

CONSPECTUS:
Since the pioneering work of Kochi in the 1970s, iron has attracted great interest for cross-coupling catalysis due to its low cost and toxicity as well as its potential for novel reactivity compared to analogous reactions with precious metals like palladium. Today there are numerous iron-based cross-coupling methodologies available, including challenging alkyl–alkyl and enantioselective methods. Furthermore, cross-couplings with simple ferric salts and additives like NMP and TMEDA (N-methylpyrrolidone and tetramethylethylenediamine) continue to attract interest in pharmaceutical applications. Despite the tremendous advances in iron cross-coupling methodologies, in situ formed and reactive iron species and the underlying mechanisms of catalysis remain poorly understood in many cases, inhibiting mechanism-driven methodology development in this field.
This lack of mechanism-driven development has been due, in part, to the challenges of applying traditional characterization methods such as nuclear magnetic resonance (NMR) spectroscopy to iron chemistry due to the multitude of paramagnetic species that can form in situ. The application of a broad array of inorganic spectroscopic methods (e.g., electron paramagnetic resonance, 57Fe Mössbauer, and magnetic circular dichroism) removes this barrier and has revolutionized our ability to evaluate iron speciation. In conjunction with inorganic syntheses of unstable organoiron intermediates and combined inorganic spectroscopy/gas chromatography studies to evaluate in situ iron reactivity, this approach has dramatically evolved our understanding of in situ iron speciation, reactivity, and mechanisms in iron-catalyzed cross-coupling over the past 5 years. This Account focuses on the key advances made in obtaining mechanistic insight in iron-catalyzed carbon–carbon cross-couplings using simple ferric salts, iron-bisphosphines, and iron-N-heterocyclic carbenes (NHCs). Our studies of ferric salt catalysis have resulted in the isolation of an unprecedented iron-methyl cluster, allowing us to identify a novel reaction pathway and solve a decades-old mystery in iron chemistry. NMP has also been identified as a key to accessing more stable intermediates in reactions containing nucleophiles with and without β-hydrogens. In iron-bisphosphine chemistry, we have identified several series of transmetalated iron(II)-bisphosphine complexes containing mesityl, phenyl, and alkynyl nucleophile-derived ligands, where mesityl systems were found to be unreliable analogues to phenyls. Finally, in iron-NHC cross-coupling, unique chelation effects were observed in cases where nucleophile-derived ligands contained coordinating functional groups. As with the bisphosphine case, high-spin iron(II) complexes were shown to be reactive and selective in cross-coupling. Overall, these studies have demonstrated key aspects of iron cross-coupling and the utility of detailed speciation and mechanistic studies for the rational improvement and development of iron cross-coupling methods.
Graphical Abstract
INTRODUCTION
In the 1970s, Kochi demonstrated that simple iron salts can catalyze the stereoselective generation of C–C bonds (Scheme 1).1,2 Subsequently, iron-catalyzed cross-coupling reactions have attracted significant interest as versatile and cost-effective alternatives to those using precious metal catalysts. In particular, work by Cahiez in the 1990s showed that the addition of N-methylpyrrolidone (NMP) in cross-couplings with iron salts resulted in a wider substrate scope than that achievable in Kochi’s original protocol (Scheme 1).3 Following Cahiez’s work, a multitude of reaction methods have been developed which expand on the use of NMP in iron chemistry4 and utilize a variety of ligands such as bisamines,5–7 N-heterocyclic carbenes (NHCs),8–11 and bisphosphines,12–16 enabling broadened substrate scopes and improved selectivity (Scheme 1).
Scheme 1.

Select Iron-Catalyzed Cross-Coupling Reactions
Despite the broad success and application of iron-catalyzed cross-coupling for organic synthesis,17 mechanistic insight into these reactions is substantially under-developed compared to the understanding of palladium cross-coupling. A significant contribution to this lack of insight arises from the fundamental challenges in studying iron-based cross-coupling systems, including the air and thermal instability of organoiron intermediates that can form in situ as well as iron’s rich redox chemistry. For example, iron can undergo both one- and two-electron reaction pathways, accessing a multitude of oxidation and spin states, many of which are paramagnetic and complicate characterization by traditional methods, including nuclear magnetic resonance (NMR) spectroscopy.17 Furthermore, multiple iron species can form in situ, speciation can evolve with time, and multiple iron complexes can be reactive toward electrophiles yet exhibit different reaction rates or selectivities. Early studies tackled these challenges by utilizing physical organic methods, single in situ spectroscopies, and theoretical studies in the absence of supporting experiments that directly probe the nature of iron intermediates. Consequently, iron(–II)/iron(0),18 iron(0)/iron- (II),5 iron(I)/iron(III),19 and iron(II)/iron(III)20 redox couples have all been proposed in the literature for iron cross-coupling mechanisms.
The recent utilization of methods that directly probe iron speciation (inorganic spectroscopies and single-crystal X-ray diffraction, XRD) with concurrent gas chromatography (GC) and kinetics studies has revolutionized our understanding of iron cross-coupling systems, providing detailed insight into iron speciation, reactivity, and mechanism. Our group has pioneered this experimental approach in cross-coupling, utilizing frozen solution 57Fe Mössbauer spectroscopy (simply called Mössbauer spectroscopy herein) to directly examine iron speciation in situ, complemented by additional inorganic spectroscopic methods and low-temperature inorganic synthesis of unstable organoiron species.21 Computational studies of electronic structure and bonding in isolated complexes have also provided insight into the nature of iron–ligand bonding and potential reactivity.22–24 While these spectroscopic and structural studies are essential to determining iron speciation, gaining insight into the mechanism ultimately requires the ability to evaluate reactivity, product distributions, and catalytic relevance of such species. Our ability to directly correlate organic product formation via GC analysis to in situ formed iron speciation with Mössbauer spectroscopy has enabled us to unambiguously define reactive iron species in this central class of reactions. Specifically, we use a combination of solid and frozen solution Mössbauer spectroscopic studies of isolated, X-ray crystallographically defined organoiron complexes to identify their presence on cycle during separate in situ reaction studies using reported or modified catalytic reaction protocols. This Account focuses on the development and evolution of mechanistic insight into iron-catalyzed cross-coupling in simple iron salt and iron–ligand systems that have resulted from the rigorous application of our multi-faceted research approach over the past 5 years.
FERRIC SALT-CATALYZED CROSS-COUPLING WITH ALKYL GRIGNARD REAGENTS
Iron-catalyzed cross-couplings utilizing simple ferric salts have attracted significant interest due to the practical utility of such facile cross-coupling methods and the fascinating organo-metallic chemistry that must underlie catalysis in the absence of well-defined supporting ligands. In situ iron speciation and mechanism of this chemistry were first explored by Kochi in the 1970s, where electron paramagnetic resonance (EPR) studies provided evidence for the reduction of iron(III) salts (i.e., FeCl3, Fe(acac)3, and Fe(dbm)3) upon treatment with alkyl Grignard reagents to generate a S = 1/2 iron species in situ, leading to the proposal of an iron(I) active species.19 In contrast, Fürstner proposed the possibility of at least two different reaction manifolds, depending upon the presence or absence of β-hydrogens in the nucleophile (Scheme 2). In support of the organoferrate reaction manifold, Fürstner and co-workers reported elegant synthetic studies which led to the isolation of the homoleptic iron(II) organoferrate complex [(FeMe4)(MeLi)][Li(Et2O)]2 using MeLi in Et2O.25 This complex exhibited a color change from red to yellow and reactivity toward activated electrophiles when dissolved in tetrahydrofuran (THF).26 It was also found to be active in both stoichiometric and catalytic ring-opening/cross-coupling reactions with MeMgBr.27 However, the color change upon dissolution in THF was highly suggestive of a structural change in this solvent; furthermore, an iron(II) center is not capable of generating the S = 1/2 EPR signal observed by Kochi, except in cases where it is part of a mixed-valence multi-nuclear system.
