

Abstract
Phosphoproteomics requires better separation of phosphopeptides to boost the coverage of the phosphoproteome. We argue that an alternative separation method that produces orthogonal phosphopeptide separation to the widely used LC needs to be considered. Capillary zone electrophoresis (CZE) is one important alternative because CZE and LC are orthogonal for phosphopeptide separation and because the migration time of peptides in CZE can be accurately predicted. In this work, we coupled strong cation exchange (SCX)-reversed-phase LC (RPLC) to CZE-MS/MS for large-scale phosphoproteomics of the colon carcinoma HCT116 cell line. The CZE-MS/MS-based platform identified 11,555 phosphopeptides. The phosphopeptide dataset is at least 100% larger than that from previous CZE-MS/MS studies and will be a valuable resource for building a model for predicting the migration time of phosphopeptides in CZE. Phosphopeptides migrate significantly slower than corresponding unphosphopeptides under acidic conditions of CZE separations and in a normal polarity. According to our modeling data, phosphorylation decreases peptide’s charge roughly by one charge unit, resulting in dramatic decrease in electrophoretic mobility. Preliminary investigations demonstrate that electrophoretic mobility of phosphopeptides containing one phosphoryl group can be predicted with the same accuracy as for non-modified peptides (R2 ~0.99). The CZE-MS/MS and LC-MS/MS were complementary in large-scale phosphopeptide identifications and produced different phosphosite motifs from the HCT116 cell line. The data highlight the value of CZE-MS/MS for phosphoproteomics as a complementary separation approach for not only improving the phosphoproteome coverage but also providing more insight into the phosphosite motifs.
Graphical Abstract
Introduction
Protein phosphorylation is a key reversible post-translational modification in nature, and it is involved in various cellular processes such as transcriptional and translational regulation, cellular signaling, metabolism, and cell differentiation.[1] Global site-specific characterization of protein phosphorylation allows us to gain insights into the regulatory role of phosphorylation in fundamental biological processes. Multi-dimensional liquid chromatography (MDLC)-tandem mass spectrometry (MS/MS) (e.g., strong cation exchange (SCX)-reversed-phase LC (RPLC)) is routinely used for large-scale phosphoproteomics and it can identify over 10,000 phosphorylation events per study.[2–10] More than 50,000 distinct phosphopeptides have been reported from a single human cancer cell line using MDLC-MS/MS.[3]
Based on statistical estimates, there are over half a million potential phosphorylation sites in the human proteome.[3,11,12] We need to boost the peptide separation to reach a deeper coverage of the human phosphoproteome. Since the proteomics community has made tremendous efforts in improving MDLC-MS/MS for phosphoproteomics in last 20 years, we argue that an alternative separation method that is complementary to the LC for phosphopeptide separation will be very useful for deep phosphoproteomics.
Capillary zone electrophoresis (CZE) is a powerful method for the separation of biomolecules (e.g., peptides and proteins) and it can have extremely high separation efficiency.[13–19] CZE-MS/MS has attracted great attention for proteomics recently because of the improvements in the CE-MS interface,[20–23] the sample stacking method, [24–26] and the high-quality coating on the inner wall of the separation capillary.[27]
CZE-MS/MS has some unique features for phosphoproteomics. First, CZE-MS/MS and RPLC-MS/MS can sample different pools of the phosphopeptides in cells due to the different separation mechanisms of CZE and RPLC (size-to-charge ratio vs. hydrophobicity).[28,30] The combination of these two methods can boost the phosphoproteome coverage significantly. Second, CZE can separate the phosphorylated and unphosphorylated forms of peptides due to their significant difference in charge. This feature reduces the interference of phosphopeptide identification (ID) from unphosphopeptides.[28,29] Third, the migration time of peptides in CZE can be predicted easily and accurately.[31] If we can generate a large phosphopeptide dataset using CZE-MS/MS, we can build a simple model to predict the migration time of phosphopeptides. This unique feature of CZE-MS/MS makes it a powerful tool for phosphoproteomics because the predicted migration time of phosphopeptides can be used to evaluate their ID confidence from a database search and even guide the database search.
Few papers have been published on using CZE-MS/MS for phosphoproteomics. We previously coupled CZE to a Q-Exactive mass spectrometer via an electro-kinetically pumped sheath flow CE-MS interface for phosphoproteomics of a human cell line.[28] 2,300 phosphopeptides were identified with single-shot CZE-MS/MS in 100 min, and the data suggested the high potential of CZE-MS/MS for large-scale phosphoproteomics. Recently, Faserl et al. investigated the sheathless CE-MS interface-based CZE-MS/MS for large-scale phosphoproteomics.[29] They identified over 5,000 phosphopeptides by coupling RPLC fractionation to the CZE-MS/MS. To boost the number of phosphopeptide IDs using the CZE-MS/MS, the loading capacity and the separation window of CZE need to be improved. Recently, we developed a novel CZE-MS/MS system with a micro-liter scale sample loading volume and hours of separation window, opening the door to using CZE-MS/MS for large-scale proteomics.[32,33] The CZE-MS/MS system employed a 1-meter separation capillary with a high-quality neutral coating on its inner wall for eliminating the electroosmotic flow, an optimized dynamic pH junction method for highly efficient online stacking of peptides and proteins, the improved electro-kinetically pumped sheath flow CE-MS interface [22] and a Q-Exactive HF mass spectrometer.
We recently coupled SCX-RPLC fractionation to the CZE-MS/MS for deep proteomics of a mouse brain, leading to extremely high peak capacity for peptide separation and 8,200 protein IDs.[34] Motivated by the high peak capacity of the SCX-RPLC-CZE system for peptide separation, in this work, we applied the SCX-RPLC-CZE-MS/MS system for large-scale phosphoproteomics of HCT116 colon cancer cells. We had three goals in this work. First, boost the number of phosphopeptide IDs from a human cell line using CZE-MS/MS. The large phosphopeptide dataset will be useful for building a model for predicting the migration time of phosphopeptides. Second, we were interested in investigating how phosphorylation influences electrophoretic mobility of peptides. Third, we wished to investigate the difference between our CZE-MS/MS data and the literature LC-MS/MS data regarding the phosphosite motifs. We speculated that the good complementarity between CZE-MS/MS and RPLC-MS/MS for peptide IDs might result in significant differences in phosphosite motifs and found that the data supported our hypothesis.
Experimental section
Materials and reagents
All reagents were bought from Sigma-Aldrich (St. Louis, MO) unless stated otherwise. LC/MS grade water, formic acid (FA), methanol, acetonitrile (ACN), HPLC grade acetic acid (AA) and hydrofluoric acid (HF) were purchased from Fisher Scientific (Pittsburgh, PA). Acrylamide was obtained from Acros Organics (NJ, USA). Fused silica capillaries (50 μm i.d./360 μm o.d.) were purchased from Polymicro Technologies (Phoenix, AZ).
