

Abstract

A series of (E)-3-(4-((2,4-bis(trifluoromethyl)benzyl)oxy)-3-methoxyphenyl)-2-cyanoacrylamide derivatives were designed and synthesized as new estrogen-related receptor α (ERRα) degraders based on the proteolysis targeting chimera (PROTAC) concept. One of the representative compounds 6c is capable of specifically degrading ERRα protein by >80% at a relatively low concentration of 30 nM, becoming one of the most potent and selective ERRα degraders to date. Compound 6c could be utilized as a new powerful research tool for further biological investigation of ERRα.
Keywords: Estrogen-related receptor α (ERRα), proteolysis targeting chimera (PROTAC), protein degradation
Estrogen-related receptors (ERRs) belong to an orphan nuclear receptor superfamily.1 Three isoforms of ERRs (i.e., ERRα,2 β, and γ3) have been identified to date. ERRβ is associated with early development,4 whereas ERRα and ERRγ are being considered as novel potential targets for metabolic disorders.5,6 ERRα is the first-discovered and most-studied member of this subgroup and was named because of its relatively high homology with estrogen receptor α (ERα). In addition to the cross-talk with ERα signals,7 ERRα also plays a critical role in regulating metabolism and energy homeostasis8 by interacting with multiple transcriptional cofactors, such as the peroxisome proliferator-activated receptor γ coactivator 1 proteins (i.e., PGC-1α and PGC-1β),9 receptor-interacting protein 140 corepressor (RIP-140),10 etc.
A small molecule up-regulating function of ERRα had been suggested as a new potential therapeutic strategy for type II diabetes and other metabolic disorders, based on its crucial roles in mitochondrial oxidative phosphorylation (OXPHOS) and fatty acid oxidation processes.6 However, whole-body ERRα knockout animals are unexpectedly lean and nondiabetic.11 Pharmacological inhibition of ERRα by using an inverse agonist also exhibited in vivo antidiabetic efficacy.12 These paradoxical results indicate complicated and diverse functions of ERRα in metabolic regulation.
ERRα/PGC-1α(β) transcriptional axis also mediates metabolic adaptations of various types of human cancer cells.13−15 For instance, metabolic alterations driven by ERRα obviously stimulate proliferation, migration, angiogenesis, metastasis, and drug resistance of human breast cancer cells.16−18 ERRα overexpression has also been frequently detected in Her2+ and triple-negative breast tumors and associated with poor clinical outcome of patients.19 Pharmacological inhibition and gene knockout of ERRα were proven to mitigate breast cancer progression both in vitro and in vivo.20,21
To facilitate biological investigation on the target, several ERRα inverse agonists and small molecules improving its transcriptional function have been discovered.12,22−32 Most recently, a proteolysis targeting chimera (PROTAC) compound was also designed and synthesized by Crews et al. to induce the degradation of ERRα (Figure 1).33
Figure 1.

Chemical structures of representative ERRα inverse agonists and previously reported ERRα degrader.
PROTAC is a new “chemical-small-molecule-induced protein knockdown” technology to recruit an E2/E3 ligase to the target protein and promote polyubiquitination and subsequent protein degradation via proteasome.34 Comparing with the classical activity inhibitors, PROTAC can mimic the natural proteasome degradation process to totally abolish functions of target protein. Additionally, obvious advantages, such as superior target specificity35 and accessibility to “undruggable” targets,36 make PROTAC as an attractive strategy for next generation drug discovery.37,38
Herein, we would like to report our effort to identify a new series of PROTAC-based ERRα degradation inducers with obviously improved potency.
Architecturally, PROTAC molecules possess two distinct ligand moieties that are able to bind with the target protein and E2/E3 ubiquitin ligase, respectively, and a covalently connecting linker.34 Therefore, the first step for designing a degrader is to identify a novel ERRα ligand.
Compound 1 (XCT790)23 is the first reported and widely investigated ERRα inverse agonist with an IC50 value of 0.37 μM in a GAL4-ERRα cell-based transfection assay. A preliminary computational study suggested that compound 1 could fit into a ligand-induced pocket of ERRα (PDB code: 3K6P, Figure 2A). The α-cyano-α, β-unsaturated carbonyl moiety is close to Cys325 residue with a distance of 3.5 Å, suggesting a covalent interaction could be formed. The diaryl ether core was embedded in a hydrophobic pocket formed by Phe382, Val369, Leu405, Val366, Val491, and Phe495 residues, whereas the thiadiazoloacylamide group of 1 seemed to extend toward solvent exposing surface of the protein and did not form any defined interaction with ERRα. It was hypothesized that the thiadiazoloacylamide group in compound 1 could be removed without affecting the binding with ERRα. Compound 4a was thus designed and synthesized based on this hypothesis (Figure 2B).
