Biochem. Biophys. Res. Commun. 467, 303–309 (2015). [DOI] [PubMed] [Google Scholar]
- 26.Pilling D., Buckley C. D., Salmon M., Gomer R. H., Inhibition of fibrocyte differentiation by serum amyloid P. J. Immunol. 171, 5537–5546 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pilling D., Fan T., Huang D., Kaul B., Gomer R. H., Identification of markers that distinguish monocyte-derived fibrocytes from monocytes, macrophages, and fibroblasts. PLOS ONE 4, e7475 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bellini A., Marini M. A., Bianchetti L., Barczyk M., Schmidt M., Mattoli S., Interleukin (IL)-4, IL-13, and IL-17A differentially affect the profibrotic and proinflammatory functions of fibrocytes from asthmatic patients. Mucosal Immunol. 5, 140–149 (2012). [DOI] [PubMed] [Google Scholar]
- 29.Schnoor M., Cullen P., Lorkowski J., Stolle K., Robenek H., Troyer D., Rauterberg J., Lorkowski S., Production of type VI collagen by human macrophages: A new dimension in macrophage functional heterogeneity. J. Immunol. 180, 5707–5719 (2008). [DOI] [PubMed] [Google Scholar]
- 30.Ling C., Yamashita M., Zhang J., Bless D. M., Welham N. V., Reactive response of fibrocytes to vocal fold mucosal injury in rat. Wound Repair Regen. 18, 514–523 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Niedermeier M., Reich B., Gomez M. R., Denzel A., Schmidbauer K., Göbel N., Talke Y., Schweda F., Mack M., CD4+ T cells control the differentiation of Gr1+ monocytes into fibrocytes. Proc. Natl. Acad. Sci. U.S.A. 106, 17892–17897 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abe R., Donnelly S. C., Peng T., Bucala R., Metz C. N., Peripheral blood fibrocytes: Differentiation pathway and migration to wound sites. J. Immunol. 166, 7556–7562 (2001). [DOI] [PubMed] [Google Scholar]
- 33.Pilling D., Vakil V., Gomer R. H., Improved serum-free culture conditions for the differentiation of human and murine fibrocytes. J. Immunol. Methods 351, 62–70 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Crawford J. R., Pilling D., Gomer R. H., Improved serum-free culture conditions for spleen-derived murine fibrocytes. J. Immunol. Methods 363, 9–20 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cox N., Pilling D., Gomer R. H., Distinct Fcγ receptors mediate the effect of serum amyloid P on neutrophil adhesion and fibrocyte differentiation. J. Immunol. 193, 1701–1708 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pilling D., Zheng Z., Vakil V., Gomer R. H., Fibroblasts secrete Slit2 to inhibit fibrocyte differentiation and fibrosis. Proc. Natl. Acad. Sci. U.S.A. 111, 18291–18296 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pilling D., Vakil V., Cox N., Gomer R. H., TNF-α–stimulated fibroblasts secrete lumican to promote fibrocyte differentiation. Proc. Natl. Acad. Sci. U.S.A. 112, 11929–11934 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shao D. D., Suresh R., Vakil V., Gomer R. H., Pilling D., Pivotal advance: Th-1 cytokines inhibit, and Th-2 cytokines promote fibrocyte differentiation. J. Leukoc. Biol. 83, 1323–1333 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Quan T. E., Bucala R., Culture and analysis of circulating fibrocytes. Methods Mol. Med. 135, 423–434 (2007). [DOI] [PubMed] [Google Scholar]
- 40.Cox J., Hein M. Y., Luber C. A., Paron I., Nagaraj N., Mann M., Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteomics 13, 2513–2526 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fabregat A., Sidiropoulos K., Garapati P., Gillespie M., Hausmann K., Haw R., Jassal B., Jupe S., Korninger F., McKay S., Matthews L., May B., Milacic M., Rothfels K., Shamovsky V., Webber M., Weiser J., Williams M., Wu G., Stein L., Hermjakob H., D’Eustachio P., The reactome pathway knowledgebase. Nucleic Acids Res. 44, D481–D487 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lorenz K., Sicker M., Schmelzer E., Rupf T., Salvetter J., Schulz-Siegmund M., Bader A., Multilineage differentiation potential of human dermal skin-derived fibroblasts. Exp. Dermatol. 17, 925–932 (2008). [DOI] [PubMed] [Google Scholar]