Cell Growth Conditions
The human colon carcinoma cell line HCT 116 was obtained from American Type Culture Collection (ATCC). The cells were grown in RPMI 1640 cell culture medium (Life Technologies) supplemented with 10% fetal bovine serum (FBS) (Thermo Scientific). The provider assured authentication of the cell line by cytogenetic analysis. In addition, the cell line was validated by short tandem repeat (STR) analysis within the last two years.
Sample Preparation and phosphorylated peptide enrichment
A lysis buffer with 8 M urea with 75 mM NaCI, 50 mM Tris-HCI (pH 8.2), 10 mM sodium pyrophosphate, 1 mM PMSF, 1 mM Na3VO4, 1 mM NaF, 1 mM β-glycerophosphate, and 1 EDTA-free protease inhibitor cocktail was prepared. HCT116 colon cancer cells were cultured to 70% confluence followed by cell lysis with the lysis buffer. A small aliquot of the cell lysate was subjected to the Bicinchoninic acid assay for protein concentration measurement. Three mg of extracted protein was subjected to denaturation at 37 °C for 1 h, reduction with 5 mM Dithiothreitol (DTT) at 37 °C for 1 h, and alkylation with 14 mM iodoacetamide (IAA) for 30 min at room temperature. The alkylation was terminated by adding 5 mM DTT for 25 min. The sample was then diluted with 25 mM Tris-HCI buffer (pH 8.2) with 1 mM CaCl2. Trypsin was added to the sample for overnight digestion at 37 °C. Phosphopeptides in the desalted digest were enriched with TiO2 beads in a 1:4 peptides to beads ratio based on the references [35] and [36]. After enrichment, the phosphopeptides were desalted, lyophilized and stored at −80 °C before use. We assumed 70% recovery during tryptic digestion and 10% recovery during phosphopeptide enrichment, resulting in about 200 μg phosphopeptides in the end.
SCX-RPLC fractionation
An SCX-RPLC online fractionation was performed based on reference [34] with some minor modifications. Briefly, a 4.6 mm i.d. × 12.5 mm length SCX trap column (Zorbax 300SCX, Agilent Technologies) and a 2.1 mm i.d. × 150 mm length C18 RP column (Zorbax 300Extend-C18, Agilent Technologies) were connected directly for online 2D-LC fractionation. An Agilent Infinity II HPLC system was used for the experiment. 0.1% formic acid (FA) in water, 0.1% FA in acetonitrile (ACN), and 890 mM ammonium acetate solution (pH = 2.88) were used as mobile phase A, B, C for separation, respectively. Mobile phase A and C were used for stepwise elution of peptides from the SCX column. Mobile phase A and B were used to generate a linear gradient for RPLC separation of peptides.
Roughly 200-μg phosphopeptides were dissolved in mobile phase A and then loaded onto the SCX column with mobile phase A at a flow rate of 0.3 mL/min for 20 min. The phosphopeptides retained on the SCX column were eluted stepwise by two different concentrations of ammonium acetate solution: 150 mM and 890 mM. Then, each SCX eluate was captured on the RPLC column. RPLC gradient separation was performed at a 0.3 mL/min flow rate for 70 min: 0–5 min, 2%B; 5–7 min, 2–8% B; 7–47 min, 8–40% B; 47–49 min, 40–80%; 49–59 min, 80% B; 59–60 min, 80–2% B; 60–70 min, 2%B. 42 fractions were collected (1 fraction/ min) from 6 to 48 min for each salt step elution and the fractions were named based on the elution order. From fraction 2 to fraction 41, fractions were combined by the following rule: fraction N + fraction (N+20). The fraction 1 was combined with the mixture of fraction 2 and fraction 22, and fraction 42 was combined with the mixture of fraction 21 and fraction 41. In total, there were 40 fractions (20 fractions/salt step x 2 salt steps) collected, and they were lyophilized and stored at −80 °C before use.
CZE-MS/MS
An ECE-001 CE autosampler (CMP Scientific, Brooklyn, NY) and a Q-Exactive HF mass spectrometer (Thermo Fisher Scientific) were coupled with the third-generation electro-kinetically pumped sheath flow CE-MS interface (an EMASS-II CE-MS interface, CMP Scientific).[22] A borosilicate glass capillary (1.0 mm o.d., 0.75 mm i.d.) was pulled with a Sutter P-1000 flaming/brown micropipette puller to make an electrospray emitter. The opening of the emitter was 20–40 μm.
A 95-cm long fused silica capillary (50 µm i.d., 360 µm o.d.) was used for CZE separation. The inner wall of the capillary was coated with linear polyacrylamide (LPA) based on reference [27]. One end of the LPA coated capillary was etched with hydrofluoric acid based on reference [16] to reduce the outer diameter to less than 100 µm. The background electrolyte (BGE) for CZE was 5% (v/v) acetic acid (AA) (pH 2.4) and the sheath buffer for electrospray was 10% (v/v) methanol and 0.2% (v/v) FA in water. The etched end of the capillary was introduced into the electrospray emitter, and the distance between the etched end and the orifice of the emitter was ~300 µm. The distance between the emitter orifice and the inlet of the mass spectrometer was ~2 mm. 2.2 kV voltage was applied for electrospray ionization.
The 40 LC fractions were redissolved in 5 µL of 50 mM ammonium bicarbonate (pH 8) for CZE-MS/MS. For sample injection, approximately 200 nL or 300 nL of each sample was injected into the capillary for analysis. Then, 30 kV voltage was applied at the injection end for 5400 seconds for CZE separation, followed by capillary flushing with the BGE for 900 seconds under a 5-psi pressure. One CZE-MS/MS run was performed for each LC fraction.
A Q-Exactive HF mass spectrometer was used in CZE-MS/MS. A data-dependent acquisition (DDA) method was employed. The mass resolution was 60,000 (at m/z 200) for both full MS scans and MS/MS scans. The automatic gain control targets were set to 3E6 and 1E5 for full MS scans and MS/MS scans, respectively. For full MS scans, the maximum injection time was 50 ms with a scan range of 300 to 1500 m/z. For MS/MS scans, the maximum injection time was set to 110 ms. Top ten most abundant ions were sequentially isolated with a 2-m/z isolation window for fragmentation with 28% normalized collision energy. The dynamic exclusion was set to 40 s. Ions with charges higher than 1 and lower than 8 were selected for fragmentation.