Figure 2.
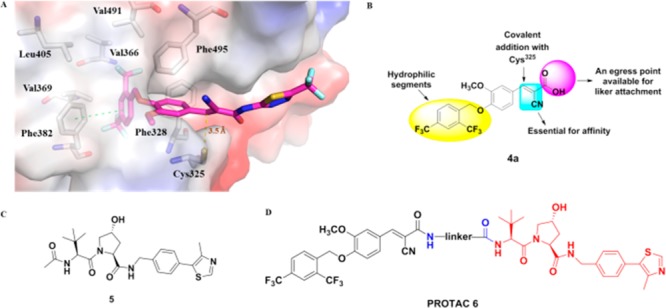
(A) Predicted binding mode of compound 1 with ERRα (PDB code: 3K6P). (B) Structure–activity relationship (SAR) overview for compound 4a. (C) Chemical structure of von-Hippel-Lindau (VHL) ligand 5. (D) Chemical structure of designed PROTACs 6.
Activity of compound 4a to block the protein–protein interaction of ERRα with PGC-1α coactivator was determined by utilizing a well-established time-resolved fluorescence resonance energy transfer (TR-FRET) assay.30 Under the experimental conditions, reference compounds 1 and 2 exhibited strong inhibitory potencies with IC50 values of 61.3 and 33.0 nM, respectively, which are comparable to the reported data.12,30 Encouragingly, the structurally simplified molecule 4a displayed a ∼11-fold improved potency with an IC50 value of 5.67 nM. Additionally, removal of the thiadiazoloacylamide group also resulted in a significant solubility improvement of 4a (data not shown). Highly consistent to our prediction, the terminal carboxylic acid in 4a could be replaced by a hydrophilic group, such as an unsubstituted (4c) or substituted (4d, 4e, and 4f) amide without affecting the binding affinity with ERRα. However, esterification of the carboxylic acid (4b) caused a ∼4-fold potency loss in the TR-FRET assay. These data collectively suggested that the terminal carboxylic acid group could be a feasible position to introduce a linker for further PROTAC molecule design (Table 1).
Table 1. In Vitro Binding Affinities to ERRα of Inverse Agonists 4a–4fa.
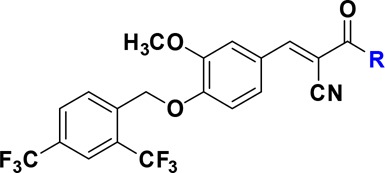
| Cpds | R | TR-FRETa IC50 (nM) |
|---|---|---|
| 4a | OH | 5.67 ± 0.58 |
| 4b | OCH3 | 20.33 ± 1.53 |
| 4c | NH2 | 4.33 ± 1.53 |
| 4d | NHCH2CH2OCH3 | 4.33 ± 0.58 |
| 4e | NHCH2CH2CH2CH2COOH | 6.00 ± 1.00 |
| 4f | NHCH2CH2OCH2CH2COOH | 4.67 ± 0.58 |
| 2 | 33.0 ± 1.73 | |
| 1 | 61.33 ± 6.11 |
Affinity to ERRα was determined by using a TR-FRET assay (Invitrogen, USA). The data are means from at least three independent experiments.
Several small-molecule E2/E3 ligase ligands,39 such as Mouse Double Minute 2 (MDM2) ligand Nutlin derivatives, cellular inhibitor of apoptosis protein 1 (cIAP1) inhibitor bestain, cereblon (CRBN) ligand thalidomide derivatives, and the von-Hippel-Lindau (VHL) ligand, have been incorporated into PROTAC molecules to mediate degradation of the target proteins. It is also clear that the E2/E3 ubiquitin ligase ligand choice40 has significant impact on efficiency and target specificity of a PROTAC molecule. The VHL ligand had been previously proven to be highly efficient to induce degradation of ERRα protein.33 Therefore, VHL ligand 5 (Figure 2C), which harbors a solvent-exposing acetamide group for conjugating a linker,41 was first utilized for our new ERRα degrader design.
