

Abstract

To evade the possible toxicity associated with the formation of quinone-imine metabolite in amodiaquine (AQ), the para-hydroxyl group was replaced with a −F atom, and the resulting 4′-fluoro-amodiaquine (FAQ) was hybridized with substituted pyrimidines. The synthesized FAQ–pyrimidines displayed better in vitro potency than chloroquine (CQ) against the resistant P. falciparum strain (Dd2), exhibiting up to 47.3-fold better activity (IC50: 4.69 nM) than CQ (IC50: 222 nM) and 2.8-fold better potency than artesunate (IC50: 13.0 nM). Twelve compounds exhibited better antiplasmodial activity than CQ against the CQ-sensitive (NF54) strain. Two compounds were evaluated in vivo against a P. berghei-mouse malaria model. Mechanistic heme-binding studies, computational docking studies against Pf-DHFR and in vitro microsomal stability studies were performed for the representative molecules of the series to assess their antimalarial efficacy.
Keywords: Aminoquinoline, fluoro-amodiaquine, pyrimidine, antiplasmodial, antimalarial, hybrid
Despite the global interventions to control malaria, it has still remained a major cause of human suffering and mortality. According to the World Health Organization’s (WHO) latest malaria report, there were an estimated 216 million cases of malaria in 2016, up from 211 million cases in the previous year, with 445,000 people succumbing to this age-old disease.1 Currently, WHO authorizes the use of artemisinins in the form of ACTs where short-acting artemisinins are coadministered with longer acting traditional antimalarial drugs. However, recent emergence of artemisinin-resistant plasmodial strains in five southeast Asian countries of Greater Mekong subregion2−4 point toward an alarming situation, where no treatment options will be available for such devastating multidrug resistant malaria infections. This situation calls for drastic measures to develop new antimalarial agents.
Amodiaquine (AQ) is a 4-aminoquinoline antimalarial similar to the blockbuster antimalarial chloroquine (CQ) but has a 4-hydroxy anilino function in its side chain instead of the linear aliphatic chain of CQ but retains four carbon atoms between the secondary and tertiary nitrogens (Figure 1). The internitrogen separation between quinoline nitrogen and tertiary nitrogen in the “active conformation” of both AQ and CQ is ∼8.30 Å as calculated by molecular modeling studies.5 AQ was found to have an excellent activity profile with better activity than CQ against CQ-resistant P. falciparum strains, but its therapeutic use was restricted due to its inherent toxicity,6,7 possibly due to the formation of quinone-imine (QI) metabolites akin to those produced during paracetamol metabolism.8,9 Lately, use of AQ has resurrected as a combination therapy partner due to its inherent advantages such as low price and effectiveness against CQ-resistant strains.
Figure 1.
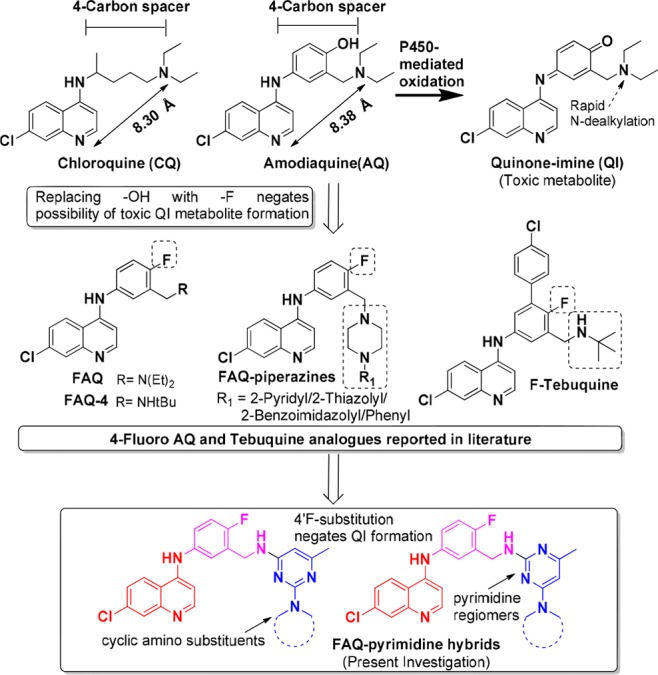
Design of FAQ–pyrimidine hybrids under present investigation through utilization of 4-fluoro substitution in AQ/tebuquine as previously reported in the literature.