Data analysis
Proteome Discoverer 2.2 software (Thermo Fisher Scientific) was used for data analysis. Sequest HT was used for the database search.[37] The human proteome database (UP000005640, 70,965 protein sequences) containing reviewed and unreviewed proteins was downloaded from UniProt (http://www.uniprot.org/). All raw files were searched against both the forward database and a decoy (reverse) database to estimate the false discovery rate (FDR). [38] Maximum two missed cleavage sites were allowed for peptide identification, and the peptide length was set to 6 to 144 amino acid residues. The mass tolerances of precursor and fragments were 20 ppm and 0.05 Da, respectively. Oxidation (methionine) and phosphorylation (serine, threonine and tyrosine) were set as dynamic modifications. Acetylation at the protein N-terminal was chosen as a dynamic modification. Carbamidomethylation (cysteine) was set as a static modification. The peptide ID was filtered with confidence as high, corresponding to a 1% FDR. Protein grouping was enabled, and the strict parsimony principle was applied. The phosphoRS that integrated into the workflow was used to evaluate the confidence of the phosphosite localization. [39] Unless specified otherwise, the numbers of protein and peptide IDs reported in this work were all from the Proteome Discoverer 2.2 software.
MaxQuant 1.5.5.1 [40] was also used for the database search to compare phosphopeptide IDs and phosphosite motifs obtained from our CZE-MS/MS data with the literature data. The Andromeda search engine was used to search the MS/MS spectra.[41] The same human database used in the Proteome Discoverer search was used. The peptide mass tolerances of the first search and main search were 20 and 4.5 ppm, respectively. The fragment ion mass tolerance was 20 ppm. Trypsin was selected as the protease. The variable and static modifications were the same as the Proteome Discoverer search. The minimum length of a peptide was set to 7. The FDRs were 1% for both peptide and protein IDs. For phosphopeptide identifications, the phosphosite localization probability should be better than 0.75.
An online available GRAVY calculator (http://www.qravv-calculator.de/) was used to calculate the grand average of hydropathy (GRAVY) values of peptides. Online version of SSRCalc (http://hs2.proteome.ca/SSRCalc/SSRCalcX.html) was used to calculate the hydrophobicity indexes for peptides. [42] Molecular weights and isoelectric points of identified peptides were calculated using the “Compute pl/Mw” tool in ExPASy (http://web.expasy.org/compute_pi/). Motif-x (http://motif-x.med.harvard.edu/motif-x.html) was used to extract motifs from the data sets, default settings were used except MS/MS was chosen as foreground format, and the human proteome was chosen as the organism. [43] Motif alignment was performed with WebLogo3 (http://weblogo.threeplusone.com/create.cgi).
Observed and predicted electrophoretic mobility of peptides
The data from LC fraction 8 was used for the electrophoretic mobility analysis. Only peptides having no variable modifications except for single phosphorylation were used for the analysis. Peptides’ observed electrophoretic mobility (µef observed) was determined using migration times (tM, min) - time of MS/MS acquisition of the most intense tandem spectra for each unique peptide identification. We assumed that the electroosmotic flow (EOF) at 5% (v/v) acetic acid (pH 2.4) in the BGE was very low and mapped tM into electrophoretic mobility (µef) using the equation for their experimental conditions (a 95 cm long capillary at 280 volts/cm):
Sequence-Specific Retention Calculator (SSRCalc) CZE model reported previously was used to predict the electrophoretic mobility of peptides. [31] While peptide charge and mass are the main parameters in determining mobility value, we introduced corrections for several sequence-specific features affecting corrected charge value (Zc) applied for calculations:
where 3.069 and 386 are empirical coefficients applied to align modeling output with experimentally measured values; Zc – peptide charge at pH 2.4, corrected using thirteen residue and sequence specific coefficients; Mc = (0.66*M + 0.34*N*110.9), corrected mass to reflect the influence of different amino acid size; M is the molecular weight of peptides; N is the peptide length; OFFSET is a polynomial empirical function of Zc/N to correct prediction for peptides with extremely high and low mobility values.
Results and discussion
As shown in Figure 1A, 3 mg of HCT-116 cell proteins were digested into peptides with trypsin, followed by phosphopeptide enrichment using TiO2 beads based on references [35] and [36]. The phosphopeptides were fractionated with online SCX-RPLC into 40 fractions based on the charge and hydrophobicity of phosphopeptides. Each LC fraction was analyzed by dynamic pH junction based CZE-MS/MS, [32] and CZE separates peptides based on their size-to-charge ratios. The SCX, RPLC, and CZE are orthogonal for peptide separation. As shown in Figure 1B, a 2-min RPLC eluate was further separated by CZE into a 50-min window. As shown in Figure 1C, the correlation between m/z and migration time of peptides from the database search is complicated but, in general, peptides with higher m/z tend to migrate slower in the capillary during the CZE separation.
Figure 1.
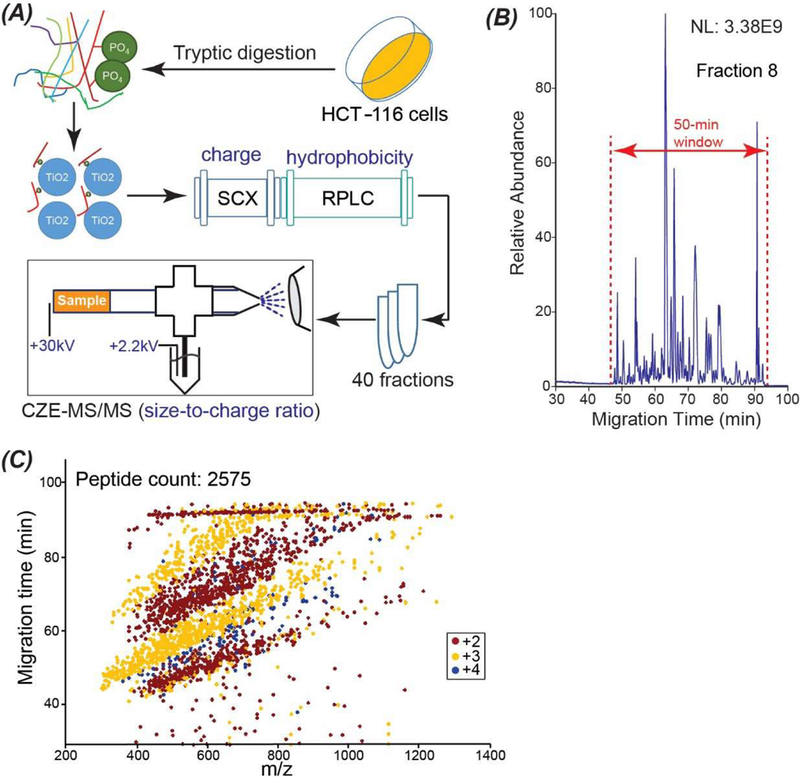
(A) The experimental design of the work. (B) Base peak electropherogram of one RPLC fraction (fraction 8) after CZE-MS/MS analysis. (C) Mass-to-charge ratio (m/z) vs. migration time of peptides identified by CZE-MS/MS from the RPLC fraction 8. The charges of peptides labelled in (C) are their gas-phase charges determined by MS.