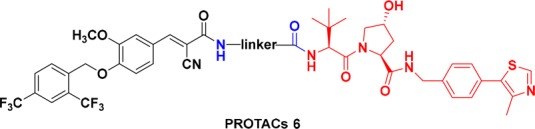
A series of PROTAC molecules were first designed and synthesized by hybridizing 4a and VHL amino building block 13 with different linkers (Scheme 1). Carboxylic acid 4a was coupled with amino ester 10 in the presence of HATU and DIPEA at room temperature in DMF to give the amide 11, which was deprotected with TFA to afford the corresponding carboxylic acid intermediate 12. Upon coupling with VHL amino building block 13 (prepared as previously described, Supporting Information), the designed PROTACs (6a–b, 6d, 6g–j) were generated. Differently, ERRα-PROTACs (6c, 6e–f) and the epimeric 6c (6c-Epi) were prepared by using an alternative protocol. VHL amino ligand 13 was coupled with Boc-protected amino acid 14, and the resulting compound 15 was treated with TFA to give 16. The final products were obtained by condensation of 16 and 4a. Compound 6c-Epi was synthesized by using an epi-VHL building block harboring a reversed (S) stereochemistry at the proline 4-position.33
Scheme 1. Synthesis of PROTACs 6a–6j.

Reagents and conditions: (a) 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo [4,5-b]-pyridinium 3-oxide hexafluorophosphate (HATU), N,N-diisopropylethylamine (DIPEA), N,N-dimethylformamide (DMF), room temperature (rt), 39–71%; (b) trifluoroacetic acid (TFA), dichloromethane (DCM), rt.
Binding affinities of the new hybrids and their function to degrade ERRα were determined by utilizing in vitro TR-FRET assay and Western blotting analysis in the MDA-MB-231 cells, respectively (Table 2 and Supporting Information). The first reported ERRα PROTAC 3 (C29_PROTAC) was utilized as a positive control in both assays. Compound 3 exhibited an IC50 value of 8.0 nM to block the protein–protein interaction of ERRα with PGC-1α peptide and induced approximately 40% protein degradation at 100 nM (D100 nM) after 4.0 h treatment, which are similar to the previously reported data.33
Table 2. Binding Affinities and Degradation Activities to ERRα of PROTACs 6a–6j.
| Cpds | linker | TR-FRETa IC50 (nM) | D30 nMb (%) | D100 nMb (%) |
|---|---|---|---|---|
| 6a | (CH2)2 | 7.33 ± 3.06 | 28 | 50 |
| 6b | (CH2)3 | 6.33 ± 1.15 | 58 | 85 |
| 6c | (CH2)4 | 12.67 ± 2.89 | 83 (D10 nM = 39) | 96 |
| 6d | (CH2)5 | 5.67 ± 1.53 | 58 | 78 |
| 6e | (CH2)6 | 12.33 ± 2.52 | 23 | 71 |
| 6f | (CH2)7 | 7.67 ± 0.58 | 3 | 9 |
| 6g | CH2CH2OCH2 | 11.33 ± 0.58 | 79 | 92 |
| 6h | CH2CH2O CH2CH2 | 9.33 ± 4.16 | 63 (D10 nM = 25) | 89 |
| 6i | (CH2CH2O)2 CH2CH2 | 5.33 ± 2.52 | 0 | 15 (D300 nM = 19) |
| 6j | (CH2CH2O)3 CH2CH2 | 7.00 ± 1.00 | 0 | 4 (D300 nM = 10) |
| 6c-Epi | (CH2)4 | 5.33 ± 1.15 | 6 | NTc |
| 3 | 8.00 ± 2.65 | 1 | 40 |
Binding affinity with ERRα was determined by using a TR-FRET assay.
D (%): % degradation is measured at indicated concentration by using Western blot assay and generated by ImageJ software analysis.46 The data are means from at least three independent experiments. Representative Western blots can be found in the Supporting Information (Figure S2).
NT: not tested.