Scheme 2.

Mechanistic Cycles Proposed by Kochi19 (Left) and Fürstner26 (Right) in Simple Ferric Salt Cross-Coupling
Over the past 5 years, our group has addressed these issues through reactivity studies of ferric salts and MeMgBr under catalytically relevant conditions, revolutionizing the understanding of these ferric salt systems. Addition of 4 equiv of MeMgBr to FeCl3 in THF at low temperature resulted in the isolation of the homoleptic iron(III) ferrate, [MgCl(THF)5]-[FeMe4] (1) (Scheme 3). Contrary to Fürstner’s iron(II) complex, 1 displayed an EPR signal indicative of S = 3/2.28 Freeze-trapped solution EPR determined that, upon warming of 1, a broad S = 1/2 EPR signal was generated, along with formation of ethane. This was suggestive that 1 was a highly reactive intermediate organoiron species along the reduction pathway to form the S = 1/2 iron species originally observed by Kochi. These studies established the important effect of solvent and nucleophile (MeMgBr vs MeLi) on the nature of iron-methyl species formed in situ.
Scheme 3.

Iron Species Formed in Reactions of FeCl3 with MeMgBr in THF
Our next task was to identify the thermally unstable S = 1/2 species formed in situ during Kochi’s original studies—a key unknown in this reaction class for more than 40 years. Through meticulous crystallographic studies combined with EPR and magnetic circular dichroism (MCD) spectroscopic analysis of isolated and in situ iron speciation, we identified the S = 1/2 species as an unprecedented multi-nuclear iron cluster, [MgCl(THF)5][Fe8Me12] (2) (Scheme 3).29 Reaction with β-bromostyrene led to consumption of 2, determined by EPR, but with no concurrent formation of cross-coupled product. However, when additional MeMgBr was added following the initial reaction of 2 with an electrophile, rapid and selective formation of the cross-coupled product was observed. This isolation of a reactive, multi-metallic organoiron species for cross-coupled product formation defined an entirely new paradigm for reactive species in iron cross-coupling. In the future, identification of the pseudo-stable intermediate formed upon reaction of 2 with an electrophile will shed more light on the one- and two-electron processes in the reaction pathway to form cross-coupled product.
In addition to the THF/Et2O solvent effects we observed, the changes in catalytic performance made possible through the addition of NMP in iron-catalyzed cross-coupling were highlighted by Cahiez and co-workers.3 At the time, NMP was named as a co-solvent, increasing catalytic performance through the common assumption that it could stabilize organoiron intermediates. Though pre-mixing experiments were assumed to form FeXn (NMP)m-type species, the molecular-level nature of this stabilization was never defined explicitly. Recently, we have identified that the inclusion of NMP in the reaction mixture of FeCl3 and MeMgBr in THF disfavors formation of 2 and leads to in situ formation of a homoleptic trimethyliron(II) ferrate, [Mg(NMP)6][FeMe3]2 (3) (Scheme 4), which incorporates six NMP molecules coordinated to a charge-balancing magnesium dication.30 Thus, NMP stabilizes the formation of this iron species via modification of the cation, not iron coordination. Reactivity studies of in situ generated 3 with β-bromostyrene combined with freeze-trapped Mössbauer analysis demonstrated rapid and selective formation of a cross-coupled product and consumption of 3. The direct reactivity of 3 with an electrophile to form a cross-coupled product is distinct from that of 2, which required an additional equivalent of MeMgBr following reaction with the electrophile for product formation, highlighting the diverse reaction pathways accessible in iron cross-coupling.
Scheme 4.
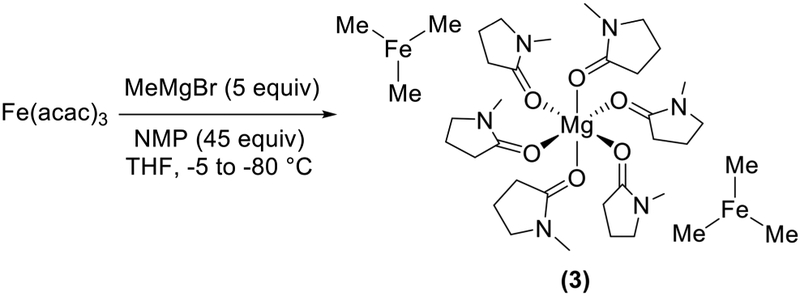
NMP’s Effect on Iron Speciation in Reactions of Fe(acac)3 and MeMgBr in THF
FERRIC SALT-CATALYZED CROSS-COUPLING WITH ARYL GRIGNARD REAGENTS
Beyond organoiron species involved in simple salt cross-coupling with MeMgBr, the reactive iron species formed with aryl Grignard reagents have also been the focus of intense scrutiny. Fürstner hypothesized that aryl nucleophiles would behave similarly to methyl reagents (organoferrate manifold, Scheme 2) which also lack β-hydrogens.26 In support of this, [Ph4Fe][Li(OEt2)]4 and [Ph4Fe][Li(Et2O)2][Li(1,4-diox-ane)] were isolated from reactions of FeCl3 or FeCl2 in Et2O at low temperature.25 While the isolation of these complexes was a useful proof of concept, their formation from lithium reagents in Et2O yields complications similar to those previously discussed for reactions with methyl nucleophiles. To circumvent these issues, in situ spectroscopic studies with catalytically relevant reagents and solvents using X-ray absorption spectroscopy (XAS) suggested the formation of a dinuclear iron species with a direct Fe-Fe contact.31 By contrast, extensive in situ NMR and EPR studies led to the proposal that a reactive iron(I) species is formed in situ in reactions of phenyl Grignard reagents and iron salts in THF.32–37 Additional studies using density functional theory (DFT) and cyclic voltammetry (CV) have further supported the possible formation of an iron(I) active species. However, none of these experimental studies have provided direct structural evidence for a reactive iron(I) complex.
More recently, electrospray ionization mass spectrometry (ESI-MS) studies suggested the formation of a multi-nuclear iron species, [Ph7Fe4]−, from reactions between Fe(acac)3 and PhMgCl in THF.38 Unfortunately, the specific ESI-MS method employed was only able to observe charged species, meaning any neutral complexes existing in solution would not be observed—a critical limitation shown by subsequent structural studies (vide infra). Despite these extensive characterization efforts, direct structural evidence and the identification of an iron species capable of selectively reacting with electrophiles were significant limitations.
Since studies that combine structural characterization, in situ spectroscopy, and organic product analysis provide such a complete set of mechanistic information, the work of Bedford and co-workers on the more sterically encumbered mesityl-magnesium bromide (MesMgBr) system with iron has been particularly insightful.32 Both [Fe(Mes)3]− and Fe2Mes4 were isolated and observed to react with bromooctane to generate cross-coupled products with 50% and 24% yields, respectively. When p-tolyl Grignard reagents were used, EPR and 1H NMR also suggested the formation of [Fe(p-tolyl)3]-; however, the S = 1/2 EPR signal observed was not quantified, obscuring the relevance of these molecules to catalysis. Despite these impressive insights into speciation and reactivity with MesMgBr, this nucleophile is not very effective or selective in iron-catalyzed cross-coupling; hence, the applicability of these structural and reactivity data to less sterically encumbered and more catalytically active aryl nucleophiles is unclear.