Large-scale phosphoproteomics of the HCT-116 cell line using SCX-RPLC-CZE-MS/MS
CZE-MS/MS analyses of the 40 SCX-RPLC fractions produced 6,502 protein IDs, 33,301 peptide IDs, and 11,555 phosphopeptides with a peptide-level 1% FDR. The corresponding raw files have been deposited to the ProteomeXchange Consortium via the PRIDE [44] partner repository with the dataset identifier PXD012255. Proteome discoverer 2.2 was used for the peptide and protein IDs. 10,029 phosphopeptides were identified with phosphosite localization probability better than 95%. To our knowledge, our phosphopeptide dataset represents the largest phosphoproteomics data so far using CZE-MS/MS. In the literature, we reached 2,300 phosphorylated peptide IDs with single-shot CZE-MS/MS in 100 min [28] and Faserl et al. approached over 5,000 phosphorylated peptide IDs using an RPLC-CZE-MS/MS system in about 60 hours. [29] In this work, we identified 11,555 phosphopeptides using the SCX-RPLC-CZE-MS/MS in 67 hours. All three studies employed the Proteome Discoverer platform for data analysis. Our system improved the number of phosphopeptide IDs by 100% compared with Faserl’s work with a comparable instrument time. We noted that the phosphopeptide identification efficiency decreased drastically from the single-shot CZE-MS/MS data (1400 phosphopeptides/hour) [28] to the RPLC-CZE-MS/MS data (90 phosphopeptides/hour) [29] and our SCX-RPLC-CZE-MS/MS data (170 phosphopeptides/hour).
We also noted that the specificity of our TiO2 enrichment was low (about 35%) regarding the ratio between phosphopeptide IDs and total peptide IDs. We believe the number of phosphopeptide IDs can be improved significantly with a better phosphopeptide enrichment procedure. The large-scale phosphopeptide dataset produced in this work will be useful for building a simple model for accurate prediction of migration time of phosphopeptides. [31] The accurately predicted migration time of phosphopeptides could be used to evaluate the confidence of their IDs from a database search and even further guide the database search. The lists of identified proteins and phosphopeptides are shown in Supporting Information I.
The SCX with two salt step elution (150 mM (salt step 1) and 890 mM (salt step 2) ammonium acetate solution, pH = 2.88) separated the phosphopeptides well, and only 618 out of the 11,555 phosphopeptides were overlapped between those two salt steps, Figure 2A. Phosphopeptides in the salt step 2 tend to have higher charge states (Figure 2B), have higher molecular weight (Figure 2C), and be more basic (Figure 2D) compared with that in the salt step 1. The number of phosphopeptide IDs per LC fraction ranges from 300 to 700 for most of the fractions, and the distribution is moderately uniform, Figure 2E. In CZE, phosphopeptides tend to migrate significantly slower than unphosphopeptides in the separation capillary, Figure 2F. This feature makes CZE-MS/MS useful for phosphoproteomics because the interference of phosphopeptide IDs from unphosphopeptides can be reduced.
Figure 2.
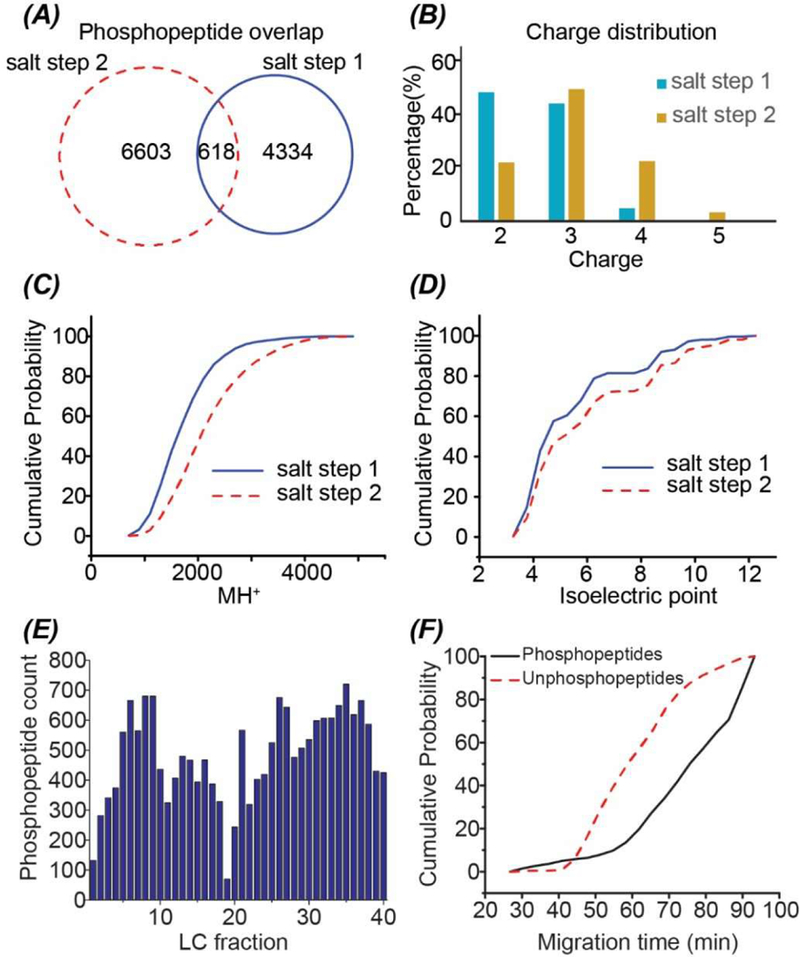
Summary of the phosphopeptide IDs using the SCX-RPLC-CZE-MS/MS. (A) Overlap of the identified phosphopeptides from the two salt steps of the SCX. Salt step 1 and 2 used 150 mM and 890 mM ammonium acetate solution (pH = 2.88) for peptide elution, respectively. (B) The charge distribution of identified phosphopeptides in the two salt steps. The phosphopeptides’ charges are their gas-phase charges determined by MS. (C) Cumulative distribution of mass of identified phosphopeptides in the two salt steps. (D) Cumulative distribution of isoelectric point (pi) of identified phosphopeptides in the two salt steps. The pi was calculated based on the peptide sequence. (E) The number of phosphopeptide IDs across the 40 LC fractions. (F) Cumulative distribution of migration time of identified phosphopeptides and unphosphopeptides in one LC fraction (fraction 8).
Investigating the effect of phosphorylation on electrophoretic mobility of peptides
Phosphopeptides tend to migrate significantly slower than their unphosphorylated forms under acidic conditions used for CZE separations and in a normal polarity. Addition of one phosphoryl group reduces overall positive charge of peptides by one charge unit, thus resulting in drastic decrease of electrophoretic mobility.