It was shown that length and chemical types of the hybrid linker had minor influence on ERRα binding affinities of the PROTAC molecules. Compounds 6a–6j exhibited strong binding with ERRα, with IC50 values ranging from 5.33 to 12.67 nM, which are comparable to the parental molecule 4a. These data further supported that our initial hypothesis that the terminal carboxylic acid group in 4a is a feasible position for E2/E3 ligase ligand linkage. However, the introduced linkers had significant impact on ERRα protein degradation capabilities of the molecules. For instance, compounds 6a and 6b, which harbor a 2-carbon or 3-carbon linker, exhibited almost the same ERRα inhibitory potencies with IC50 values of 7.3 and 6.3 nM, respectively. However, they displayed significantly varied capabilities to induce degradation of ERRα protein, with D30 nM and D100 nM values of 28%, 50% and 58%, 85%, respectively. Although a 4-carbon linker caused an approximately 2-fold ERRα binding affinity loss, the resulting compound 6c exhibited the strongest protein degradation potency, with D30 nM and D100 nM values of 83% and 96%, respectively. It could even induce approximately 39% of ERRα degradation at a concentration as low as 10 nM (D10 nM). These results strongly support the hypothesis that each PROTAC causes a unique conformational change and that protein degradation requires certain plasticity in binding.42 The compound with a 5-carbon linker (6d) also displayed strong ERRα degradation potency, whereas a further longer carbon linker (e.g., 6- or 7- carbon linker) caused an obvious protein-degrading potency loss (6e and 6f). The investigation also revealed that the linker could be tolerated by an oxygen atom-containing moiety to retain the strong ERRα degrading potency. Compounds 6g and 6h exhibited D100 nM values of 92% and 89%, respectively. It was noteworthy that compound 6h harbors the same alkyl ether linker with that of previously reported ERRα degrader 3,33 but its potency is significantly stronger than that of compound 3. For instance, the ERRα degrading potency of compound 3 was almost abolished at 30 nM (with a D30 nM value of approximately 1%), but compound 6h induced 25% and 63% ERRα degradation at concentrations of 10 and 30 nM, respectively. Similar to observation on the compounds with pure carbon linkers, longer polyethylene glycol (PEG) linkers also caused significant protein degrading potency loss of the resulting compounds (6i and 6j). Not surprisingly, although 6c-Epi exhibited strong binding with the target protein ERRα, its ERRα degrading function was totally abolished because the epi-VHL building block almost completely prevented the binding with VHL E3 ligase.33
Molecules obtained by hybridizing compound 4a with small molecular ligands of other E2/E3 ligases (e.g., MDM2, cIAP1 and CRBN) were also designed and synthesized. However, none of them exhibited superior ERRα degradation potency to that of molecule 6c (Supporting Information).
The selectivity of compound 6c to induce ERRα degradation was further validated in MDA-MB-231 breast cancer cells (Figures 3 and 4). It was confirmed that compound 6c dose-dependently induced ERRα degradation with an efficacious dose as low as 3.0 nM at 4.0 h (Figure 3A), whereas 6c did not show obvious effect on ERRβ and ERRγ homologues at relatively high concentrations (up to 10.0 μM, Figure 3B) at 4.0 h. These data are highly consistent with the specific binding of 6c with ERRα protein (Supporting Information). A time-course investigation further revealed that compound 6c could initiate ERRα degradation after 30 min exposure and demonstrated a maximum effect at approximately 4.0 h after treatment. However, the level of ERRα could be recovered after 48 h (Figure 3C), which might be due to resynthesis of the protein and/or other intracellular feedback response. Further investigation also showed that 6c potently decreased protein levels of ERRα downstream target genes, e.g., ATP5B, medium-chain acyl CoA dehydrogenase (MCAD), and pyruvate dehydrogenase kinase 4 (PDK4) in the MDA-MB-231 cells after a 24 h treatment (Supporting Information).
Figure 3.

Degradation effects of PROTAC 6c on ERRα, ERRβ, and ERRγ proteins in MDA-MB-231 cells by Western blot analysis. (A) PROTAC 6c dose-dependently degrades ERRα protein. (B) PROTAC 6c has no effect on the levels of ERRβ and ERRγ protein at indicated concentration. (C) Time-points degradation analysis of PROTAC 6c at 30 nM.
Figure 4.

Mechanism study of PROTAC 6c in MDA-MB-231 cells by Western blotting analysis. (A) PROTAC 6c-mediated ERRα degradation is abrogated by pretreatment with proteasome inhibitor Epoxomicin. (B) PROTAC 6c-mediated ERRα degradation is abrogated by pretreatment with ERRα ligand 4f or VHL ligand 5.
Proteasome is importantly involved in the E2/E3 ligase mediated protein degradation.43,44 In order to elucidate the mechanism of 6c-induced ERRα degradation, we further investigated the contribution of proteasome in ERRα degrading process. MDA-MB-231 cells were preincubated with 1.0 M proteasome inhibitor Epoxomicin,45 then treated with 6c at 30 nM for 4.0 h. Western blot analysis suggested that pretreatment of proteasome inhibitor successfully rescued the ERRα degradation induced by 6c, validating the crucial role of proteasome system in VHL-mediated ERRα degradation (Figure 4A). However, no protein degradation was observed for direct treatments of ERRα ligand 4f and VHL amino building block 13, either in a single agent or in a combination (Figure 4A), whereas pretreatment with ERRα ligand 4f or VHL ligand 5 successfully abrogated the ERRα degradation mediated by 6c (Figure 4B). These data collectively supported the necessity of ternary complex formation for ERRα degradation.