Various strategies to reduce the toxicity associated with AQ/AQ-analogues through structural modifications have been reported10 but often on the expense of its antimalarial potency. O’Neill et al.11 replaced the para-OH group by a −F atom in the aniline ring of AQ to generate fluoro-amodiaquine (FAQ), which cannot form the reactive QI metabolites through P450-mediated oxidation (Figure 1). However, it was found to possess a 2- to 2.5-fold reduced activity than AQ against the CQ-sensitive and resistant strains. Similar strategy was adopted to synthesize fluoro-tebuquine analogues, but it resulted in diminished antimalarial activity compared to both AQ and tebuquine.12,13 FAQ-4 (4′-fluoro-N-tert-butylamodiaquine) was later identified as a candidate for further development studies based on its activity profile (comparable to AQ), low toxicity, and an acceptable safety profile.14 More recently, Sousa et al.15 reported the synthesis of FAQ-piperazines (Figure 1), but these analogues exhibited a 16–106-fold reduction in antiplasmodial activity.
Recently, molecular hybridization strategy has been utilized to discover new antimalarial agents16 and provides a way for recycling classical antimalarial pharmacophores.17 Our group has successfully utilized this approach and synthesized novel hybrids including 4-aminoquinoline-triazines,18,19 4-amino- quinoline-purines,20 and 4-aminoquinoline-pyrimidines21,22 exhibiting better antiplasmodial activities than the parent drugs, with the latter class of hybrids displaying the best antimalarial potencies. In continuation of our efforts to synthesize novel antimalarial hybrids and in order to retain the advantages associated with AQ, it was decided to utilize the FAQ scaffold, and FAQ–pyrimidines were designed (Figure 1), synthesized, and assessed for their antiplasmodial activities against CQ-sensitive and CQ-resistant P. falciparum strains.
To introduce the desired 3-(aminomethyl)-4-fluoroaniline linker to the 4-aminoquinoline scaffold, commercially available 4,7-dichloroquinoline (1) was reacted with 5-amino-2-fluorobenzonitrile (2), wherein the SNAr coupling product viz. 5-((7-chloroquinolin-4-yl)amino)-2-fluorobenzonitrile (3) was formed (Scheme 1). The nitrile group of intermediate 3 was then reduced to yield the corresponding benzyl amine (4) using CoCl2·6H2O and NaBH4 (which generates cobalt boride in situ)23 as the reducing agent. Subsequent coupling of intermediate-4 with 2,4-dichloro-6-methylpyrimidine afforded the regiomers 6a (major) and 6b (minor). On examining the characteristic 1HNMR peak of the lone pyrimidine ring’s proton (at its fifth position), the respective structures of regiomers 6a and 6b were assigned (see Figure S1 and detailed discussion in the Supporting Information). In 6a, this proton appeared at δ 6.37 ppm, while in the case of regiomer 6b, it appeared slightly downfield at δ 6.65 ppm because of the deshielding effect of the adjacent electronegative −Cl atom (at pyrimidine’s fourth position). Various secondary cyclic amines were then appended to both the regiomers 6a and 6b to yield the desired FAQ–pyrimidine hybrids (7a–h and 8a–h) as shown in Scheme 1.
Scheme 1.

Reaction conditions: (i) t-BuOH, 110 °C, 6 h, 90%; (ii) CoCl2·6H2O, NaBH4, MeOH, 0 °C to rt, 2 h, 83%; (iii) Et3N, DMF, 60 °C, 14 h, 57% (6a) and 16% (6b); (iv) secondary cyclic amines, K2CO3, DMF, 120 °C, 14 h, 79–92%.