Recently, our group has expanded the knowledge about iron-phenyl species and their reactivity using a combination of air-and temperature-sensitive XRD, inorganic spectroscopy, and reaction studies in catalytically relevant solvents. In one example, [Mg(acac)(THF)4]2[FePh2(μ-Ph)]2.4THF (4) was isolated and characterized from the reaction of Fe(acac)3 and PhMgBr in THF at −80 °C (Figure 1A).39 Solid Mössbauer spectroscopy of 4 was consistent with a high-spin iron(II) center containing highly covalent Fe-C bonds.21,40 4 proved to be extremely unstable, but with the addition of NMP, [Mg(NMP)6][FePh2(μ-Ph)]2.3.5THF (4-NMP) was accessible at slightly warmer temperatures and used for characterization. Unfortunately, this dinuclear species was shown to be unreactive toward electrophiles. This was in stark contrast to the Fe2Mes4 dimer, which produced 24% cross-coupled product at room temperature upon reaction with an electrophile, further highlighting the challenge in comparing model mesityl systems with iron cross-couplings involving slimmer aryl Grignard reagents.
Figure 1.

Multi-nuclear iron species formed in reactions of PhMgBr and Fe(acac)3 in THF. (A) [Mg(acac)(THF)4]2[Fe(Ph)2(μ-Ph)]2. 4THF (4) with counterions and THF molecules omitted for clarity. (B) Fe4(μ-Ph)6(THF)4 (5) and its derivatives, Fe4(μ-p-tolyl)6-(THF)3, Fe4(μ-p-tolyl)6(THF)3, and Fe4(μ4-F-Ph)6(THF)4. Note that THF* indicates a labile THF determined crystallographically for the p-tolyl cluster.
The instability of 4 and 4-NMP led to the hypothesis that a more thermally stable iron-phenyl species must exist in the absence of NMP. Consequently, Fe4(μ-Ph)6(THF)4.2THF (5) and several other aryl derivatives (Figure 1B) were isolated at −30 °C and characterized by XRD.39 These tetranuclear complexes were more reduced than the dinuclear species, formally containing two iron(I) and two iron(II) centers. As expected, 5 was found to be thermally stable at room temperature for up to 5 min in THF, enabling stoichiometric reactions to be performed with electrophiles under catalytically relevant conditions. Reactions of 5 with bromocyclohexane resulted in the rapid and selective formation of phenylcyclo-hexane with kobs ≈ 12 min−1. This was in contrast to iron cross-coupling systems previously reported by Nakamura and coworkers which performed poorly in the absence of another popular additive, tetramethylethylenediamine (TMEDA).7 In fact, a modified catalytic protocol was generated to target in situ formation of 5, enabling formation of >95% cross-coupled product in the absence of any additives. This is a key demonstration of how insight into in situ iron speciation can be utilized for the development of effective reaction protocols.
This work showed the first direct syntheses, structural characterizations, and reactivity studies of iron-phenyl species formed upon the reaction of Fe(acac)3 and PhMgBr in THF. By varying the temperature, both an unreactive dimer and a more reduced, reactive iron-phenyl cluster could be isolated for further study. It was also possible to vary substitution of the phenyl rings using different Grignard reagents to aid in the identification of the pathway to iron cluster formation from iron salts by stepwise addition experiments and consequent crystallographic identification. This formation pathway and the confirmed reactivity of the iron cluster will aid in the future development of cross-coupling with iron salts.
Despite the success of these initial iron-phenyl studies, there are limitations to fully understanding the mechanism of iron salt-catalyzed cross-coupling reactions involving aryl Grignard reagents. For example, additional unstable iron species could also be accessible in situ, such as the mononuclear iron(I) that has been suggested by in situ EPR or a [Fe(p-tolyl)3] − previously hypothesized by Bedford and co-workers,32 depending on the reaction conditions and protocols employed. Additionally, other additives to iron salt catalysis, like TMEDA, remain essential for promoting certain cross-coupling reactions, but little direct evidence exists apart from our work as to their potential interaction with iron centers and/or counterions, despite recent efforts such as those of Cahiez and Bedford.32,41 Future studies should be directed toward increasing the library of known iron-aryl species formed in situ during catalysis in addition to studying the broader effects of additives on iron cross-coupling reactions.
IRON BISPHOSPHINE CATALYSIS
Beyond simple ferric salt catalysis, some of the most significant methodological advances in iron-catalyzed cross-coupling have been in iron-bisphosphine systems. For example, Bedford and co-workers investigated the use of mono- and bidentate phosphines as additives to FeCl3 to catalyze the coupling of alkyl halides bearing β-hydrogens with aryl Grignard reagents (Scheme 5a).8 Subsequent studies have employed ligands such as dpbz, dppe, SciOPP, and Xantphos to extend the substrate scope to include organozinc and organoboron nucleophiles. More challenging sp3 substrates12–16,42 have also been successfully used in reactions including Kumada, Suzuki– Miyaura, and Negishi reactions as well as the first enantio-selective cross-coupling with iron (Scheme 5f).43
Scheme 5.

Select Iron-Bisphosphine Cross-Couplings
Mechanistic investigations of these cross-coupling systems have primarily focused on well-defined iron-bisphosphine complexes as pre-catalysts. For example, using FeCl2(dpbz)2 as a pre-catalyst, Bedford and co-workers used inorganic synthetic and spectroscopic techniques to suggest an iron(I) reaction pathway in the Negishi coupling of benzyl halides and phosphates with organozinc reagents (Scheme 5e).16 Stable five-coordinate iron(I) complexes, FeX(dpbz)2 and FeX-(dppe)2 (X = halide), were identified as easily accessible in situ formed iron species by EPR, but without spin quantification or direct reactivity studies, their role on- or off-cycle from catalysis remained ambiguous.
Contrary to this iron(I) pathway, Nakamura and co-workers proposed an iron(II)/iron(III) catalytic cycle for the iron- SciOPP Suzuki–Miyaura coupling of primary and secondary alkyl halides with aryl boron compounds based on radical clock substrate experiments and the presence of homocoupled biaryls.12 Similar mechanisms were also proposed for Kumada-type reactions using aryl and alkynyl nucleo-philes.8,13,14,44 In the absence of direct structural evidence, in situ characterization studies by Nakamura and Koszinowski used solution-phase XAS and ESI-MS to show the significance of iron(II)-SciOPP complexes to catalysis.44,45 However, complicated reaction mixtures and the inability of ESI-MS to observe neutral species still challenged the unambiguous assignment of in situ formed and reactive iron species. Extensive DFT studies have also been performed on Nakamura’s enantioselective bisphosphine system, suggesting that enantioselectivity originates from radical addition to a mono-transmetalated iron(II) to form iron(III), which will subsequently reductively eliminate and form cross-coupled products.46,47
Due to the complexity of iron speciation in iron-bisphosphine cross-coupling systems, the use of multiple inorganic spectroscopic methods, XRD, and detailed reaction/ kinetic studies of all iron species formed in situ has been required to provide the most definitive mechanistic information. Initial studies by our group focused on iron-SciOPP-catalyzed Kumada cross-couplings with MesMgBr, demonstrating that reaction of FeCl2(SciOPP) (6) with MesMgBr can generate mono-, bis-, and tris-mesitylated iron(II) species in situ.21 XRD and solid and frozen solution Mössbauer parameters of isolated crystalline material combined with in situ solution Mössbauer spectroscopy showed that the addition of 1 and 2 equiv of MesMgBr to 6 generated FeBrMes-(SciOPP) (7) and FeMes2(SciOPP) (8), respectively. [MgX3-(THF)6][FeMes3] (X = halide) or FeMes3 − (9) was generated only in the presence of excess Grignard reagent, displacing the SciOPP ligand. Chemically quenched GC with concurrent freeze-trapped Mössbauer studies established that, while both 8 and 9 produced the desired heterocoupled product with 1-iododecane (Figure 2) along with 6, 9 was much less selective. No additional iron species were observed to form in solution after addition of alkyl iodide. This reaction also demonstrated that slow addition of Grignard reagent during catalysis was essential to avoid formation of 9 and favor formation of 8 as the active species during catalytic turnover.