- 43.Hanson S. E., Kim J., Quinchia Johnson B. H., Bradley B., Breunig M. J., Hematti P., Thibeault S. L., Characterization of mesenchymal stem cells from human vocal fold fibroblasts. Laryngoscope 120, 546–551 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chesney J., Bacher M., Bender A., Bucala R., The peripheral blood fibrocyte is a potent antigen-presenting cell capable of priming naive T cells in situ. Proc. Natl. Acad. Sci. U.S.A. 94, 6307–6312 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Medina A., Brown E., Carr N., Ghahary A., Circulating monocytes have the capacity to be transdifferentiated into keratinocyte-like cells. Wound Repair Regen. 17, 268–277 (2009). [DOI] [PubMed] [Google Scholar]
- 46.Medina A., Kilani R. T., Carr N., Brown E., Ghahary A., Transdifferentiation of peripheral blood mononuclear cells into epithelial-like cells. Am. J. Pathol. 171, 1140–1152 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Robertson N. J., Fairchild P. J., Waldmann H., Ectopic transplantation of tissues under the kidney capsule. Methods Mol. Biol. 380, 347–353 (2007). [DOI] [PubMed] [Google Scholar]
- 48.Progatzky F., Dallman M. J., Lo Celso C., From seeing to believing: Labelling strategies for in vivo cell-tracking experiments. Interface Focus 3, 20130001 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Curnow S. J., Fairclough M., Schmutz C., Kissane S., Denniston A. K. O., Nash K., Buckley C. D., Lord J. M., Salmon M., Distinct types of fibrocyte can differentiate from mononuclear cells in the presence and absence of serum. PLOS ONE 5, e9730 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kleaveland K. R., Velikoff M., Yang J., Agarwal M., Rippe R. A., Moore B. B., Kim K. K., Fibrocytes are not an essential source of type I collagen during lung fibrosis. J. Immunol. 193, 5229–5239 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kleaveland K. R., Moore B. B., Kim K. K., Paracrine functions of fibrocytes to promote lung fibrosis. Expert Rev. Respir. Med. 8, 163–172 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang J. F., Jiao H., Stewart T. L., Shankowsky H. A., Scott P. G., Tredget E. E., Fibrocytes from burn patients regulate the activities of fibroblasts. Wound Repair Regen. 15, 113–121 (2007). [DOI] [PubMed] [Google Scholar]
- 53.Mitra S. K., Hanson D. A., Schlaepfer D. D., Focal adhesion kinase: In command and control of cell motility. Nat. Rev. Mol. Cell Biol. 6, 56–68 (2005). [DOI] [PubMed] [Google Scholar]
- 54.Bertini R., Allegretti M., Bizzarri C., Moriconi A., Locati M., Zampella G., Cervellera M. N., Di Cioccio V., Cesta M. C., Galliera E., Martinez F. O., Di Bitondo R., Troiani G., Sabbatini V., D’Anniballe G., Anacardio R., Cutrin J. C., Cavalieri B., Mainiero F., Strippoli R., Villa P., Di Girolamo M., Martin F., Gentile M., Santoni A., Corda D., Poli G., Mantovani A., Ghezzi P., Colotta F., Noncompetitive allosteric inhibitors of the inflammatory chemokine receptors CXCR1 and CXCR2: Prevention of reperfusion injury. Proc. Natl. Acad. Sci. U.S.A. 101, 11791–11796 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gordon S., Taylor P. R., Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 5, 953–964 (2005). [DOI] [PubMed] [Google Scholar]
- 56.Hwang J. H., Shim S. S., Seok O. S., Lee H. Y., Woo S. K., Kim B. H., Song H. R., Lee J. K., Park Y. K., Comparison of cytokine expression in mesenchymal stem cells from human placenta, cord blood, and bone marrow. J. Korean Med. Sci. 24, 547–554 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.He Q., Wan C., Li G., Concise review: Multipotent mesenchymal stromal cells in blood. Stem Cells 25, 69–77 (2007). [DOI] [PubMed] [Google Scholar]