The synthesized FAQ–pyrimidine hybrids (6a, 6b, 7a–h, and 8a–h) along with the intermediates 3 and 4 were then evaluated against CQ-sensitive NF54 strain (an African strain isolated from a patient in Netherlands) and CQ-resistant Dd2 strain (a South-East Asian strain, which is CQ and mefloquine-resistant) of P. falciparum. The results (IC50 values) are graphically illustrated in Figure 2 (also refer Table S1 in the Supporting Information for numerical IC50 values). While the intermediate 3 was moderately active (IC50 > 3.35 μM against NF54 strain and 3.20 μM against Dd2 strain of P. falciparum), intermediate 4 (4′-fluoro-bis-desethyl-amodiaquine) was found to display good activity with IC50 values of 46.06 and 58.72 nM against the CQ-sensitive and CQ-resistant strains, respectively. When pyrimidine was coupled with the intermediate-4, a 4–5-fold drop in the antiplasmodial activity of the resultant intermediates 6a and 6b (bearing a −Cl substituent) was observed. However, when the −Cl substituent of pyrimidine was replaced with the cyclic amino substituents to generate the final FAQ–pyrimidine hybrids (7a–h and 8a–h), antiplasmodial activity was greatly improved with compound 8b being the best active molecule with IC50 = 4.69 nM against the CQ-resistant Dd2 strain, a 47.3-fold better activity than CQ and 2.8-fold better than artemisinin. A similar trend of increasing antiplasmodial activity on the substitution of pyrimidine’s −Cl substituent with cyclic amines was observed in our previous studies21,22 reconfirming the importance of these substitutions on aminoquinoline–pyrimidine hybrids. On comparing the activities of regiomers 6a, 7a, 7c, and 7f–h (pyrimidine ring attached from its fourth-position to the FAQ scaffold) with their corresponding regiomeric counterparts 6b, 8a, 8c, and 8f–h (pyrimidine ring attached from its second-position to the FAQ moiety), it can be reasoned that the former hybrids showed better antiplasmodial activity than the latter regiomers. However, this trend was reversed in hybrids having a piperidin-1-yl, morpholin-1-yl, or thiomorpholin-1-yl substitution (7b, 7d, and 7e vs 8b, 8d, and 8e). These results indicate that cyclic amine substituents may have a greater impact on activity profile than the point of attachment of FAQ scaffold to the pyrimidine ring.
Figure 2.

Antiplasmodial activity pattern of FAQ–pyrimidine hybrids.
While no obvious trend in the antiplasmodial activity with the change in the cyclic amino substitution was observed; however, hybrids containing the piperidine and methyl/ethyl piperazine cyclic amine substituents were found to be more active. The changes in antiplasmodial activity were more evident in regiomers 8a–h where moving from piperidin-1-yl substituent (8b) to 5-membered pyrrolidin-1-yl (8a) or 7-membered azepan-1-yl (8c) and from methylpiperazin-1-yl (8f) to ethylpiperazin-1-yl (8g) and phenylpiperazin-1-yl (8h) showed a marked reduction in the antiplasmodial potency of FAQ–pyrimidine hybrids.
All the synthesized FAQ–pyrimidines exhibited better antiplasmodial potency against the resistant strain than the reference drug (CQ), except one intermediate (6b). The RI (resistance index; ratio of antiplasmodial IC50 of resistant/sensitive strains) values for almost all hybrids were near the ideal value (RI ≅1) and better than CQ (RICQ = 8.22). Eleven representative compounds were further assessed for their in vitro cytotoxicity against CHO cells (Table S1, Supporting Information). It was found that intermediate 4 was itself cytotoxic to CHO cells at 31.32 μM. FAQ–pyrimidine hybrids 7a, 8b, 8d, and 8e exhibited cytotoxicity against CHO cells in the micromolar range. However, it appeared that the cytotoxicity of the FAQ–pyrimidines may depend on the cyclic amino substitution, and hybrids 8f and 8g with 4-methylpiperazin-1-yl and 4-ethylpiperazin-1-yl substituents, respectively, were found to possess low cytotoxicities (IC50 = 58.23 and 80.33 μM) against CHO cells. Although some compounds were cytotoxic to the CHO cells, the selectivity index (SI) values of the FAQ–pyrimidines were high (SINF54 range, 237–3798; SIDd2 range, 169–3432) suggesting acceptable therapeutic index for the synthesized hybrids [SI = (IC50 (μM) for cytotoxicity to VERO cells) × 1000]/(IC50 (nM) for antiplasmodial activity)].
Two potent molecules 7f and 8b were selected for further in vivo evaluation in a P. berghei mouse malaria model. The compounds were administered to the P. berghei-infected mice through intraperitoneal route, once daily on days 0, 1, and 2 days postinfection, and monitored for apparent signs of toxicity, parasitemia, and survival until day 28 postinfection (Table 1). Compound 8b displayed better antimalarial activity than 7f and cured 5 out of 5 mice, while 7f cured 3 out of 5 mice, respectively. At the same dose (oral), CQ displayed curing of 1 out of 5 mice and AQ cured 5/5 mice, respectively. At lower dose (11.1 mg/kg), only 2 out of 5 7f-dosed mice survived with mean survival time of 19.8 days. In comparison, at 11.1 mg/kg dose, hybrid 8b is able to suppress parasitemia for the duration of the dosing period with 5/5 survival of mice, but it was unable to completely clear the parasites from the infected mice. However, at the same oral dose, AQ cured 4/5 mice, while CQ led to no cure but survival of 5/5 mice.
