

Abstract
Rhabdomyoma is a rare benign tumor with skeletal muscle differentiation. Rhabdomyoma is further classified into cardiac, adult, fetal, and genital subtypes. Out of these, fetal type rhabdomyoma (FTR) is the rarest. Only a small number of cases have been recorded in the literature. FTR typically affects male infants and young children and occurs predominantly in the head and neck region. FTR is exceedingly rare in the adult, with less than 30 cases reported. The classic FTR is composed of primitive undifferentiated spindle cells with scant eosinophilic cytoplasm embedded in a myxoid stroma. Immunohistochemically, the tumor cells are positive for desmin, muscle specific actin, and myogenin. Awareness and proper recognition of this rare entity is of considerable importance to avoid misdiagnosis of embryonal rhabdomyosarcoma. In this study, we report one case of FTR in an adult patient and reviewed the literature about the clinical and pathologic presentation of FTR in the adult.
Keywords: Rhabdomyoma, Rhabdomyosarcoma, Benign spindle cell tumor
Introduction
Skeletal muscle is the largest organ in the body by weight and volume. It is developed from myotomes arising from primitive mesodermal tissue. At the earliest stage of muscle development, small primitive spindle-shaped mesodermal cells differentiate into myoblasts, which are round to oval cells with centrally located nuclei and abundant eosinophilic cytoplasm rich in myofibrils. Upon further development, these individual myoblasts align up and fuse into myotubes. With longitudinal proliferation, thickening of myofibrils and peripheral position of the nuclei, myotubes develop into muscle fiber, which first appears in human embryo at about the tenth week [1].
Tumors of skeletal muscle are predominantly malignant, e.g., rhabdomyosarcoma, which is the most common malignant soft tissue tumor of children and young adults. The benign counterpart, rhabdomyoma, is very rare, representing only 2% of skeletal muscle tumors [2]. Rhabdomyoma is further classified into different categories based on clinical and pathologic presentation. Some subtypes are extremely uncommon and not familiar to many practicing pathologists [2].
Rhabdomyoma is classified into cardiac and extracardiac categories. The cardiac type occurs predominantly in the hearts of infants and young children in the setting of tuberous sclerosis. Morphologically, the cardiac type is composed of large polygonal cardiac muscle cells with cytoplasmic vacuolization (spider cells) [2]. The extracardiac type is distinct from the cardiac type both clinically and pathologically. The specific genetic alteration for extracardiac rhabdomyoma is still unknown. It is further classified into adult, fetal and genital subtypes. The adult and fetal types are predominantly seen in the head and neck region, with adult type seen more commonly in adult patients and fetal type predominantly in infants and young children. The genital subtype exclusively occurs in the vagina and vulva of young to middle-aged female patients. Fetal and genital rhabdomyomas share morphological similarity, both composed of primitive spindle cells and more differentiated myoblasts with centrally located nuclei, prominent nucleoli and abundant eosinophilic cytoplasm with cross-striation. The adult type is composed exclusively of myoblasts; the primitive spindle cell component is not seen [2]. Out of these, fetal type is the rarest subtype and is also the one that can cause significant diagnostic challenge due to its resemblance to embryonal rhabdomyosarcoma, especially in adult patients.
In this study, we report one case of FTR in an adult patient. We reviewed the clinical and pathologic presentations, and discussed differential diagnosis for this rare entity.
Case Presentation
A 37-year-old female with no significant past medial history presented with a small soft palate polyp. According to the patient, the polyp had been present for 20 years. The polyp had increased in size during the past month and caused nasal congestion and trouble swallowing. Thus the patient decided to have it removed. Computed tomography scan showed a polyp of the soft palate without any worrisome features. Physical examination showed a 1.7 cm pedunculated polyp with smooth glistening mucosa (Fig. 1). Clinical impression was a vascular malformation.
Fig. 1.