In summary, a series of (E)-3-(4-((2,4-bis(trifluoromethyl)benzyl)oxy)-3-methoxyphenyl)-2-cyanoacrylamide derivatives were designed and synthesized as new ERRα degraders based on a proteolysis targeting chimera concept. Compound 6c is capable of specifically degrading ERRα protein by >80% at a relatively low concentration of 30 nM, representing one of the most potent and selective ERRα degraders to date. Mechanism investigation further validated that 6c-mediated ERRα degradation requires the ternary complex formation and ubiquitin proteasome participation. PROTAC 6c could be utilized as a new powerful chemical tool for further investigating functions of ERRα in physiological and pathological states.
Acknowledgments
The authors appreciate the financial support from National Natural Science Foundation of China (81502907, 21572230, 81673285, and 81425021), Guangdong Province (2014TQ01R341, 2015A030306042, 2015A030312014, 2016A050502041, and Nanyue-Baijie Award), China Postdoctoral Science Foundation (2017M622660), and Jinan University. The authors also thank Prof. Moshe Levi and Dr. Andrew Libby from Georgetown University (USA) for the proofreading of this manuscript.
Glossary
ABBREVIATIONS
- ERR
estrogen-related receptor
- ER
estrogen receptor
- OXPHOS
oxidative phosphorylation
- PROTAC
proteolysis targeting chimera
- TR-FRET
time-resolved fluorescence resonance energy transfer
- VHL
von-Hippel-Lindau
- MDM2
Mouse Double Minute 2
- cIAP1
cellular inhibitor of apoptosis protein 1
- CRBN
cereblon
- DIPEA
N,N-diisopropylethylamine
- HATU
1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]-pyridinium 3-oxide hexafluorophosphate
- TFA
trifluoroacetic acid
- DMF
N,N-dimethylformamide
- DCM
dichloromethane
- rt
room temperature
- IC50
half maximal (50%) inhibitory concentration of a substance
- PEG
polyethylene glycol
- MCAD
medium-chain acyl CoA dehydrogenase
- PDK4
pyruvate dehydrogenase kinase 4.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.9b00025.
Author Contributions
# These authors contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Giguère V. Orphan Nuclear Receptors: From Gene to Function. Endocr. Rev. 1999, 20 (5), 689–725. 10.1210/er.20.5.689. [DOI] [PubMed] [Google Scholar]
- Giguère V.; Yang N.; Segui P.; Evans R. M. Identification of a New Class of Steroid Hormone Receptors. Nature 1988, 331 (6151), 91–94. 10.1038/331091a0. [DOI] [PubMed] [Google Scholar]
- Eudy J. D.; Yao S.; Weston M. D.; Ma-Edmonds M.; Talmadge C. B.; Cheng J. J.; Kimberling W. J.; Sumegi J. Isolation of a Gene Encoding a Novel Member of the Nuclear Receptor Superfamily from the Critical Region of Usher Syndrome Type IIa at 1q41. Genomics 1998, 50 (3), 382–384. 10.1006/geno.1998.5345. [DOI] [PubMed] [Google Scholar]
- Luo J.; Sladek R.; Bader J.-A.; Matthyssen A.; Rossant J.; Giguère V. Placental abnormalities in mouse embryos lacking the orphannuclear receptor ERR-beta. Nature 1997, 388, 778–782. 10.1038/42022. [DOI] [PubMed] [Google Scholar]
- Audet-Walsh E.; Giguère V. The multiple universes of estrogen-related receptor α and γ in metabolic control and related diseases. Acta Pharmacol. Sin. 2015, 36 (1), 51–61. 10.1038/aps.2014.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handschin C.; Mootha V. K. Estrogen-related receptor α (ERRα): A novel target in type 2 diabetes. Drug Discovery Today: Ther. Strategies 2005, 2 (2), 151–156. 10.1016/j.ddstr.2005.05.001. [DOI] [Google Scholar]
- Giguère V. To ERR in the estrogen pathway. Trends Endocrinol. Metab. 2002, 13 (5), 220–225. 10.1016/S1043-2760(02)00592-1. [DOI] [PubMed] [Google Scholar]
- Giguère V. Transcriptional control of energy homeostasis by the estrogen-related receptors. Endocr. Rev. 2008, 29, 677–696. 10.1210/er.2008-0017. [DOI] [PubMed] [Google Scholar]