Table 1. In Vivo Antimalarial Activity of FAQ–Pyrimidine Hybrids in the P. berghei-mouse Malaria Model.
| % parasitemia
suppressiona |
||||||
|---|---|---|---|---|---|---|
| treatment | dose (mg/kg x no. of days postinfection) | day 5 | day 7 | survivalb | MSTc | cured |
| vehicle | NA x 3 | - | - | 0/5 | 13.2 | 0/5 |
| CQ1 | 33.3 × 3 | 100.0 | 100.0 | 5/5 | 28 | 1/5 |
| CQ1 | 11.1 × 3 | 100.0 | 99.2 | 5/5 | 28 | 0/5 |
| AQ1 | 33.3 × 3 | 100.0 | 100.0 | 5/5 | 28 | 5/5 |
| AQ1 | 11.1 × 3 | 100.0 | 100.0 | 5/5 | 28 | 4/5 |
| 7f2 | 33.3 × 3 | 100.0 | 100.0 | 5/5 | 28 | 3/5 |
| 7f2 | 11.1 × 3 | 91.9 | 92.9 | 2/5 | 19.8 | 0/5 |
| 8b2 | 33.3 × 3 | 100.0 | 100.0 | 5/5 | 28 | 5/5 |
| 8b2 | 11.1 × 3 | 100.0 | 100.0 | 5/5 | 28 | 0/5 |
The % suppression in parasitemia is calculated by considering the mean parasitemia in the vehicle control as 100%. Parasitemia suppression <80% is considered as nonsignificant.
Number of animals that survived on day 28/total animals in the group (the day of the death postinfection).
MST, mean survival time (days).
Number of mice without parasitemia (cured) until day 28 postinfection;1 oral-administration; 2intraperitoneal-administration
CQ and AQ are weak bases and are believed to diffuse across the food vacuole membrane of the parasite in a neutral form. However, inside the acidic food vacuole (pH ≈ 5.6) they get protonated and are accumulated thereby exerting their antiplasmodial action24 by interfering with the parasite’s heme-detoxification mechanism. Mechanistic heme binding studies were conducted with the active FAQ–pyrimidine hybrid 7f to validate heme-binding as a mode of action of these hybrids using the standard methods reported in the literature.25−27 The increasing concentrations of 7f led to a decrease in the Soret band (λmax 402 nm) of the monomeric heme at both pH 5.6 (corresponding to parasite’s acidic digestive vacuole) and pH 7.4 (physiological pH) as shown in Figure 3A,C. Utilizing a continuous variation method (Job’s plot), the binding stoichiometry of 7f with heme was determined. The Job’s plot showed maxima at 0.5 mol-fraction of 7f indicating a 1:1 binding stoichiometry with monomeric heme at both pH 5.6 (Figure 3B) and 7.4 (Figure 3D).
Figure 3.

Spectrophotometric titration of monomer heme with increasing concentrations of compound 7f at pH 5.6 [A] and pH 7.4 [C]. Job’s plot of (monomeric heme + 7f) complex formation at pH 5.6 [B] and pH 7.4 [D] (with CQ as reference drug).
Similar binding interactions were observed for the interaction of 7f with dimeric heme as shown in Figure 4, and a 1:1 binding stoichiometry of 7f with dimer heme was detected. CQ was used as a reference for all these experiments and showed a 1:1 binding stoichiometry.
Figure 4.
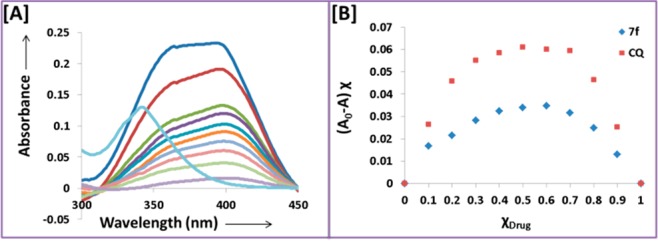
[A] Spectrophotometric titration of μ-oxodimeric heme with increasing mole fractions of compound 7f at pH 5.8. [B] Job’s plot of (μ-oxodimeric heme + 7f) complex formation at pH 5.8 (with CQ as reference drug).