Figure 2.

Reactivity of FeMes2(SciOPP) (8). Adapted with permission from ref 21. Copyright 2014 American Chemical Society.
Since mesityl and phenyl nucleophiles can result in significantly different iron speciation, as shown in the case of ferric salt reactions, subsequent studies were extended to the Kumada and Suzuki–Miyaura cross-couplings originally reported by Nakamura and co-workers using phenyl nucleophiles and secondary alkyl halides.12,13 Consistent with mesityl studies,21 mono- and bis-phenylated iron(II)-SciOPP centers were shown to be accessible in both the Kumada and Suzuki–Miyaura reactions (Scheme 6).40 FeBrPh(SciOPP) (10) was generated and studied using XRD from treatment of 6 with 1 equiv of phenylmagnesium bromide. Upon addition of another equivalent of PhMgBr, freeze-trapped solution MCD and Mössbauer spectroscopy identified the formation of the S = 2 iron(II) Fe(Ph)2(SciOPP) complex (11) with distorted tetrahedral geometry, which was only able to be generated in situ. Upon reaction of 6 with excess phenyl nucleophile, Fe(n6-biphenyl)(SciOPP) (12) was formed, retaining SciOPP coordination. This formal iron(0) complex was found to release biphenyl and generate cycloheptene upon reaction with excess bromocycloheptane, regenerating 6. These results indicated that it is an off-cycle iron species.
Scheme 6.

Reactivity of Iron(II)-SciOPP Species with PhMgBr or PhMgCl in THF (Chp = cycloheptyl)
GC reactivity studies of in situ generated mono- and bis-phenyl species with bromocycloheptane showed that both were reactive at catalytically relevant rates (kobs >12 min−1). As in the mesityl case, no additional iron species were observable by in situ Mössbauer spectroscopy after addition of an alkyl electrophile. However, Fe(Ph)X(SciOPP) was selective for exclusively cross-coupled product, suggesting that it was the dominant reactive species in catalysis (Scheme 6). In situ Mössbauer spectroscopy of the Kumada system during catalytic turnover revealed the presence of FeX2-(SciOPP) and Fe(n6-biphenyl)(SciOPP), and showed FeX2-(SciOPP) with FeXPh(SciOPP) in the Suzuki–Miyaura reaction (X = halide), highlighting the catalytic relevance of these complexes and the importance of slow nucleophile addition to disfavor formation of the less selective bis-phenyl species. Additionally, while a S = 1/2 species was also observed in this chemistry as a very minor species (<5% all iron), it was found to be unreactive or significantly less reactive than the mono- or bis-phenyl complexes.
The final iron-SciOPP system studied by our group was another Kumada coupling reaction, developed by Nakamura and co-workers, which coupled alkyl halides and (triisopropyl-silyl)ethynylmagenisum bromide (TIPS-CC-MgBr)—our first report on a system using sp-hybridized nucleophiles.14,48 Analogous to the results of the previously discussed iron-SciOPP systems, slow addition of Grignard reagent was important for productive catalysis, and transmetalated iron-SciOPP complexes, including a tris-alkynylated iron-SciOPP, were observed by XRD, frozen solution Mössbauer spectroscopy, and MCD.49 Additionally, these transmetalated iron complexes experienced rapid, complex ligand redistribution pathways in solution, generating a tris-alkynylated ferrate even in the absence of excess nucleophile (Figure 3).
Figure 3.

Reactivity of FeBr2(SciOPP) with alkynyl nucleophile. Adapted with permission from ref 49. Copyright 2017 American Chemical Society.
The reactivity of each of these complexes was assessed by addition of excess cycloheptyl bromide in toluene at 70 °C using in situ freeze-trapped Mössbauer spectroscopy and concurrent chemically quenched GC. While the tris-alkynylated iron(II)-SciOPP species was found to be catalytically inactive, the mono- and bis-iron(II)-SciOPP alkynes were kinetically competent for catalysis (kobs ≈ 0.1 min−1). Overall, this suggested an iron(II)/iron(III) mechanistic cycle similar to that present for iron-SciOPP phenyl reactions, where lower degrees of transmetalation resulted in more productive catalysis.
While the study of these iron-SciOPP systems by our group has provided detailed insight into the accessible, catalytically relevant iron species and oxidation state trends (i.e., iron(II) reactive species over iron(I)), the question remains as to the broader relevance of these findings across other bisphosphine ligands. For example, more detailed studies of systems like those developed by Bedford and Nakamura using dpbz and Xantphos ligands might show varied oxidation states or binding modes in reactive iron-bisphosphine complexes compared with the iron(II) complexes described here. The steric and electronic effects of ligands cannot be overlooked in iron catalysis, and small changes could result in diverse iron speciation and alternative reaction pathways.
IRON N-HETEROCYCLIC CARBENE CATALYSIS
NHCs have proven to be a popular and successful class of ligands for iron cross-couplings, enabling challenging alkyl–alkyl and aryl–aryl Kumada cross-coupling reactions as well as Suzuki–Miyaura cross-couplings (Scheme 7).8,9,11,50–52 Despite this broad success of iron-NHCs, these systems have seen fewer mechanistic studies performed relative to the studies of the iron salt and iron-bisphosphine systems previously discussed in this Account.
Scheme 7.

Representative Iron-NHC Cross-Couplings
One approach that has been taken involved synthesizing well-defined model complexes and testing their reactivity. These methods have been used recently in work by the groups of Deng and Tonzetich with a series of iron-NHC complexes (vide infra).23,52,56,58
For example, Deng and co-workers have synthesized numerous iron(II)-NHC complexes from reactions of well-defined iron(II)-NHC-dihalide complexes with organolithium and Grignard reagents to explore electronic structure and bonding trends that could provide insight into catalytic intermediates.53,54 It was found that mono- and bidentate NHCs both acted successfully as supporting ligands for the formation of transmetalated iron complexes at low temperature. These data suggested that these bis-transmetalated iron complexes could act as intermediates in catalysis. In another series of detailed reports, many iron(II)-NHC complexes bearing bis-aryl (phenyl, mesityl), -methyl trimethylsilyl, -alkynyl, and -alkenyl moieties were isolated and structurally characterized (Figure 4).55–58 These were demonstrated to be reactive toward alkyl halides, although the reactions required elevated temperatures or long reaction times of 4–36 h. The bis-aryl complexes were also proven to be catalytically competent for cross-coupling with reasonable yields even when formed in situ. This catalytic performance suggested that iron-NHC-aryl species could be involved in the C–C bond formation step in heteroaryl coupling reactions.
Figure 4.