- Puigserver P.; Spiegelman B. M. Peroxisome proliferator-activated receptor-γ coactivator 1α (PGC-1α): transcriptional coactivator and metabolic regulator. Endocr. Rev. 2003, 24, 78–90. 10.1210/er.2002-0012. [DOI] [PubMed] [Google Scholar]
- Christian M.; White R.; Parker M. G. Metabolic regulation by the nuclear receptor corepressor RIP140. Trends Endocrinol. Metab. 2006, 17, 243–250. 10.1016/j.tem.2006.06.008. [DOI] [PubMed] [Google Scholar]
- Luo J.; Sladek R.; Carrier J.; Bader J.; Richard D.; Giguère V. Reduced fat mass in mice lacking orphan nuclear receptor estrogen-related receptor γ. Mol. Cell. Biol. 2003, 23, 7947–7956. 10.1128/MCB.23.22.7947-7956.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patch R. J.; Searle L. L.; Kim A. J.; De D.; Zhu X.; Askari H. B.; O’Neill J. C.; Abad M. C.; Rentzeperis D.; Liu J.; Kemmerer M.; Lin L.; Kasturi J.; Geisler J. G.; Lenhard J. M.; Player M. R.; Gaul M. D. Identification of Diaryl Ether-Based Ligands for Estrogen-Related Receptor α as Potential Antidiabetic Agents. J. Med. Chem. 2011, 54, 788–808. 10.1021/jm101063h. [DOI] [PubMed] [Google Scholar]
- DeBerardinis R. J.; Lum J. J.; Hatzivassiliou G.; Thompson C. B. The Biology of Cancer: Metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008, 7, 11–20. 10.1016/j.cmet.2007.10.002. [DOI] [PubMed] [Google Scholar]
- Tam I. S.; Giguère V. There and back again: The journey of the estrogen-related receptors in the cancer realm. J. Steroid Biochem. Mol. Biol. 2016, 157, 13–19. 10.1016/j.jsbmb.2015.06.009. [DOI] [PubMed] [Google Scholar]
- Chang Ch.-Y.; McDonnell D. P. Molecular Pathways: The metabolic regulator estrogen-related receptor as a therapeutic target in cancer. Clin. Cancer Res. 2012, 18 (22), 6089–6095. 10.1158/1078-0432.CCR-11-3221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianco S.; Lanvin O.; Tribollet V.; Macari C.; North S.; Vanacker J. M. Modulating estrogen receptor-related receptor-α activity inhibits cell proliferation. J. Biol. Chem. 2009, 284, 23286–23292. 10.1074/jbc.M109.028191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S.; Chang Ch.-Y.; Safi R.; Liu X.; Baldi R.; Jasper J. S.; Anderson G. R.; Liu T.; Rathmell J. C.; Dewhirst M. W.; Wood K. C.; Locasale J. W.; McDonnell D. P. ERRα-regulated lactate metabolism contributes to resistance to targeted therapies in breast cancer. Cell Rep. 2016, 15, 323–335. 10.1016/j.celrep.2016.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deblois G.; Smith H. W.; Tam I. S.; Gravel S. P.; Caron M.; Savage P.; Labbé D. P.; Bégin L. R.; Tremblay M. L.; Park M.; Bourque G.; St-Pierre J.; Muller W. J.; Giguère V. ERRα mediates metabolic adaptations driving lapatinib resistance in breast cancer. Nat. Commun. 2016, 7, 12156. 10.1038/ncomms12156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T.; Miki Y.; Moriya T.; Shimada N.; Ishida T.; Hirakawa H.; Ohuchi N.; Sasano H. Estrogen-Related Receptor α in Human Breast Carcinoma as a Potent Prognostic Factor. Cancer Res. 2004, 64 (13), 4670–4676. 10.1158/0008-5472.CAN-04-0250. [DOI] [PubMed] [Google Scholar]
- Stein R. A.; Chang C. Y.; Kazmin D. A.; Way J.; Schroeder T.; Wergin M.; Dewhirst M. W.; McDonnell D. P. Estrogen-related receptor α is critical for the growth of estrogen receptor-negative breast cancer. Cancer Res. 2008, 68, 8805–8812. 10.1158/0008-5472.CAN-08-1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwyer M. A.; Joseph J. D.; Wade H. E.; Eaton M. L.; Kunder R. S.; Kazmin D.; Chang C. Y.; McDonnell D. P. WNT11 expression is induced by estrogen-related receptor α and β-catenin and acts in an autocrine manner to increase cancer cell migration. Cancer Res. 2010, 70, 9298–9308. 10.1158/0008-5472.CAN-10-0226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deuschle U.; Heck S.; Kober l.; Bauer U.; Balogh I.. NR3B1 nuclear receptor binding 3-substituted pyrazole derivatives. EP Patent 1398029, 2004.