To gain further insights about the plausible hybrid action of the FAQ–pyrimidines and considering the fact that pyrimidine-based antifolates target Pf-DHFR enzyme (P. falciparum dihydrofolate reductase), molecular docking studies of the best active compounds (7a, 7f, 8b, and 8e) were performed in the binding pocket of both the wild type Pf-DHFR-TS (PDB ID: 3QGT) and quadruple mutant Pf-DHFR-TS (N51I, C59R, S108 N, I164L; PDB ID: 3QG2) structures using Glide 5.8 (Schrodinger, LLC, New York, NY, 2012). The binding energies of FAQ–pyrimidines toward the wild and quadruple mutant Pf-DHFR-TS structures were comparable to the standard Pf-DHFR inhibitors (pyrimethamine, cycloguanil, and WR99210) and the native substrate, dihydrofolate (Table S2, Supporting Information). Compound 8b binds deep in the mutant Pf-DHFR’s binding site forming a major hydrogen bond interaction between the side chain oxygen atom of Asp54 residue of the plasmodial DHFR and the linker −NH group between fluorobenzene and pyrimidine of 8b. The oxygen atom of side chain of Asp54 is also H-bonded with the ring −NH group (charged conformer) of the pyrimidine moiety (Figure 5B). Further, the aromatic phenyl ring of Phe58 contributes in forming two π–π interactions between pyrimidine ring and another with the fluorobenzene ring of the compound, which is significant as this residue is known to cause steric hindrance to classical Pf-DHFR inhibitors such as pyrimethamine in the mutant enzyme. Similar interaction pattern was observed for compound 8b in the binding site of wild type Pf-DHFR, but the π–π interactions were not observed (Figure 5A).
Figure 5.
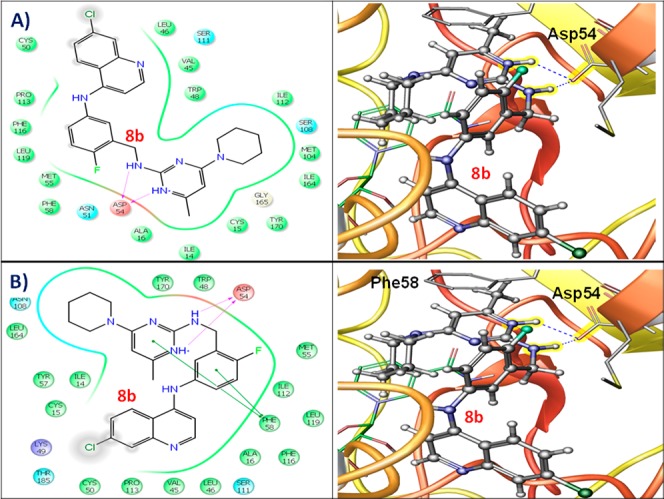
Two- and three-dimensional docking poses of compound 8b showing the interactions in the binding sites of (A) wild type Pf-DHFR-TS (PDB ID: 3QGT) and (B) quadruple mutant Pf-DHFR-TS (PDB ID: 3QG2).
The metabolic stability of compound 7f was also examined by measuring its half-life (t1/2) and intrinsic clearance (CLint) upon incubation with human liver microsomes in the presence and in the absence of NADPH. Verapamil served as the positive control for the experiment. Compound 7f displayed a t1/2 of 16.7 min and an intrinsic clearance of 82.8 μL·min–1·mg–1. In comparison, AQ was found to have the literature values of t1/2 = 5.4 min and CLint = 431 μL·min–1·mg–1.28
As mentioned earlier, protonation of CQ and AQ inside the parasite’s food vacuole is important for antimalarial activity. We predicted the pKa and protonation states (at pH 5.6) of the FAQ–pyrimidines in order to correlate them with the observed antiplasmodial activity. ChemAxon29 was used for predicting pKa and protonation states of the test compounds, and these are presented in Table S3 of the Supporting Information. The diminished antiplasmodial activity of intermediate 3 having a single protonable quinolinyl N atom in comparison to the diprotic bases CQ and AQ can be attributed to this hypothesis. Intermediate 4 also has two protonable sites and hence shows better activity. Intermediates 6a and 6b have a quinolinyl N atom, which is protonable but the pyrimidine’s N atom has a low pKa of 3.38–3.92 (which means it remains unprotonated) and hence their activity reduces. After cyclic amine substitutions, pKa values of pyrimidine’s N atom are improved (pKa range 7.66–7.82) in the resulting FAQ–pyrimidines (7a–h and 8a–h), and they show better antiplasmodial activities. Compounds 7f–g and 8f–g with N-methyl/ethyl piperazine substituents have three protonable nitrogens, and this can be a reason for their increased antiplasmodial activity. However, nitrogen of the N-phenyl piperazine substituent in 7h and 8h has a low pKa (3.00 and 3.12) and is not protonated, which may have led to their decreased activity. However, the same hypothesis does not hold true for FAQ–pyrimidines having other cyclic amine substituents, and there may be other factors involved in the putative accumulation of FAQ–pyrimidines or they may have other synergistic mode(s) of action.