As shown in Figure 3A and 3B, the phosphorylated forms of peptides QGGGGGGGSVPGIER and AGELTEDEVER migrate much slower than their unphosphorylated forms and their migration time difference (∆ time) is about 20 min. We noted that the ∆ time should be larger than 20 min because we started to flush the capillary by applying a 5-psi pressure at 90 min. As shown in Figure 3C, the doubly phosphorylated form of the peptide AAKLSEGSQPAEEEEDQETPSR migrates slower than the singly phosphorylated form due to the one more negative charge. Their ∆ time should be much larger than 3 min because they were both pushed out of the capillary by the pressure.
Figure 3.
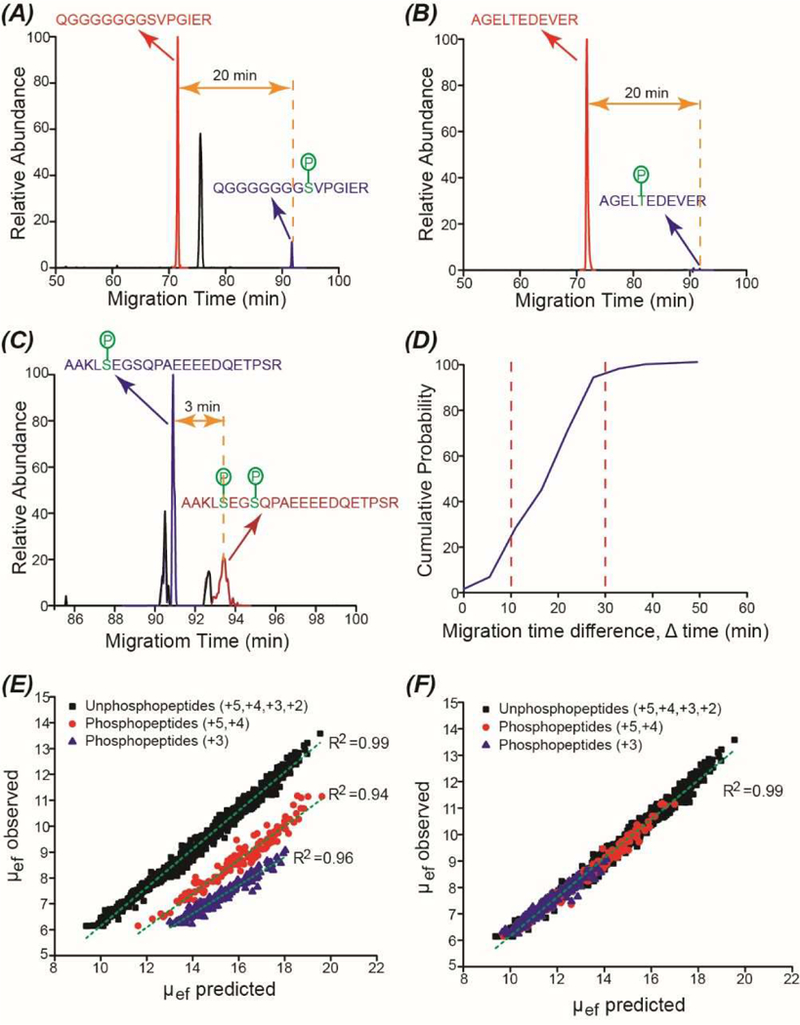
(A) Extracted ion electropherogram (EIE) of phosphorylated and unphosphorylated forms of the peptide QGGGGGGGSVPGIER. (B) EIE of phosphorylated and unphosphorylated forms of the peptide AGELTEDEVER. (C) EIE of singly phosphorylated and doubly phosphorylated forms of the peptide AAKLSEGSQPAEEEEDQETPSR. (D) Cumulative distribution of the migration time difference (∆ time) between unphosphorylated and singly phosphorylated forms of peptides. The figure was based on the data from six LC fractions. (E) Correlations between observed and predicted electrophoretic mobility (µef) of unphosphopeptides and phosphopeptides with one phosphoryl group. The non-modified SSRCalc CZE model31 was used to highlight the effect of phosphorylation. (F) Correlation between observed and predicted µef of peptides using the modified SSRCalc CZE model. µef × 105 (cm2*V−1*S−1) is shown in (E) and (F). The peptides’ charges in (E) and (F) are shown for non-modified peptide sequences (counting the number of lysine, arginine, and histidine residues, plus positively charged N-terminus).
We manually analyzed the data from six LC fractions regarding the ∆ time between unphosphorylated and singly phosphorylated forms of peptides. We obtained 200 pairs of peptides and their altered migration (∆) time in CZE. As shown in Figure 3D, for all the 200 pairs of peptides, the singly phosphorylated forms migrate slower than the corresponding unphosphorylated forms, which is demonstrated by the positive ∆ time values. For about 70% of the peptide pairs, the ∆ time ranges from 10 to 30 min. We reached two conclusions here. First, for the majority of peptides studied, the addition of one phosphoryl group onto the peptide can drastically slow down its migration in the capillary during CZE. Second, adding one phosphoryl group to different peptides influences their migration to various extents.
We further investigated the effect of phosphorylation on electrophoretic mobility of peptides through comparing the observed and predicted mobility values of phosphopeptides and unphosphopeptides. We used the data from one LC fraction (fraction 8) for this task. The observed and predicted electrophoretic mobility of peptides were calculated using the methods described in the “Experimental section”. Application of non-modified SSRCalc CZE model (without considering the effect of negatively charged phosphoryl groups) was used to illustrate the effect of phosphorylation, Figure 3E and Figure S1 in supporting information II. Mobility of unphosphopeptides follows SSRCalc prediction (R2 =0.99), [31] whereas addition of one phosphoryl group decreases mobility dramatically, Figure 3E and Figure S1. Phosphopeptides carrying two positive charges prior to the modification (+2) and a small fraction of phosphopeptides with three positive charges (+3) migrated extremely slow in the capillary and were pushed out by the pressure in the end of the CZE-MS/MS run, Figure S1. The information on electrophoretic mobility of peptides is listed in supporting information I. After removing all the phosphopeptides (+2) and some of the phosphopeptides (+3) with mobility lower than 6.2×10−5 cm2*V−1*S−1, we obtained reasonably good linear correlations between observed and predicted electrophoretic mobility values within each group of phosphopeptides (R2 ≥0.94), Figure 3E. We noted that the observed mobilities of peptides were obviously lower than their predicted mobility. We attributed the phenomenon to the dynamic pH junction sample stacking method used in the CZE experiments, which slowed down the mobility of peptides in the capillary.