- Busch B. B.; Stevens W. C. Jr.; Martin R.; Ordentlich P.; Zhou S.; Sapp D. W.; Horlick R. A.; Mohan R. Identification of a Selective Inverse Agonist for the Orphan Nuclear Receptor Estrogen-Related Receptor α. J. Med. Chem. 2004, 47, 5593–5596. 10.1021/jm049334f. [DOI] [PubMed] [Google Scholar]
- Kallen J.; Lattmann R.; Beerli R.; Blechschmidt A.; Blommers M. J. J.; Geiser M.; Ottl J.; Schlaeppi J.-M.; Strauss A.; Fournier B. Crystal Structure of Human Estrogen-related Receptor α in Complex with a Synthetic Inverse Agonist Reveals Its Novel Molecular Mechanism. J. Biol. Chem. 2007, 282, 23231–23239. 10.1074/jbc.M703337200. [DOI] [PubMed] [Google Scholar]
- Chisamore M. J.; Mosley R. T.; Cai S.-J.; Birzin E. T.; O’Donnell G.; Zuck P.; Flores O.; Schaeffer J.; Rohrer S. P.; Don Chen J.; Wilkinson H. A. Identification of Small Molecule Estrogen-Related Receptor α-Specific Antagonists and Homology Modeling To Predict the Molecular Determinants as the Basis for Selectivity over ERRβ and ERRγ. Drug Dev. Res. 2008, 69 (4), 203–218. 10.1002/ddr.20246. [DOI] [Google Scholar]
- Wang J.; Fang F.; Huang Z.; Wang Y.; Wong C. Kaempferol Is an Estrogen-Related Receptor α and γ Inverse Agonist. FEBS Lett. 2009, 583 (4), 643–647. 10.1016/j.febslet.2009.01.030. [DOI] [PubMed] [Google Scholar]
- Xu S.; Zhuang X.; Pan X.; Zhang Z.; Duan L.; Liu Y.; Zhang L.; Ren X.; Ding K. 1-Phenyl-4-benzoyl-1H-1,2,3-triazoles as orally bioavailable transcriptional function suppressors of estrogen-related receptor α. J. Med. Chem. 2013, 56 (11), 4631–4640. 10.1021/jm4003928. [DOI] [PubMed] [Google Scholar]
- Patch R. J.; Huang H.; Patel S.; Cheung W.; Xu G.; Zhao B. P.; Beauchamp D. A.; Rentzeperis D.; Geisler J. G.; Askari H. B.; Liu J.; Kasturi J.; Towers M.; Gaul M. D.; Player M. R. Indazole-based ligands for estrogen-related receptor α as potential anti-diabetic agents. Eur. J. Med. Chem. 2017, 138, 830–853. 10.1016/j.ejmech.2017.07.015. [DOI] [PubMed] [Google Scholar]
- Zhang L.; Liu P.; Chen H.; Li Q.; Chen L.; Qi H.; Shi X.; Du Y. Characterization of a selective inverse agonist for estrogen related receptor α as a potential agent for breast cancer. Eur. J. Pharmacol. 2016, 15 (789), 439–448. 10.1016/j.ejphar.2016.08.008. [DOI] [PubMed] [Google Scholar]
- Du Y.; Song L.; Zhang L.; Ling H.; Zhang Y.; Chen H.; Qi H.; Shi X.; Li Q. The discovery of novel, potent ERR-alpha inverse agonists for the treatment of triple negative breast cancer. Eur. J. Med. Chem. 2017, 136, 457–467. 10.1016/j.ejmech.2017.04.050. [DOI] [PubMed] [Google Scholar]
- Peng L.; Gao X.; Duan L.; Ren X.; Wu D.; Ding K. Identification of Pyrido[1,2-α]pyrimidine-4-ones as New Molecules Improving the Transcriptional Functions of Estrogen-Related Receptor α. J. Med. Chem. 2011, 54 (21), 7729–7733. 10.1021/jm200976s. [DOI] [PubMed] [Google Scholar]
- Wei W.; Schwaid A. G.; Wang X.; Wang X.; Chen S.; Chu Q.; Saghatelian A.; Wan Y. Ligand Activation of ERRα by Cholesterol Mediates Statin and Bisphosphonate Effects. Cell Metab. 2016, 23, 479–491. 10.1016/j.cmet.2015.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondeson D. P.; Mares A.; Smith I. E. D.; Ko E.; Campos S.; Miah A. H.; Mulholland K. E.; Routly N.; Buckley D. L.; Gustafson J. L.; Zinn N.; Grandi P.; Shimamura S.; Bergamini G.; Faelth-Savitski M.; Bantscheff M.; Cox C.; Gordon D. A.; Willard R. R.; Flanagan J. J.; Casillas L. N.; Votta B. J.; den Besten W.; Famm K.; Kruidenier L.; Carter P. S.; Harling J. D.; Churcher I.; Crews C. M. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem. Biol. 2015, 11 (8), 611–617. 10.1038/nchembio.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neklesa T. K.; Winkler J. D.; Crews C. M. Targeted protein degradation by PROTACs. Pharmacol. Ther. 2017, 174, 138–144. 10.1016/j.pharmthera.2017.02.027. [DOI] [PubMed] [Google Scholar]