Prediction of ADME parameters for the six most active compounds (IC50Dd2-strain < 18 nM) were performed, and the results are presented in Table S4 of the Supporting Information. Except QPlogHERG descriptor (prediction for HERG-K+ channel inhibition), all other ADME parameters were found to lie within the acceptable range.
To summarize, a set of 18 FAQ–pyrimidine hybrids were designed and synthesized in order to discover new antimalarial agents effective against drug-resistant and drug-sensitive plasmodial strains. Out of the 18 synthesized FAQ–pyrimidines (including intermediates 6a and 6b), all compounds, except one (6b), were found to display a better potency than CQ against the resistant strain (Dd2), whereas 12 compounds were found to possess better potency than CQ against the CQ-sensitive NF54 strain. Two molecules (7f and 8b) were further assessed in vivo against P. berghei mouse malaria model and cured mice at 33.3 mg/kg dose. Mechanistic heme binding studies were performed that showed good heme-binding interactions comparable to the reference 4-aminoquinoline drug (CQ). Based on the combined inputs from the structure–activity relationships, molecular docking studies on Pf-DHFR-TS, pKa, and ADME predictions, it is intended to further optimize FAQ–pyrimidines to generate promising antimalarial agents.
Acknowledgments
M.T. and P.P. are thankful to DST and UGC for the award of INSPIRE-senior research fellowship and D. S. Kothari postdoctoral fellowship, respectively. The authors are thankful to USIC-CIF and DU-DST Mass facility, University of Delhi, Delhi for analytical data. The authors would like to express their appreciation to DST-FIST for providing the high precision bioanalytical facility (HPBAF) at Ocular Pharmacology and Pharmacy Division, Dr. Rajendra Prasad Centre for Ophthalmic Sciences, All India Institute of Medical Sciences, New Delhi.
Glossary
ABBREVIATIONS
- FAQ
4′-fluoro-amodiaquine
- FAQ-4
4′-fluoro-N-tert-butylamodiaquine
- Pf-DHFR-TS
P. falciparum dihydrofolate reductase thymidylate synthase
- NADPH
β-nicotinamide adenine dinucleotide 2′-phosphate reduced tetrasodium salt hydrate
- HERG-K+
Human Ether-a-go-go-related gene potassium channel
- ADME
absorption, distribution, metabolism, and excretion
- QP
Quikprop (Schrödinger, LLC, New York, NY)
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.8b00496.
Full experimental details (including 1HNMR/13CNMR spectra and HPLC chromatograms) for the synthesized compounds, procedures for their heme-binding experiments and computational studies along with Table S1 for antiplasmodial activity and cytotoxicity, Table S2 for docking studies, Table S3 for pKa and protonation state prediction, and Table S4 for ADME predictions (PDF)
Author Contributions
All authors have given approval to the final version of the manuscript.
This research work was supported by grants from SERB (Science and Engineering Research Board) and Govt. of India through its EMR project [EMR/2014/001127].
The authors declare no competing financial interest.
Supplementary Material
References
- World Malaria Report: 2017. https://www.who.int/malaria/publications/world-malaria-report-2017/en/ (accessed Jan 1, 2018).