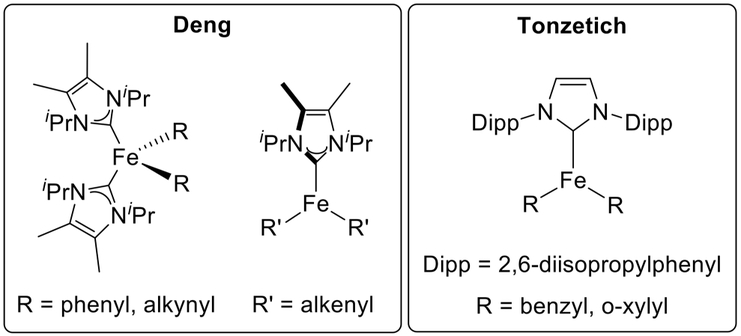
Example model organometallic iron(II)-NHC complexes studied by the groups of Deng and Tonzetich.52,55,56,58
In addition to stoichiometric reaction studies, Tonzetich and co-workers demonstrated the efficacy of discrete iron(II)-NHC complexes as pre-catalysts (Figure 4) for the cross-coupling of bromocycloheptane with phenyl and benzyl Grignard reagents (Scheme 7d).52 Model intermediate bis-transmetalated iron- (II)-NHC complexes using these nucleophile types were isolated and characterized, and their stoichiometric reactivity was evaluated. It was also noted that radical clock substrates indicated not only a single-electron process but also that the distribution of products was dependent on the concentration of the pre-catalyst. This was attributed to a radical, generated from reaction with the organohalide coupling partner, dissociating from the catalyst solvent cage rather than undergoing a rapid radical rebound.
In contrast, mechanistic studies of catalytic iron-NHC systems have been poorly defined, often carried out as part of the development of the catalytic protocol itself. For example, Nakamura and co-workers proposed a heteroleptic metalate complex for their iron fluoride-catalyzed aryl–aryl cross-coupling system (Scheme 7b).9 This consisted of an iron(II)-NHC mono-aryl species with two fluorides bridging a magnesium counterion. However, these studies were based purely on DFT, with no experimental evidence to guide the theoretical modeling of such species.59,60 More recently, Bedford and co-workers conducted preliminary mechanistic investigations into their Suzuki–Miyaura cross-coupling system (Scheme 7e).51 This reaction was proposed to be substrate-directed through coordination to the pyrrole motif supported by isotope-labeling studies, though this was not verified by any direct spectroscopic or structure studies of the iron species.
The most thorough study of iron-NHC cross-coupling originates from recent studies by our group on the alkyl–alkyl cross-coupling system reported by Cárdenas and co-workers (Scheme 7c).11 Initial investigations focused on analysis of the pre-catalyst activation step, which was originally proposed to generate an iron(I) reactive species. However, following EPR spin quantitation of the reaction mixture, a S = 1/2 iron(I) species was found to be present, amounting to <0.5% of all iron in solution.61 Concurrent Mössbauer spectroscopy was consistent with formation of S = 2 iron(II) centers identified as (IMes)Fe((1,3-dioxan-2-yl)ethyl)2 (13-IMes), which contained bis-chelating alkyl moieties oriented to disfavor β-H elimination (Figure 5A), and its THF adduct. Complex 13-IMes was shown to be reactive and selective for cross-coupled product as a pre-catalyst for the reported reaction. Further reactivity studies using isolated 13-IMes showed that it turned over twice in the presence of excess electrophile, producing 80% cross-coupled product with respect to iron and (IMes)-FeBr2(THF) (14) (Figure 5A) at a rate of >24 min−1. In addition, sub-stoichiometric reactions with electrophile indicated that the first turnover was only ~60% selective and the second ~100% selective for cross-coupled product. Reactivity studies were also performed using SIPr, resulting in only 23% cross-coupled product formation during catalysis. Additionally, characterization of (SIPr)Fe((1,3-dioxan-2-yl)ethyl)2 (13-SlPr) by XRD showed loss of chelation of one alkyl moiety in the presence of the SIPr ligand (Figure 5A). Combined with the synthesis and spectroscopic characterization of a series of iron-NHC bis-chelates,62 this suggested that the selectivity was dependent on NHC N-substituents, backbone, and substrate chelation.
Figure 5.

(A) Isolated iron-NHC complexes relevant to alkyl–alkyl cross-coupling. (B) Reactivity summary of observed iron(II) species and their transformations in the Cardenas iron-NHC-catalyzed alkyl–alkyl cross-coupling.
During catalytic turnover, in situ Mössbauer spectroscopy showed the presence of (IMes)FeBr((1,3-dioxan-2-yl)ethyl) (15, Figure 5A) and 14.61 Each of these species was isolated and characterized by XRD and solid and frozen solution Mössbauer spectroscopy to correlate their parameters to those of the observed iron species in catalysis. This mixture was not reactive with electrophiles but easily transmetalated to re-form 13-IMes with the addition of excess Grignard reagent, as summarized in Figure 5B. This is consistent with the presence of under-transmetalated iron(II) complexes in solution during catalysis for previously discussed iron-SciOPP systems and slow Grignard reagent addition being crucial for high selectivity.40
Overall, this study indicated that there may be a broader trend, beyond iron-bisphosphine chemistry, of iron(II) reactive species and slow nucleophile addition for selective product formation in catalysis. Additionally, the NHC ligand’s steric bulk and the ability of the nucleophile to interact with the iron center through an oxygen of the alkyl moiety were also shown to be crucial to selectivity in this reaction through synthetic and in situ reactivity studies. The future expansion of such rigorous studies to iron cross-coupling systems using different NHC ligands and cross-coupling partners will be critical to defining the effect of NHC structural variations and nucleophiles on iron speciation and mechanism.
SUMMARY AND FUTURE OUTLOOK
Evaluating in situ iron speciation, reactivity, and mechanism in iron-catalyzed cross-coupling is extremely challenging. Multiple iron species can form in situ, speciation can evolve with time, and multiple iron complexes can be reactive toward electrophiles yet exhibit different reaction rates or product distributions. Further complications include the assignment of iron structures from in situ spectroscopic analyses due to the multiple spin and oxidation states that might be present as well as the diverse coordination numbers and structures that iron can access. Unsurprisingly, early studies focused on physical organic, single spectroscopic, and theoretical methods, leading to diverse and often conflicting mechanistic proposals. However, the recent utilization of in situ inorganic spectroscopies and XRD methods that directly probe iron speciation, combined with GC reaction studies and kinetics analyses, have revolutionized our understanding of these systems, as outlined in this Account.
In ferric salt-catalyzed cross-coupling, the isolation and characterization of reactive iron species formed in situ have evolved our understanding of reactive iron species from simple mononuclear hypotheses to multi-nuclear reactive species with both alkyl and aryl nucleophiles. Additionally, our understanding of the role of NMP in these reactions (interactions with cations to modify speciation) has also been greatly advanced, but moving forward, several key challenges remain. First, the definition of the iron-alkyl species formed with alkyl nucleophiles containing β-hydrogens will be key to defining the breadth of iron-alkyl clusters in cross-coupling and determining whether similar structures are conserved across all alkyl nucleophiles. Furthermore, additional experimental definition of the reaction coordinates of iron clusters with electrophiles is required in order to continue to refine our understanding of the underlying reaction mechanisms. Continued isolation and structural definition of the iron “ate” species formed in this reaction class will continue to broaden our understanding of what iron species can form and their relative reactivities. More importantly, such studies will allow for reaction protocol development to favor formation of specific iron “ate” species to achieve improved cross-coupling methodologies.