- Adjei A. A. What is the right dose? The elusive optimal biologic dose in phase I clinical trials. J. Clin. Oncol. 2006, 24, 4054–4055. 10.1200/JCO.2006.07.4658. [DOI] [PubMed] [Google Scholar]
- Lazo J. S.; Sharlow E. R. Drugging undruggable molecular cancer targets. Annu. Rev. Pharmacol. Toxicol. 2016, 56, 23–40. 10.1146/annurev-pharmtox-010715-103440. [DOI] [PubMed] [Google Scholar]
- Tinworth C. P.; Lithgow H.; Churcher I. Small moleculemediated protein knockdown as a new approach to drug discovery. MedChemComm 2016, 7, 2206–2216. 10.1039/C6MD00347H. [DOI] [Google Scholar]
- Lai A. C.; Crews C. M. Induced protein degradation: an emerging drug discovery paradigm. Nat. Rev. Drug Discovery 2017, 16, 101–114. 10.1038/nrd.2016.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulatov E.; Ciulli A. Targeting Cullin–RING E3 ubiquitin ligases for drug discovery: structure, assembly and small-molecule modulation. Biochem. J. 2015, 467 (3), 365–386. 10.1042/BJ20141450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai A. C.; Toure M.; Hellerschmied D.; Salami J.; Jaime-Figueroa S.; Ko E.; Hines J.; Crews C. M. Modular PROTAC Design for the Degradation of Oncogenic BCR-ABL. Angew. Chem., Int. Ed. 2016, 55, 807–810. 10.1002/anie.201507634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galdeano C.; Gadd M. S.; Soares P.; Scaffidi S.; Van Molle I.; Birced I.; Hewitt S.; Dias D. M.; Ciulli A. Structure-guided design and optimization of small molecules targeting the protein-protein interaction between the von Hippel-Lindau (VHL) E3 ubiquitin ligase and the hypoxia inducible factor (HIF) alpha subunit with in vitro nanomolar affinities. J. Med. Chem. 2014, 57 (20), 8657–8663. 10.1021/jm5011258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak R. P.; DeAngelo S. L.; Buckley D.; He Z.; Donovan K. A.; An J.; Safaee N.; Jedrychowski M. P.; Ponthier C. M.; Ishoey M.; Zhang T.; Mancias J. D.; Gray N. S.; Bradner J. E.; Fischer E. S. Plasticity in binding confers selectivity in ligand induced protein degradation. Nat. Chem. Biol. 2018, 14 (7), 706–714. 10.1038/s41589-018-0055-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciechanover A. Proteolysis: from the lysosome to ubiquitin and the proteasome. Nat. Rev. Mol. Cell Biol. 2005, 6, 79–87. 10.1038/nrm1552. [DOI] [PubMed] [Google Scholar]
- Kleiger G.; Mayor T. Perilous journey: a tour of the ubiquitin proteasome system. Trends Cell Biol. 2014, 24, 352–359. 10.1016/j.tcb.2013.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng L.; Mohan R.; Kwok B.; Elofsson M.; Sin N.; Crews C. M. Epoxomicin, a potent and selective proteasome inhibitor, exhibits in vivo antiinflammatory activity. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 10403–10408. 10.1073/pnas.96.18.10403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider G. A.; Rasband W. S.; Eliceiri K. W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9 (7), 671–675. 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.