First attempts have been made to adapt SSRCalc CZE model to prediction of phosphopeptides’ mobility values. The corrected charge (Zc) of phosphopeptides has been modified to improve correlation for entire set of peptides shown in Figure 3E (phosphopeptides and unphosphopeptides). We found that Zc values had to be adjusted by −0.91 and by −1.0 for +5/+4 and +3 phosphopeptides, respectively. Figure 3F shows prediction accuracy (R2 ~0.99) for combined set of peptides, identical to the collection of unphosphopeptides in Figure 3E. This data indicate that the charge shift is indeed very close to the expected contribution from one phosphoryl group. The information on electrophoretic mobility of peptides after charge adjustment is provided in supporting information I. We need to note that much larger phosphopeptide datasets with confident assignment of modification site is needed for the development of sequence-dependent model for mobility prediction and for better understanding of how phosphorylation influences electrophoretic mobility of peptides. Similar to the effect of acidic Asp/Glu residues reported before, [31] we anticipate that N-terminal positioning of phosphate (or in close proximity to other positively charged groups) will result in a larger decrease in mobility.
Comparing our phosphoproteome dataset from CZE-MS/MS with an LC-MS/MS dataset in literature
Recently, Kubiniok et al. performed deep phosphoproteomics of HCT116 cells using TiO2 enrichment, SCX-RPLC-MS/MS and MaxQuant software for data analysis.[45] We compared the HCT116 phosphoproteomics datasets from our SCX-RPLC-CZE-MS/MS with Kubiniok’s SCX-RPLC-MS/MS. In order to make a fair comparison, we reanalyzed our data with MaxQuant software and filtered the data with the same criteria as Kubiniok et al. 6,221 phosphopeptides were identified using MaxQuant software, and only 45% of these phosphopeptides were covered by that identified in the Kubiniok’s work, suggesting good complementarity between those two platforms for phosphopeptide IDs. The result here agrees well with the data in the literature that CZE-MS/MS and RPLC-MS/MS are well complementary for peptide and phosphopeptide IDs. [28,29,34] Further analyses of the physicochemical properties of identified phosphopeptides demonstrated that CZE-MS/MS tended to identify basic, small and hydrophilic phosphopeptides compared with LC-MS/MS (Figure S2 in supporting information II); these data agree with reports in the literature. [34,46,47] The data highlights that CZE-MS/MS can make a significant contribution to phosphoproteomics by improving the phosphoproteome coverage.
We further analyzed the phosphopeptides exclusively identified in our work or Kubiniok’s work regarding the phosphosite motifs using the Motif-x, Figure 4. Interestingly, we observed significantly different motif logos between those two datasets for both phosphoserine and phosphothreonine. The corresponding phosphosites from the phosphopeptides exclusively identified in our work tend to be surrounded by acidic amino acids (glutamic acid and aspartic acid) after the phosphosites and basic amino acids (lysine and arginine) before the phosphosites compared to that in Kubiniok’s work, Figure 4. The data further highlights the value of CZE-MS/MS for phosphoproteomics for not only improving the phosphoproteome coverage but also providing more insight into the phosphosite motifs.
Figure 4.
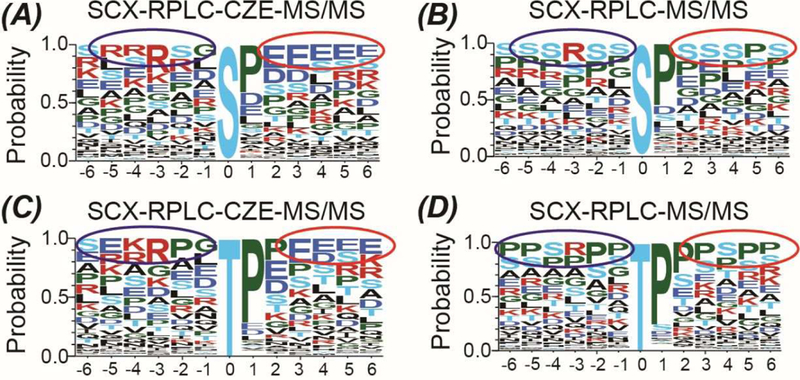
Summary of the phosphosite motif data from the SCX-RPLC-CZE-MS/MS in this work and from the SCX-RPLC-MS/MS in reference [45]. Motif-x (http://motif-x.med.harvard.edu/motif-x.html) was used to extract motifs from the data sets. Motif alignment was performed with WebLogo3 (http://webloqo.threeplusone.com/create.cgi). Motif logo of the phosphoserine (A) and phosphothreonine (C) based on the phosphopeptides exclusively identified in the SCX-RPLC-CZE-MS/MS data. Motif logo of the phosphoserine (B) and phosphothreonine (D) based on the phosphopeptides exclusively identified in the SCX-RPLC-MS/MS data.
Conclusions
An SCX-RPLC-CZE-MS/MS platform was employed for large-scale phosphoproteomics of the HCT116 cell line with the production of 11,555 phosphopeptide IDs. The dataset represents the largest phosphoproteome data so far using CZE-MS/MS. We are working on building a simple model based on the phosphoproteome dataset generated here for accurate prediction of phosphopeptide migration time in CZE. Our preliminary modeling attempts demonstrate that, similar to the unmodified tryptic peptides, the electrophoretic mobility of phosphopeptides can be accurately predicted. We expect that the predicted migration time of phosphopeptides will be useful to improve the confidence of phosphopeptide IDs from the database search and even guide the database search.
We expect that the number of phosphopeptide IDs from biological samples using CZE-MS/MS can be significantly boosted via several improvements. First, the phosphopeptide enrichment procedure can be dramatically improved. In this work, the specificity of phosphopeptide enrichment was only 35%. Over 80% and even 90% phosphopeptide enrichment specificity should be approachable with an optimized procedure based on the data in the literature.[48,49] Second, the separation system can be improved. Recently, we developed a high-resolution nanoflow RPLC-CZE-MS/MS system for deep and highly sensitive bottom-up proteomics with the production of 60,000 peptide IDs with only 5-µg of peptides as the starting material.[50] We expect significant improvements in both the number of phosphopeptide IDs and sensitivity will be achieved by using the nanoRPLC-CZE-MS/MS platform.
Supplementary Material
Acknowledgments
Daoyang Chen and Liangliang Sun thank the support from the Michigan State University and the National Institute of General Medical Sciences, National Institutes of Health (NIH), through Grant R01GM125991. Amanda B. Hummon thanks the support from the National Institutes of Health (R01GM110406), and the National Science Foundation (CAREER Award, CHE-1351595). Modeling studies were supported by grant from the Natural Sciences and Engineering Research Council of Canada (RGPIN-2016-05963 – Oleg Krokhin).
Footnotes
Supporting Information
Lists of identified phosphopeptides and proteins (XLSX)
Correlations between observed and predicted electrophoretic mobility of unphosphopeptides and phosphopeptides; Physicochemical properties of phosphopeptides (PDF)
Conflicts of interest
The authors declare no competing financial interest.