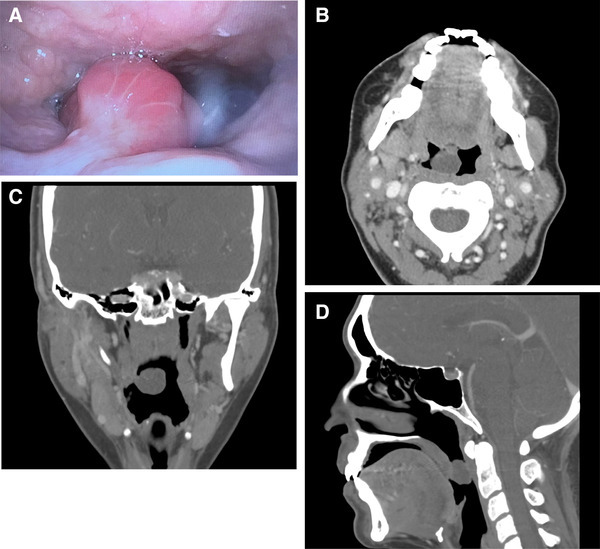
Photograph of the polyp by flexible laryngoscopy (a). Three-dimensional computed tomography scan of the polyp (b–d) showing that the polyp is attached to the nasal side of soft palate
Microscopic examination of the resected polyp showed a pauci-cellular polyp with overlying intact respiratory mucosa (Fig. 2). The lesion was composed predominantly of small spindle cells with fine chromatin, inconspicuous nucleoli, and delicate finely tapered bipolar or unipolar eosinophilic cytoplasm (Fig. 2). The background had abundant myxoid stroma with admixed chronic inflammatory cells. Scattered myoblasts with centrally located nuclei, prominent nucleoli, and abundant eosinophilic cytoplasm with cross-striations were seen at the edge of the polyp. This type of gradient of maturation has been described in fetal rhabdomyoma by Thompson [3]. Mitotic figures were not identified. Increased cellularity, atypia, or necrosis was absent. Immunohistochemically, the lesion was diffusely strongly positive for desmin. The spindle cells and myoblasts were also positive for myogenin (Fig. 3). Other tested markers, such as cytokeratin, S100, smooth muscle actin, and CD34 were negative. Diagnosis of fetal rhabdomyoma, classic (myxoid) variant, was made.
Fig. 2.
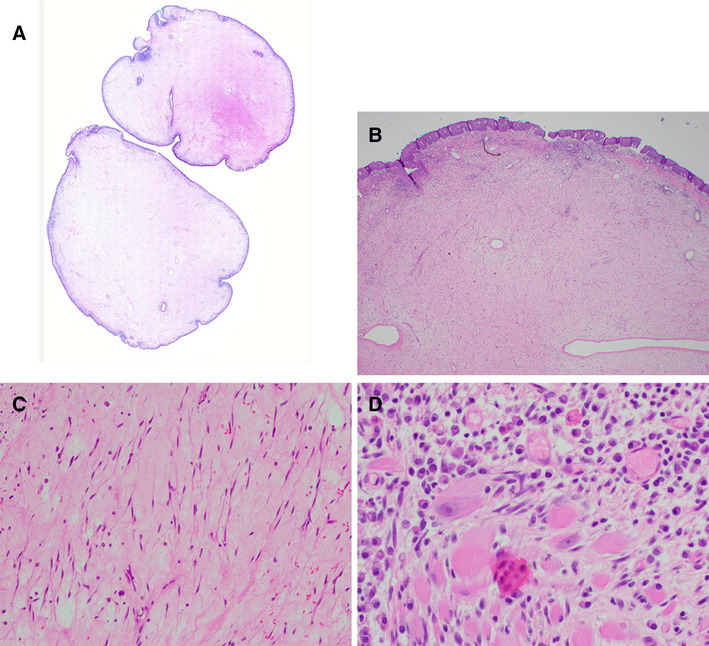
Whole mount picture of the resected polyp (a). Mass with intact overlying mucosa (b, ×40). Primitive spindle cells with delicate bipolar and unipolar eosinophilic cytoplasm (c, ×400). Scant myoblasts are located at the periphery of the lesion (d, ×400)
Fig. 3.