Regarding cross-couplings using ligand additives or iron–ligand pre-catalysts, at present, only iron-SciOPP and iron-NHC cross-coupling systems have been studied in sufficient detail to unambiguously define all iron speciation and reactivity. While these studies each identified reactive, high-spin iron(II) species for the formation of cross-coupled product, it remains unclear if this trend is common across all iron-ligand cross-coupling systems. In fact, for systems with less bulky bisphosphines (i.e., dpbz, dppe), it has been observed that iron(I) species can more readily form, likely a result of differing reaction coordinates for reductive elimination (i.e., mono- vs multi-nuclear). As such, there is no reason to believe that all iron–ligand cross-coupling systems undergo a conserved mechanism in the fields of bisphosphines and NHCs. It is imperative that analogous, rigorous studies of these systems be performed in order to define the mechanistic diversity in iron–ligand-catalyzed cross-coupling.
The evolution of the mechanistic understanding of iron-catalyzed cross-couplings over the past 5 years has truly been outstanding—answering decades-old questions of reactive iron species in ferric salt cross-couplings as well as defining key reactive pathways in more modern cross-coupling systems with bisphosphine and NHC ligands. Despite this tremendous progress, we are still only at the tip of the iceberg in terms of our understanding of these systems, and much work remains. The continued advancement of fundamental mechanistic insight is essential to achieve the full potential of iron in cross-coupling chemistry.
Acknowledgments
Funding
This work was supported by the National Institutes of Health (R01GM111480).
Biography
Michael L. Neidig received his Ph.D. from Stanford University under the supervision of Professor Edward I. Solomon in 2007. Following research and postdoctoral positions at Dow Chemical and Los Alamos National Laboratory, Michael joined the Department of Chemistry at the University of Rochester in 2011.
Stephanie H. Carpenter received her B.S. in chemistry from the University of Kentucky in 2014. Her Ph.D. research focuses on iron-catalyzed C–H functionalization.
Daniel J. Curran received his B.S. in chemistry from The College of New Jersey in 2016. His Ph.D. research focuses on electronic structure and bonding in transient metal complexes.
Joshua C. DeMuth received his B.A. in chemistry from Rollins College in 2014. His Ph.D. research includes mechanisms in iron cross-coupling and C–H functionalization reactions.
Valerie E. Fleischauer received her B.A. in chemistry from Buffalo State College in 2013 and Ph.D. in chemistry from the University of Rochester in 2018, studying the mechanism of iron-catalyzed cross-coupling reactions.
Theresa E. Iannuzzi received her B.S. in chemistry from the University of Scranton in 2014. Her Ph.D. research focuses on iron and cobalt C–H functionalization.
Peter G. N. Neate received his M.Chem. in 2012 and Ph.D. in chemistry from the University of Edinburgh in 2017. He is now a postdoctoral fellow at the University of Rochester studying the mechanism of iron-catalyzed reactions.
Jeffrey D. Sears received his B.S. in chemistry in 2014 and M.S. in chemistry at Georgia State University in 2015. His Ph.D. research focuses on iron-catalyzed coupling reactions.
Nikki J. Wolford received her B.S. in chemistry from Millersville University in 2016. Her Ph.D. research focuses on the synthesis of homoleptic metal complexes.
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Tamura M; Kochi J Iron Catalysis in the Reaction of Grignard Reagents with Alkyl Halides. J. Organomet. Chem. 1971, 31, 289–309. [Google Scholar]
- (2).Tamura M; Kochi JK Vinylation of Grignard Reagents. Catalysis by Iron. J. Am. Chem. Soc. 1971, 93, 1487–1489. [Google Scholar]
- (3).Cahiez G; Avedissian H Highly Stereo- and Chemoselective Iron-Catalyzed Alkenylation of Organomagnesium Compounds. Synthesis 1998, 1998, 1199. [Google Scholar]
- (4).Gärtner D; Stein AL; Grupe S; Arp J; Jacobi von Wangelin A Iron-Catalyzed Cross-Coupling of Alkenyl Acetates. Angew. Chem. Int. Ed. 2015, 54, 10545–10549. [DOI] [PubMed] [Google Scholar]
- (5).Cahiez G; Duplais C; Moyeux A Iron-Catalyzed Alkylation of Alkenyl Grignard Reagents. Org. Lett. 2007, 9, 3253–3254. [DOI] [PubMed] [Google Scholar]
- (6).Guérinot A; Reymond S; Cossy J Iron-Catalyzed Cross-Coupling of Alkyl Halides with Alkenyl Grignard Reagents. Angew. Chem, Int. Ed. 2007, 46, 6521–6524. [DOI] [PubMed] [Google Scholar]
- (7).Nakamura M; Matsuo K; Ito S; Nakamura E Iron-Catalyzed Cross-Coupling of Primary and Secondary Alkyl Halides with Aryl Grignard Reagents. J. Am. Chem. Soc. 2004, 126, 3686–3687. [DOI] [PubMed] [Google Scholar]
- (8).Bedford RB; Betham M; Bruce DW; Danopoulos AA; Frost RM; Hird M Iron–Phosphine, – Phosphite, – Arsine, and – Carbene Catalysts for the Coupling of Primary and Secondary Alkyl Halides with Aryl Grignard Reagents. J. Org. Chem. 2006, 71, 1104–1110. [DOI] [PubMed] [Google Scholar]
- (9).Hatakeyama T; Nakamura M Iron-Catalyzed Selective Biaryl Coupling: Remarkable Suppression of Homocoupling by the Fluoride Anion. J. Am. Chem. Soc. 2007, 129, 9844–9845. [DOI] [PubMed] [Google Scholar]
- (10).Ghorai SK; Jin M; Hatakeyama T; Nakamura M Cross-Coupling of Non-Activated Chloroalkanes with Aryl Grignard Reagents in the Presence of Iron/N-Heterocyclic Carbene Catalysts. Org. Lett. 2012, 14, 1066–1069. [DOI] [PubMed] [Google Scholar]
- (11).Guisán-Ceinos M; Tato F; Buñuel E; Calle P; Cárdenas DJ Fe-Catalysed Kumada-Type Alkyl-Alkyl Cross-Coupling. Evidence for the Intermediacy of Fe(I) Complexes. Chem. Sci. 2013, 4, 1098–1104. [Google Scholar]
- (12).Hatakeyama T; Hashimoto T; Kondo Y; Fujiwara Y; Seike H; Takaya H; Tamada Y; Ono T; Nakamura M Iron-Catalyzed Suzuki-Miyaura Coupling of Alkyl Halides. J. Am. Chem. Soc. 2010, 132, 10674–10676. [DOI] [PubMed] [Google Scholar]