References
- [1].Graves JD; Krebs EG Protein phosphorylation and signal transduction. Pharmacol. Ther 1999, 82, 111–121. [DOI] [PubMed] [Google Scholar]
- [2].Yue X; Lukowski JK; Weaver EM; Skube SB; Hummon AB Quantitative Proteomic and Phosphoproteomic Comparison of 2D and 3D Colon Cancer Cell Culture Models. J. Proteome Res 2016, 15, 4265–4276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Sharma K; D’Souza RC; Tyanova S; Schaab C; Wiśniewski JR; Cox J; Mann M Ultradeep human phosphoproteome reveals a distinct regulatory nature of Tyr and Ser/Thr-based signaling. Cell Rep 2014, 8, 1583–94. [DOI] [PubMed] [Google Scholar]
- [4].Yue XS; Hummon AB Combination of multistep IMAC enrichment with high-pH reverse phase separation for in-depth phosphoproteomic profiling. J. Proteome Res 2013, 12, 4176–86. [DOI] [PubMed] [Google Scholar]
- [5].Zhou H; Di Palma S; Preisinger C; Peng M; Polat AN; Heck AJ; Mohammed S Toward a comprehensive characterization of a human cancer cell phosphoproteome. J. Proteome Res 2013, 12, 260–71. [DOI] [PubMed] [Google Scholar]
- [6].Song C; Ye M; Han G; Jiang X; Wang F; Yu Z; Chen R; Zou H Reversed-phase-reversed-phase liquid chromatography approach with high orthogonality for multidimensional separation of phosphorylated peptides. Anal. Chem 2010, 82, 53–6. [DOI] [PubMed] [Google Scholar]
- [7].Erickson BK; Jedrychowski MP; McAlister GC; Everley RA; Kunz R; Gygi SP Evaluating multiplexed quantitative phosphorylated peptide analysis on a hybrid quadrupole mass filter/linear ion trap/orbitrap mass spectrometer. Anal. Chem 2015, 87, 1241–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Phanstiel DH; Brumbaugh J; Wenger CD; Tian S; Probasco MD; Bailey DJ; Swaney DL; Tervo MA; Bolin JM; Ruotti V; Stewart R; Thomson JA; Coon JJ Proteomic and phosphoproteomic comparison of human ES and iPS cells. Nat. Methods 2011, 8, 821–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Peuchen EH; Cox OF; Sun L; Hebert AS; Coon JJ; Champion MM; Dovichi NJ; Huber PW Phosphorylation Dynamics Dominate the Regulated Proteome during Early Xenopus Development. Sci. Rep 2017, 7, 15647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Wang F; Song C; Cheng K; Jiang X; Ye M; Zou H Perspectives of comprehensive phosphoproteome analysis using shotgun strategy. Anal. Chem 2011, 83, 8078–85. [DOI] [PubMed] [Google Scholar]
- [11].Boersema PJ; Foong LY; Ding VM; Lemeer S; van Breukelen B; Philp R; Boekhorst J; Snel B; den Hertog J; Choo AB; Heck AJ In-depth qualitative and quantitative profiling of tyrosine phosphorylation using a combination of phosphorylated peptide immunoaffinity purification and stable isotope dimethyl labeling. Mol. Cell. Proteomics 2010, 9, 84–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Ubersax JA; Ferrell JE Jr. Mechanisms of specificity in protein phosphorylation. Nat. Rev. Mol. Cell Biol 2007, 8, 530–41. [DOI] [PubMed] [Google Scholar]
- [13].Jorgenson JW; Lukacs KD Capillary zone electrophoresis. Science 1983, 222, 266–72. [DOI] [PubMed] [Google Scholar]
- [14].Han X; Wang Y; Aslanian A; Fonslow B; Graczyk B; Davis TN; Yates JR 3rd. In-line separation by capillary electrophoresis prior to analysis by top-down mass spectrometry enables sensitive characterization of protein complexes. J. Proteome Res 2014, 13, 6078–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Zhang Z; Hebert AS; Westphall MS; Qu Y; Coon JJ; Dovichi NJ Production of Over 27 000 Peptide and Nearly 4400 Protein Identifications by Single-Shot Capillary-Zone Electrophoresis-Mass Spectrometry via Combination of a Very-Low-Electroosmosis Coated Capillary, a Third-Generation Electrokinetically-Pumped Sheath-Flow Nanospray Interface, an Orbitrap Fusion Lumos Tribrid Mass Spectrometer, and an Advanced-Peak-Determination Algorithm. Anal. Chem 2018, doi: 10.1021/acs.analchem.8b02991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Sun L; Zhu G; Zhao Y; Yan X; Mou S; Dovichi NJ Ultrasensitive and fast bottom-up analysis of femtogram amounts of complex proteome digests. Angew. Chem. Int. Ed 2013, 52, 13661–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Lombard-Banek C; Moody SA; Nemes P Single-Cell Mass Spectrometry for Discovery Proteomics: Quantifying Translational Cell Heterogeneity in the 16-Cell Frog (Xenopus) Embryo. Angew. Chem. Int. Ed 2016, 55, 2454–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Busnel JM; Schoenmaker B; Ramautar R; Carrasco-Pancorbo A; Ratnayake C; Feitelson JS; Chapman JD; Deelder AM; Mayboroda OA High capacity capillary electrophoresis-electrospray ionization mass spectrometry: coupling a porous sheathless interface with transient-isotachophoresis. Anal. Chem 2010, 82, 9476–83. [DOI] [PubMed] [Google Scholar]
- [19].Cheng YF; Wu SL; Chen DY; Dovichi NJ Interaction of capillary zone electrophoresis with a sheath-flow cuvette detector. Anal. Chem 1990, 62, 496–503. [Google Scholar]
- [20].Moini M Simplifying CE-MS operation. 2. Interfacing low-flow separation techniques to mass spectrometry using a porous tip. Anal. Chem 2007, 79, 4241–6. [DOI] [PubMed] [Google Scholar]
- [21].Wojcik R; Dada OO; Sadilek M; Dovichi N Simplified capillary electrophoresis nanospray sheath-flow interface for high efficiency and sensitive peptide analysis. J. Rapid Commun. Mass Spectrom 2010, 24, 2554–60. [DOI] [PubMed] [Google Scholar]
- [22].Sun L; Zhu G; Zhang Z; Mou S; Dovichi NJ Third-generation electrokinetically pumped sheath-flow nanospray interface with improved stability and sensitivity for automated capillary zone electrophoresis-mass spectrometry analysis of complex proteome digests. J. Proteome Res 2015, 14, 2312–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Maxwell EJ; Zhong X; Zhang H; van Zeijl N; Chen DD Decoupling CE and ESI for a more robust interface with MS. Electrophoresis 2010, 31, 1130–7. [DOI] [PubMed] [Google Scholar]
- [24].Aebersold R; Morrison HD Analysis of dilute peptide samples by capillary zone electrophoresis. J. Chromatogr 1990, 516, 79–88. [DOI] [PubMed] [Google Scholar]
- [25].Britz-McKibbin P; Chen DD Selective focusing of catecholamines and weakly acidic compounds by capillary electrophoresis using a dynamic pH junction. Anal. Chem 2000, 72, 1242–52. [DOI] [PubMed] [Google Scholar]