- Noedl H.; Se Y.; Schaecher K.; Smith B. L.; Socheat D.; Fukuda M. M. Evidence of artemisinin-resistant malaria in western Cambodia. N. Engl. J. Med. 2008, 359, 2619–2620. 10.1056/NEJMc0805011. [DOI] [PubMed] [Google Scholar]
- Dondorp A. M.; Nosten F.; Yi P.; Das D.; Phyo A. P.; Tarning J.; Lwin K. M.; Ariey F.; Hanpithakpong W.; Lee S. J.; Ringwald P.; Silamut K.; Imwong M.; Chotivanich K.; Lim P.; Herdman T.; An S. S.; Yeung S.; Singhasivanon P.; Day N. P.; Lindegardh N.; Socheat D.; White N. J. Artemisinin resistance in Plasmodium falciparum malaria. N. Engl. J. Med. 2009, 361, 455–467. 10.1056/NEJMoa0808859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairhurst R. M.; Dondorp A. M. Artemisinin-resistant Plasmodium falciparum malaria. Microbiol. Spectr. 2016, 4, 409. 10.1128/microbiolspec.EI10-0013-2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh H. L.; Go M. L.; Ngiam T. L.; Mak J. W. Conformational and structural features determining in vitro antimalarial activity in some indolo[3,2-c]quinolines, anilinoquinolines and tetrahydroindolo[3,2-d]benzazepines. Eur. J. Med. Chem. 1994, 29, 107–113. 10.1016/0223-5234(94)90206-2. [DOI] [Google Scholar]
- Hatton C. S.; Peto T. E.; Bunch C.; Pasvol G.; Russell S. J.; Singer C. R.; Edwards G.; Winstanley P. Frequency of severe neutropenia associated with amodiaquine prophylaxis against malaria. Lancet 1986, 1, 411–414. 10.1016/S0140-6736(86)92371-8. [DOI] [PubMed] [Google Scholar]
- Neftel K. A.; Woodtly W.; Schmid M.; Frick P. G.; Fehr J. Amodiaquine induced agranulocytosis and liver damage. Br. Med. J. (Clin. Res. Ed.) 1986, 292, 721–723. 10.1136/bmj.292.6522.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinson J. A.; Roberts D. W.; James L. P.. Mechanisms of acetaminophen-induced liver necrosis. In ; Adverse Drug Reactions. Handbook of Experimental Pharmacology; Uetrecht J., Ed; Springer: Berlin, Heidelberg, 2010; Vol. 196, pp 369–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jewell H.; Ruscoe J. E.; Maggs J. L.; O’Neill P. M.; Storr R. C.; Ward S. A.; Park B. K. The effect of chemical substitution on the metabolic activation, metabolic detoxication, and pharmacological activity of amodiaquine in the mouse. J. Pharmacol. Exp. Ther. 1995, 273, 393–404. [PubMed] [Google Scholar]
- O’Neill P. M.; Barton V. E.; Ward S. A.; Chadwick J.. 4-Aminoquinolines: chloroquine, amodiaquine and next-generation analogues. In Treatment and Prevention of Malaria: Antimalarial Drug Chemistry, Action and Use; Staines H. M., Krishna S., Eds.; Springer: Basel, 2012; pp 19–44. [Google Scholar]
- O’Neill P. M.; Harrison A. C.; Storr R. C.; Hawley S. R.; Ward S. A.; Park B. K. The effect of fluorine substitution on the metabolism and antimalarial activity of amodiaquine. J. Med. Chem. 1994, 37, 1362–1370. 10.1021/jm00035a017. [DOI] [PubMed] [Google Scholar]
- O’Neill P. M.; Hawley S. R.; Storr R. C.; Ward S. A.; Park B. K. The effect of fluorine substitution on the antimalarial activity of tebuquine. Bioorg. Med. Chem. Lett. 1996, 6, 391–392. 10.1016/0960-894X(96)00040-6. [DOI] [Google Scholar]
- O’Neill P. M.; Willock D. J.; Hawley S. R.; Bray P. G.; Storr R. C.; Ward S. A.; Park B. K. Synthesis, antimalarial activity, and molecular modeling of tebuquine analogues. J. Med. Chem. 1997, 40, 437–448. 10.1021/jm960370r. [DOI] [PubMed] [Google Scholar]
- O’Neill P. M.; Shone A. E.; Stanford D.; Nixon G.; Asadollahy E.; Park B. K.; Maggs J. L.; Roberts P.; Stocks P. A.; Biagini G.; Bray P. G. Synthesis, antimalarial activity, and preclinical pharmacology of a novel series of 4′-fluoro and 4′-chloro analogues of amodiaquine. Identification of a suitable “back-up” compound for N-tert-butyl isoquine. J. Med. Chem. 2009, 52, 1828–1844. 10.1021/jm8012757. [DOI] [PubMed] [Google Scholar]