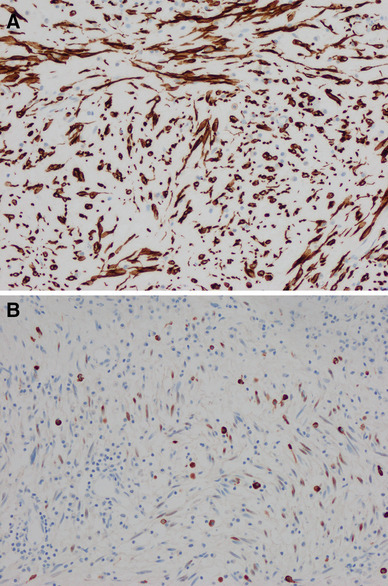
Tumor cells are diffusely positive for desmin (a, ×200). Myogenin stains the nuclei of spindle cells and myoblast. Inflammatory cells show nonspecific cytoplasmic stain (b, ×200)
Discussion
Rhabdomyoma is a group of heterologous benign tumors with skeletal muscle differentiation. These tumors show distinct yet overlapping clinical and pathologic features. The cardiac type is predominantly seen in infants and young children with tuberous sclerosis. These tumors can occur anywhere in the heart and are well-demarcated, non-encapsulated nodular masses ranging in size from millimeters to several centimeters. They usually present as multiple masses in the heart, with solitary mass seen in only about 10% of cases. Microscopically the tumor is composed of large polygonal cells with clear cytoplasmic glycogen vacuoles separated by strands of eosinophilic cytoplasm containing contractile myofilaments (spider cells) [2]. Immunohistochemically, the typical skeletal muscle immunophenotype, e.g., positivity for desmin, myogenin, and muscle specific actin, is present. Expression of HMB-45 and loss of expression of tuberin and hamartin have also been documented in some case, supporting a relation with PEComa as a component of tuberous sclerosis complex [4, 5]. Clinically, cardiac rhabdomyoma is characterized by high possibility of spontaneous regression, especially in infants. Surgery is reserved for masses that cause cardiac symptoms and is curative. Extracardiac rhabdomyomas are not associated with tuberous sclerosis. Although rare cases arise in the setting nevoid basal cell carcinoma syndrome (Gorlin syndrome), and other rare cases of multi-centric rhabdomyomas suggest a genetic origin, the association of extracardiac rhabdomyoma with PTCH gene or other specific gene alteration has not been firmly established [6, 7]. Morphologically, adult rhabdomyoma is composed of myoblasts, which are differentiated large cells with abundant myofilaments in the cytoplasm; while fetal and genital types have component of primitive spindle cells with scant cytoplasm. Myoblasts are usually also seen, but can be scant in number, especially in fetal type. All three subtypes of rhabdomyoma are treated by surgical resection, with adult type with frequent recurrence, and fetal and genital type generally cured by complete resection [2].
FTR was first described in 1972 in a series of nine cases [8]. Since then, more than 100 cases have been reported, with the largest case series containing 24 cases [9]. Much of the earlier literature did not separate genital rhabdomyoma from fetal rhabdomyoma. It was found later that genital rhabdomyoma occurs exclusively in young to middle aged female and represents a distinct variant [10]. Majority of FTR occur in the head and neck region in patients < 3 years of age; however, it can be seen in patients of wide age range and in wide anatomic locations including skin, extremities, mediastinum, retroperitoneum, etc. Excluding genital rhabdomyoma, less than 30 cases of FTR have been reported in adults of > 20 years of age (Table 1). Patients’ age ranged from 20 to 87 years, with majority being male patients (male: female ratio is 16:6). Not uncommonly, the tumor, including the case in this study, was reported to have been present for a long time, raising the possibility that the tumor actually arose during childhood [9]. All except two tumors were located in the head and neck region (Table 1). The two tumors outside of head and neck region were from thigh of a 54 year old male and bladder of an 87 year old male. Clinically, majority patients with fetal rhabdomyoma presented with asymptomatic nodule/mass or painless swelling, with the exception of masses located at vocal cord, which often caused hoarseness, and the one tumor located in bladder, which presented with hematuria. Clinical diagnosis included submucosal cyst [11], vascular malformation, or unspecified benign lesion [12]. However, clinical differential diagnosis was not mentioned in the majority of the literature. For all of those 22 cases from adult patients, surgical removal was curative with no recurrence or malignant transformation reported.