- [26].Zhu G; Sun L; Yan X; Dovichi NJ Bottom-up proteomics of Escherichia coli using dynamic pH junction preconcentration and capillary zone electrophoresis-electrospray ionization-tandem mass spectrometry. Anal. Chem 2014, 86, 6331–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Zhu G; Sun L; Dovichi NJ Thermally-initiated free radical polymerization for reproducible production of stable linear polyacrylamide coated capillaries, and their application to proteomic analysis using capillary zone electrophoresis-mass spectrometry. Talanta 2016, 146, 839–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Ludwig KR; Sun L; Zhu G; Dovichi NJ; Hummon AB Over 2300 phosphorylated peptide identifications with single-shot capillary zone electrophoresis-tandem mass spectrometry in a 100 min separation. Anal. Chem 2015, 87, 9532–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Faserl K; Sarg B; Gruber P; Lindner HH Investigating capillary electrophoresis-mass spectrometry for the analysis of common post-translational modifications. Electrophoresis 2018, 39, 1208–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Sarg B; Faserl K; Kremser L; Halfinger B; Sebastiano R; Lindner HH Comparing and combining capillary electrophoresis electrospray ionization mass spectrometry and nano-liquid chromatography electrospray ionization mass spectrometry for the characterization of post-translationally modified histones. Mol. Cell. Proteomics 2013, 12, 2640–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Krokhin OV; Anderson G; Spicer V; Sun L; Dovichi NJ Predicting Electrophoretic Mobility of Tryptic Peptides for High-Throughput CZE-MS Analysis. Anal. Chem 2017, 89, 2000–2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Chen D; Shen X; Sun L Capillary zone electrophoresis-mass spectrometry with microliter-scale loading capacity, 140 min separation window and high peak capacity for bottom-up proteomics. Analyst 2017, 142, 2118–2127. [DOI] [PubMed] [Google Scholar]
- [33].Lubeckyj RA; McCool EN; Shen X; Kou Q; Liu X; Sun L Single-Shot Top-Down Proteomics with Capillary Zone Electrophoresis-Electrospray Ionization-Tandem Mass Spectrometry for Identification of Nearly 600 Escherichia coli Proteoforms. Anal. Chem 2017, 89, 12059–12067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Chen D; Shen X; Sun L Strong cation exchange-reversed phase liquid chromatography-capillary zone electrophoresis-tandem mass spectrometry platform with high peak capacity for deep bottom-up proteomics. Anal. Chim. Acta 2018, 1012, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Li QR; Ning ZB; Tang JS; Nie S; Zeng R Effect of peptide-to-TiO2 beads ratio on phosphorylated peptide enrichment selectivity. J. Proteome Res 2009, 8, 5375–81. [DOI] [PubMed] [Google Scholar]
- [36].Yue X; Schunter A; Hummon AB Comparing multistep immobilized metal affinity chromatography and multistep TiO2 methods for phosphorylated peptide enrichment. Anal. Chem 2015, 87, 8837–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Eng JK; McCormack AL; Yates JR An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom 1994, 5, 976–89. [DOI] [PubMed] [Google Scholar]
- [38].Elias JE; Gygi SP Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 2007, 4, 207–14. [DOI] [PubMed] [Google Scholar]
- [39].Taus T; Köcher T; Pichler P; Paschke C; Schmidt A; Henrich C; Mechtler K Universal and confident phosphorylation site localization using phosphoRS. J. Proteome Res 2011, 10, 5354–62. [DOI] [PubMed] [Google Scholar]
- [40].Cox J; Mann M MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol 2008, 26, 1367–72. [DOI] [PubMed] [Google Scholar]
- [41].Cox J; Neuhauser N; Michalski A; Scheltema RA; Olsen JV; Mann M Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res 2011, 10, 1794–805. [DOI] [PubMed] [Google Scholar]
- [42].Krokhin OV Sequence-specific retention calculator. Algorithm for peptide retention prediction in ion-pair RP-HPLC: application to 300- and 100-A pore size C18 sorbents. Anal. Chem 2006, 78, 7785–95. [DOI] [PubMed] [Google Scholar]
- [43].Schwartz D; Gygi SP An iterative statistical approach to the identification of protein phosphorylation motifs from large-scale data sets. Nat. Biotechnol 2005, 23, 1391–8. [DOI] [PubMed] [Google Scholar]
- [44].Vizcaíno JA; Csordas A; del-Toro N; Dianes JA; Griss J; Lavidas I; Mayer G; Perez-Riverol Y; Reisinger F; Ternent T; Xu QW; Wang R; Hermjakob H 2016 update of the PRIDE database and its related tools. Nucleic Acids Res 2016, 44(D1), D447–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Kubiniok P; Lavoie H; Therrien M; Thibault P Time-resolved Phosphoproteome Analysis of Paradoxical RAF Activation Reveals Novel Targets of ERK. Mol. Cell. Proteomics 2017, 16, 663–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Li Y; Champion MM; Sun L; Champion PA; Wojcik R; Dovichi NJ Capillary zone electrophoresis-electrospray ionization-tandem mass spectrometry as an alternative proteomics platform to ultraperformance liquid chromatography-electrospray ionization-tandem mass spectrometry for samples of intermediate complexity. Anal. Chem 2012, 84, 1617–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Faserl K; Sarg B; Kremser L; Lindner H Optimization and evaluation of a sheathless capillary electrophoresis-electrospray ionization mass spectrometry platform for peptide analysis: comparison to liquid chromatography-electrospray ionization mass spectrometry. Anal. Chem 2011, 83, 7297–305. [DOI] [PubMed] [Google Scholar]
- [48].Zhou H; Ye M; Dong J; Corradini E; Cristobal A; Heck AJ; Zou H; Mohammed S Robust phosphoproteome enrichment using monodisperse microsphere-based immobilized titanium (IV) ion affinity chromatography. Nat. Protoc 2013, 8, 461–80. [DOI] [PubMed] [Google Scholar]
- [49].lliuk AB; Martin VA; Alicie BM; Geahlen RL; Tao WA In-depth analyses of kinase-dependent tyrosine phosphoproteomes based on metal ion-functionalized soluble nanopolymers. Mol. Cell. Proteomics 2010, 9, 2162–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Yang Z; Shen X; Chen D; Sun L Microscale Reversed-Phase Liquid Chromatography/Capillary Zone Electrophoresis-Tandem Mass Spectrometry for Deep and Highly Sensitive Bottom-Up Proteomics: Identification of 7500 Proteins with Five Micrograms of an MCF7 Proteome Digest. Anal. Chem 2018, 90, 10479–10486. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.