- (13).Hatakeyama T; Fujiwara Y.-i.; Okada Y; Itoh T; Hashimoto T; Kawamura S; Ogata K; Takaya H; Nakamura M Kumada-, Tamao-, and Corriu- Coupling of Alkyl Halides Catalyzed by an Iron-Bisphosphine Complex. Chem. Lett. 2011, 40, 1030–1032. [Google Scholar]
- (14).Hatakeyama T; Okada Y; Yoshimoto Y; Nakamura M Tuning Chemoselectivity in Iron-Catalyzed Sonogashira-Type Re-actions Using a Bisphosphine Ligand with Peripheral Steric Bulk: Selective Alkynylation of Nonactivated Alkyl Halides. Angew. Chem., Int. Ed. 2011, 50, 10973–10976. [DOI] [PubMed] [Google Scholar]
- (15).Hatakeyama T; Hashimoto T; Kathriarachchi KKADS; Zenmyo T; Seike H; Nakamura M Iron-Catalyzed Alkyl–Alkyl Suzuki–Miyaura Coupling. Angew. Chem., Int. Ed. 2012, 51, 8834–8837. [DOI] [PubMed] [Google Scholar]
- (16).Adams CJ; Bedford RB; Carter E; Gower NJ; Haddow MF; Harvey JN; Huwe M; Cartes MÁ; Mansell SM; Mendoza C; Murphy DM; Neeve EC; Nunn J Iron(I) in Negishi Cross-Coupling Reactions. J. Am. Chem. Soc. 2012, 134, 10333–10336. [DOI] [PubMed] [Google Scholar]
- (17).Fürstner A Iron Catalysis in Organic Synthesis: A Critical Assessment of What It Takes To Make This Base Metal a Multitasking Champion. ACS Cent. Sci 2016, 2, 778–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Sherry BD; Fürstner A The Promise and Challenge of Iron-Catalyzed Cross Coupling. Acc. Chem. Res. 2008, 41, 1500–1511. [DOI] [PubMed] [Google Scholar]
- (19).Smith RS; Kochi JK Mechanistic Studies of Iron Catalysis in the Cross Coupling of Alkenyl Halides and Grignard Reagents. J. Org. Chem. 1976, 41, 502–509. [Google Scholar]
- (20).Noda D; Sunada Y; Hatakeyama T; Nakamura M; Nagashima H Effect of TMEDA on Iron-Catalyzed Coupling Reactions of ArMgX with Alkyl Halides. J. Am. Chem. Soc. 2009, 131, 6078–6079. [DOI] [PubMed] [Google Scholar]
- (21).Daifuku SL; Al-Afyouni MH; Snyder BER; Kneebone JL; Neidig ML A Combined Mössbauer, Magnetic Circular Dichroism, and Density Functional Theory Approach for Iron Cross-Coupling Catalysis: Electronic Structure, In Situ Formation, and Reactivity of Iron-Mesityl-Bisphosphines. J.Am. Chem. Soc. 2014, 136, 9132–9143. [DOI] [PubMed] [Google Scholar]
- (22).Kneebone JL; Fleischauer VE; Daifuku SL; Shaps AA; Bailey JM; Iannuzzi TE; Neidig ML Electronic Structure and Bonding in Iron(II) and Iron(I) Complexes Bearing Bisphosphine Ligands of Relevance to Iron-Catalyzed C–C Cross-Coupling. Inorg. Chem. 2016, 55, 272–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Fillman KL; Przyojski JA; Al-Afyouni MH; Tonzetich ZJ; Neidig ML A Combined Magnetic Circular Dichroism and Density Functional Theory Approach for the Elucidation of Electronic Structure and Bonding in Three- and Four-Coordinate Iron(II)–N-Heterocyclic Carbene Complexes. Chem. Sci. 2015, 6, 1178–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Mo Z; Deng L Open-Shell Iron Hydrocarbyls. Coord. Chem. Rev. 2017, 350, 285–299. [Google Scholar]
- (25).Fürstner A; Krause H; Lehmann CW Unusual Structure and Reactivity of a Homoleptic “Super-Ate” Complex of Iron: Implications for Grignard Additions, Cross-Coupling Reactions, and the Kharasch Deconjugation. Angew. Chem., Int. Ed. 2006, 45, 440–444. [DOI] [PubMed] [Google Scholar]
- (26).Fürstner A; Martin R; Krause H; Seidel G; Goddard R; Lehmann CW Preparation, Structure, and Reactivity of Non-stabilized Organoiron Compounds. Implications for Iron-Catalyzed Cross Coupling Reactions. J. Am. Chem. Soc. 2008, 130, 8773–8787. [DOI] [PubMed] [Google Scholar]
- (27).Sun C-L; Fürstner A Formal Ring-Opening/Cross-Coupling Reactions of 2-Pyrones: Iron-Catalyzed Entry into Stereodefined Dienyl Carboxylates. Angew. Chem. Int. Ed. 2013, 52, 13071–13075. [DOI] [PubMed] [Google Scholar]
- (28).Al-Afyouni MH; Fillman KL; Brennessel WW; Neidig ML Isolation and Characterization of a Tetramethyliron(III) Ferrate: An Intermediate in the Reduction Pathway of Ferric Salts with MeMgBr. J. Am. Chem. Soc. 2014, 136, 15457–15460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Muñoz SB; Daifuku SL; Brennessel WW; Neidig ML Isolation, Characterization, and Reactivity of Fe8Men12−: Kochi’s S = 1/2 Species in Iron-Catalyzed Cross-Couplings with MeMgBr and Ferric Salts. J. Am. Chem. Soc. 2016, 138, 7492–7495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Muñoz SB III; Daifuku SL; Sears JD; Baker TM; Carpenter SH; Brennessel WW; Neidig ML The N-Methylpyrrolidone (NMP) Effect in Iron-Catalyzed Cross-Coupling with Simple Ferric Salts and MeMgBr. Angew. Chem., Int. Ed. 2018, 57, 6496–6500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Schoch R; Desens W; Werner T; Bauer M X-ray Spectroscopic Verification of the Active Species in Iron-Catalyzed Cross-Coupling Reactions. Chem. - Eur. J. 2013, 19, 15816–15821. [DOI] [PubMed] [Google Scholar]
- (32).Bedford RB; Brenner PB; Carter E; Cogswell PM; Haddow MF; Harvey JN; Murphy DM; Nunn J; Woodall CH TMEDA in Iron-Catalyzed Kumada Coupling: Amine Adduct versus Homoleptic “ate” Complex Formation. Angew. Chem., Int. Ed. 2014, 53, 1804–1808. [DOI] [PubMed] [Google Scholar]
- (33).Hedstroem A; Izakian Z; Vreto I; Wallentin C-J; Norrby P-O On the Radical Nature of Iron-Catalyzed Cross-Coupling Reactions. Chem. - Eur. J. 2015, 21, 5946–5953. [DOI] [PubMed] [Google Scholar]
- (34).Hedstroem A; Lindstedt E; Norrby P-O On the Oxidation State of Iron in Iron-Mediated C-C Couplings. J. Organomet. Chem. 2013, 748, 51–55. [Google Scholar]
- (35).Lefevre G; Jutand A Activation of Aryl and Heteroaryl Halides by an Iron(I) Complex Generated in the Reduction of [Fe(acac)3] by PhMgBr: Electron Transfer versus Oxidative Addition. Chem. - Eur. J. 2014, 20, 4796–4805. [DOI] [PubMed] [Google Scholar]
- (36).Zhurkin FE; Wodrich MD; Hu X A Monometallic Iron(I) Organoferrate. Organometallics 2017, 36, 499–501. [Google Scholar]
- (37).Clemancey M; Cantat T; Blondin G; Latour J-M; Dorlet P; Lefevre G Structural Insights into the Nature of Fe0 and FeI Low-Valent Species Obtained upon the Reduction of Iron Salts by Aryl Grignard Reagents. Inorg. Chem. 2017, 56, 3834–3848. [DOI] [PubMed] [Google Scholar]