- 58.Li S., Huang K.-J., Wu J.-C., Hu M. S., Sanyal M., Hu M., Longaker M. T., Lorenz H. P., Peripheral blood-derived mesenchymal stem cells: Candidate cells responsible for healing critical-sized calvarial bone defects. Stem Cells Transl. Med. 4, 359–368 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dominici M., Le Blanc K., Mueller I., Slaper-Cortenbach I., Marini F. C., Krause D. S., Deans R. J., Keating A., Prockop D. J., Horwitz E. M., Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 8, 315–317 (2006). [DOI] [PubMed] [Google Scholar]
- 60.McLeod C. M., Mauck R. L., On the origin and impact of mesenchymal stem cell heterogeneity: New insights and emerging tools for single cell analysis. Eur. Cell. Mater. 34, 217–231 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Driskell R. R., Lichtenberger B. M., Hoste E., Kretzschmar K., Simons B. D., Charalambous M., Ferron S. R., Herault Y., Pavlovic G., Ferguson-Smith A. C., Watt F. M., Distinct fibroblast lineages determine dermal architecture in skin development and repair. Nature 504, 277–281 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Philippeos C., Telerman S. B., Oulès B., Pisco A. O., Shaw T. J., Elgueta R., Lombardi G., Driskell R. R., Soldin M., Lynch M. D., Watt F. M., Spatial and single-cell transcriptional profiling identifies functionally distinct human dermal fibroblast subpopulations. J. Invest. Dermatol. 138, 811–825 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yaghi A., Zaman A., Dolovich M., Primary human bronchial epithelial cells grown from explants. J. Vis. Exp., e1789 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kim J., Hematti P., Mesenchymal stem cell-educated macrophages: A novel type of alternatively activated macrophages. Exp. Hematol. 37, 1445–1453 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dunlevy J. R., Couchman J. R., Interleukin-8 induces motile behavior and loss of focal adhesions in primary fibroblasts. J. Cell Sci. 108, 311–321 (1995). [DOI] [PubMed] [Google Scholar]
- 66.Lin S.-C., Hsiao K.-Y., Chang N., Hou P.-C., Tsai S.-J., Loss of dual-specificity phosphatase-2 promotes angiogenesis and metastasis via up-regulation of interleukin-8 in colon cancer. J. Pathol. 241, 638–648 (2017). [DOI] [PubMed] [Google Scholar]
- 67.Erde J., Loo R. R. O., Loo J. A., Enhanced FASP (eFASP) to increase proteome coverage and sample recovery for quantitative proteomic experiments. J. Proteome Res. 13, 1885–1895 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cox J., Mann M., MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 (2008). [DOI] [PubMed] [Google Scholar]
- 69.Tyanova S., Temu T., Sinitcyn P., Carlson A., Hein M. Y., Geiger T., Mann M., Cox J., The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 13, 731–740 (2016). [DOI] [PubMed] [Google Scholar]
- 70.Schneider C. A., Rasband W. S., Eliceiri K. W., NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kuleshov M. V., Jones M. R., Rouillard A. D., Fernandez N. F., Duan Q., Wang Z., Koplev S., Jenkins S. L., Jagodnik K. M., Lachmann A., McDermott M. G., Monteiro C. D., Gundersen G. W., Ma’ayan A., Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90–W97 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.R Development Core Team, R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing, 2007). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/5/eaav7384/DC1
Fig. S1. Additional characterization of human FC differentiation in vitro.
Fig. S2. Bioenergetic profiles of human FCs at 1 and 7 days.
Fig. S3. Characterization of monocytes following epithelial cell-oriented culture.
Table S1. Reactome pathways enriched in protein sets associated with FC differentiation.
Table S2. Reactome pathways enriched in the protein set overrepresented in VFFs, DFs, and MSCs compared to FCs.
Table S3. Reactome pathways enriched in the protein set overrepresented in FCs compared to VFFs, DFs, and MSCs.
Table S4. Cytokine production by FCs and VFFs, cultured under basal conditions for 24 hours.
Table S5. Antibodies and isotype controls used for flow cytometry.
Table S6. Antibodies and phallotoxin used for immunocytochemistry and immunohistochemistry.