- Sousa A. C.; Viana G. M.; Diaz N. C.; Rezende M. G.; Oliveira F. F.; Nunes R. P.; Pereira M. F.; Areas A. L.; Zalis M. G.; Frutuoso Vda S.; Faria H. C.; Domingos T. F.; Pádula Md.; Cabral L. M.; Rodrigues C. R. Design synthesis and evaluation of new fluoroamodiaquine analogues. Chem. Pharm. Bull. 2016, 64, 594–601. 10.1248/cpb.c15-01001. [DOI] [PubMed] [Google Scholar]
- Meunier B. Hybrid molecules with a dual mode of action: dream or reality?. Acc. Chem. Res. 2008, 41, 69–77. 10.1021/ar7000843. [DOI] [PubMed] [Google Scholar]
- Teixeira C.; Vale N.; Pérez B.; Gomes A.; Gomes J. R.; Gomes P. Recycling” classical drugs for malaria. Chem. Rev. 2014, 114, 11164–11220. 10.1021/cr500123g. [DOI] [PubMed] [Google Scholar]
- Manohar S.; Khan S. I.; Rawat D. S. Synthesis, antimalarial activity and cytotoxicity of 4-aminoquinoline-triazine conjugates. Bioorg. Med. Chem. Lett. 2010, 20, 322–325. 10.1016/j.bmcl.2009.10.106. [DOI] [PubMed] [Google Scholar]
- Manohar S.; Khan S. I.; Rawat D. S. 4-aminoquinoline-triazine-based hybrids with improved in vitro antimalarial activity against CQ-sensitive and CQ-resistant strains of Plasmodium falciparum. Chem. Biol. Drug Des. 2013, 81, 625–630. 10.1111/cbdd.12108. [DOI] [PubMed] [Google Scholar]
- Kumar D.; Khan S. I.; Tekwani B. L.; Ponnan P.; Rawat D. S. 4-Aminoquinoline-pyrimidine hybrids: synthesis, antimalarial activity, heme binding and docking studies. Eur. J. Med. Chem. 2015, 89, 490–502. 10.1016/j.ejmech.2014.10.061. [DOI] [PubMed] [Google Scholar]
- Manohar S.; Rajesh U. C.; Khan S. I.; Tekwani B. L.; Rawat D. S. Novel 4-aminoquinoline-pyrimidine based hybrids with improved in vitro and in vivo antimalarial activity. ACS Med. Chem. Lett. 2012, 3, 555–559. 10.1021/ml3000808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manohar S.; Pavan V. S.; Taylor D.; Kumar D.; Ponnan P.; Wiesner L.; Rawat D. S. Highly active 4-aminoquinoline–pyrimidine based molecular hybrids as potential next generation antimalarial agents. RSC Adv. 2015, 5, 28171–28186. 10.1039/C4RA16032K. [DOI] [Google Scholar]
- Osby J. O.; Heinzman S. W.; Ganem B. Studies on the mechanism of transition-metal-assisted sodium borohydride and lithium aluminum hydride reductions. J. Am. Chem. Soc. 1986, 108, 67–72. 10.1021/ja00261a011. [DOI] [Google Scholar]
- Warhurst D. C.; Craig J. C.; Adagu I. S.; Meyer D. J.; Lee S. Y. The relationship of physico-chemical properties and structure to the differential antiplasmodial activity of the cinchona alkaloids. Malar. J. 2003, 2, 26. 10.1186/1475-2875-2-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huy N. T.; Mizunuma K.; Kaur K.; Nhien N. T. T.; Jain M.; Uyen D. T.; Harada S.; Jain R.; Kamei K. 2-tert-butyl-8-quinolinamines exhibit potent blood schizontocidal antimalarial activity via inhibition of heme crystallization. Antimicrob. Agents Chemother. 2007, 51, 2842–2847. 10.1128/AAC.00288-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan T. J.; Mavuso W. W.; Ross D. C.; Marques H. M. Thermodynamic factors controlling the interaction of quinoline antimalarial drugs with ferriprotoporphyrin IX. J. Inorg. Biochem. 1997, 68, 137–145. 10.1016/S0162-0134(97)00086-X. [DOI] [PubMed] [Google Scholar]
- Xu Kelly J.; Winter R.; Riscoe M.; Peyton D. H. A spectroscopic investigation of the binding interactions between 4, 5-dihydroxyxanthone and heme. J. Inorg. Biochem. 2001, 86, 617–625. 10.1016/S0162-0134(01)00217-3. [DOI] [PubMed] [Google Scholar]
- Rajapakse C. S.; Lisai M.; Deregnaucourt C.; Sinou V.; Latour C.; Roy D.; Schrével J.; Sánchez-Delgado R. A. Synthesis of new 4-aminoquinolines and evaluation of their in vitro activity against chloroquine-sensitive and chloroquine-resistant plasmodium falciparum. PLoS One 2015, 10, e0140878 10.1371/journal.pone.0140878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calculator Plugins, Marvin 19.3.0; ChemAxon, 2019.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.