- (38).Parchomyk T; Koszinowski K Ate Complexes in Iron-Catalyzed Cross-Coupling Reactions. Chem. - Eur. J. 2016, 22, 15609–15613. [DOI] [PubMed] [Google Scholar]
- (39).Carpenter SH; Baker TM; Munoz SB; Brennessel WW; Neidig ML Multinuclear Iron-Phenyl Species in Reactions of Simple Iron Salts with PhMgBr: Identification of Fe4(μ-Ph)6(THF)4 as a Key Reactive Species for Cross-Coupling Catalysis. Chem. Sci. 2018, 9, 7931–7939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Daifuku SL; Kneebone JL; Snyder BER; Neidig ML Iron(II) Active Species in Iron-Bisphosphine Catalyzed Kumada and Suzuki–Miyaura Cross-Couplings of Phenyl Nucleophiles and Secondary Alkyl Halides. J. Am. Chem. Soc. 2015, 137, 11432–11444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Cahiez G; Habiak V; Duplais C; Moyeux A Iron-Catalyzed Alkylations of Aromatic Grignard Reagents. Angew. Chem. Int. Ed. 2007, 46, 4364–4366. [DOI] [PubMed] [Google Scholar]
- (42).Dongol KG; Koh H; Sau M; Chai CLL Iron-Catalysed sp3–sp3 Cross-Coupling Reactions of Unactivated Alkyl Halides with Alkyl Grignard Reagents. Adv. Synth. Catal 2007, 349, 1015–1018. [Google Scholar]
- (43).Jin M; Adak L; Nakamura M Iron-Catalyzed Enantio-selective Cross-Coupling Reactions of α-Chloroesters with Aryl Grignard Reagents. J. Am. Chem. Soc. 2015, 137, 7128–7134. [DOI] [PubMed] [Google Scholar]
- (44).Takaya H; Nakajima S; Nakagawa N; Isozaki K; Iwamoto T; Imayoshi R; Gower NJ; Adak L; Hatakeyama T; Honma T; Takagaki M; Sunada Y; Nagashima H; Hashizume D; Takahashi O; Nakamura M Investigation of Organoiron Catalysis in Kumada-Tamao-Corriu-type Cross-Coupling Reaction Assisted by Solution- Phase X-ray Absorption Spectroscopy. Bull. Chem. Soc. Jpn. 2015, 88, 410–418. [Google Scholar]
- (45).Parchomyk T; Demeshko S; Meyer F; Koszinowski K Oxidation States, Stability, and Reactivity of Organoferrate Complexes. J. Am. Chem. Soc. 2018, 140, 9709–9720. [DOI] [PubMed] [Google Scholar]
- (46).Lee W; Zhou J; Gutierrez O Mechanism of Nakamura’s Bisphosphine-Iron-Catalyzed Asymmetric C(sp2)–C(sp3) Cross-Coupling Reaction: The Role of Spin in Controlling Arylation Pathways. J. Am. Chem. Soc. 2017, 139, 16126–16133. [DOI] [PubMed] [Google Scholar]
- (47).Sharma AK; Sameera WMC; Jin M; Adak L; Okuzono C; Iwamoto T; Kato M; Nakamura M; Morokuma K DFT and AFIR Study on the Mechanism and the Origin of Enantioselectivity in Iron-Catalyzed Cross-Coupling Reactions. J. Am. Chem. Soc. 2017, 139, 16117–16125. [DOI] [PubMed] [Google Scholar]
- (48).Nakagawa N; Hatakeyama T; Nakamura M Iron-catalyzed Suzuki–Miyaura Coupling Reaction of Unactivated Alkyl Halides with Lithium Alkynylborates. Chem. Lett. 2015, 44, 486–488. [Google Scholar]
- (49).Kneebone JL; Brennessel WW; Neidig ML Intermediates and Reactivity in Iron-Catalyzed Cross-Couplings of Alkynyl Grignards with Alkyl Halides. J. Am. Chem. Soc. 2017, 139, 6988–7003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Chua Y-Y; Duong HA Selective Kumada Biaryl Cross-Coupling Reaction Enabled by an Iron(III) Alkoxide-N-Heterocyclic Carbene Catalyst System. Chem. Commun. 2014, 50, 8424–8427. [DOI] [PubMed] [Google Scholar]
- (51).O’Brien HM; Manzotti M; Abrams RD; Elorriaga D; Sparkes HA; Davis SA; Bedford RB Iron-Catalysed Substrate- Directed Suzuki Biaryl Cross-Coupling. Nat. Catal. 2018, 1, 429–437. [Google Scholar]
- (52).Przyojski JA; Veggeberg KP; Arman HD; Tonzetich ZJ Mechanistic Studies of Catalytic Carbon-Carbon Cross-Coupling by Well-Defined Iron NHC Complexes. ACS Catal. 2015, 5, 5938–5946. [Google Scholar]
- (53).Liu Y; Shi M; Deng L Iron(II) Dihydrocarbyls Supported by a Biphenyl-Linked Bis(benzimidazol-2-ylidene) Ligand: Syntheses and Characterization. Organometallics 2014, 33, 5660–5669. [Google Scholar]
- (54).Ouyang Z; Du J; Wang L; Kneebone JL; Neidig ML; Deng L Linear and T-Shaped Iron(I) Complexes Supported by N-Heterocyclic Carbene Ligands: Synthesis and Structure Characterization. Inorg. Chem. 2015, 54, 8808–8816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Liu Y; Wang L; Deng L Three-Coordinate Iron(II) Dialkenyl Compound with NHC Ligation: Synthesis, Structure, and Reactivity. Organometallics 2015, 34, 4401–4407. [Google Scholar]
- (56).Liu Y; Xiao J; Wang L; Song Y; Deng L Carbon-Carbon Bond Formation Reactivity of a Four-Coordinate NHC-Supported Iron(II) Phenyl Compound. Organometallics 2015, 34, 599–605. [Google Scholar]
- (57).Wang X; Zhang J; Wang L; Deng L High-Spin Iron(II) Alkynyl Complexes with N-Heterocyclic Carbene Ligation: Synthesis, Characterization, and Reactivity Study. Organometallics 2015, 34, 2775–2782. [Google Scholar]
- (58).Xiang L; Xiao J; Deng L Synthesis, Structure, and Reactivity of Organo-Iron(II) Complexes with N-Heterocyclic Carbene Ligation. Organometallics 2011, 30, 2018–2025. [Google Scholar]
- (59).Hatakeyama T; Hashimoto S; Ishizuka K; Nakamura M Highly Selective Biaryl Cross-Coupling Reactions between Aryl Halides and Aryl Grignard Reagents: A New Catalyst Combination of N-Heterocyclic Carbenes and Iron, Cobalt, and Nickel Fluorides. J. Am. Chem. Soc. 2009, 131, 11949–11963. [DOI] [PubMed] [Google Scholar]
- (60).Hatakeyama T; Ishizuka K; Nakamura M Cross-Coupling Reactions Catalyzed by Iron Group Metals and N-Heterocyclic Carbenes via Nonconventional Reaction Mechanisms. Yuki Gosei Kagaku IKyokaishi 2011, 69, 1282–1298. [Google Scholar]
- (61).Fleischauer VE; Muñoz SB III; Neate PGN; Brennessel WW; Neidig ML NHC and Nucleophile Chelation Effects on Reactive Iron(II) Species in Alkyl-Alkyl Cross-Coupling. Chem. Sci. 2018, 9, 1878–1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Muñoz SB; Fleischauer VE; Brennessel WW; Neidig ML Combined Effects of Backbone and N-Substituents on Structure, Bonding, and Reactivity of Alkylated Iron(II)-NHCs. Organometallics 2018, 37, 3093–3101. [DOI] [PMC free article] [PubMed] [Google Scholar]