Table 1.
Reported cases of fetal type rhabdomyoma in adult patients
| Author | Histologic type | Age | Sex | Location | |
|---|---|---|---|---|---|
| Case 1 | Misch [13] | Intermediate | 21 | F | Tongue |
| Case 2 | Nath [14] | Not specified | 38 | F | Orbit |
| Case 3 | Sobel [15] | Not specified | 39 | F | Larynx |
| Case 4 | Dehner [8] | Myxoid | 56 | M | Parotid |
| Case 5 | Ferlito [16] | Myxoid | 50 | M | Larynx |
| Case 6 | Fu [17] | Myxoid | 60 | M | Nasopharynx |
| Case 7 | Di Sant’Agnese [18] | Intermediate | 54 | M | Thigh |
| Case 8 | Di Sant’Agnese [18] | Intermediate | 53 | M | Vocal cord |
| Case 9 | Di Sant’Agnese [18] | Intermediate | 37 | M | Tongue |
| Case 10 | Di Sant’Agnese [18] | Myxoid | 65 | F | Vocal cord |
| Case11 | Di Sant’Agnese [18] | Intermediate | 56 | M | Neck |
| Case 12 | Kapadia [9] | Intermediate | 30 | M | Eyebrow |
| Case 13 | Kapadia [9] | Myxoid | 20 | F | Neck |
| Case 14 | Kapadia [9] | Intermediate | 58 | M | Tongue |
| Case 15 | Kapadia [9] | Intermediate | 45 | M | Face |
| Case 16 | Kapadia [9] | Intermediate | 20 | F | Buccal mucosa |
| Case 17 | Kapadia [9] | Intermediate | 37 | M | Soft palate |
| Case 18 | Kapadia [9] | Intermediate | 48 | F | Larynx, vocal cord |
| Case 19 | Wang [12] | Myxoid | 57 | M | Tonsil |
| Case 20 | Sharma [11] | Intermediate | 42 | M | Larynx |
| Case 21 | Hansen [19] | Not specified | 55 | M | Larynx |
| Case 22 | Gonzalez-Perez [20] | Not specified | 87 | M | Bladder |
Fetal rhabdomyoma is considered a benign tumor. Surgical removal is curative with very rare recurrences even when the tumor is incompletely resected. However, two cases of malignant transformation have been reported in pediatric patients. One patient was 3 months and the second patient was 18 months of age at initial resection, and the tumor recurred as rhabdomyosarcoma at 22 and 23 months respectively after initial resection [2]. Although no malignant transformation or recurrence has been reported in adult patients, one case of mixed fetal rhabdomyoma and embryonal rhabdomyosarcoma has been reported in a 31 year old female [21]. Yet in another study, differentiation of rhabdomyosarcoma into rhabdomyoma was seen in a 4 year old boy [22]. All these are rare case reports and the relationship between rhabdomyosarcoma and rhabdomyoma is poorly understood.
Morphologically, FTR typically presents as a well circumscribed mass or polyp of 2–6 cm in greatest dimension in average. Some rare case can be more than 10 cm [9]. FTR is further classified into classic or myxoid type and intermediate or juvenile type based on morphology. The classic or myxoid type is typically pauci-cellular with abundant myxoid stroma. The tumor cells have small spindle nuclei with fine chromatin and usually scant bipolar eosinophilic cytoplasm. Cross-striations are rare and often difficult to identify. The intermediate or juvenile variant is more cellular, composed of numerous more differentiated myoblasts and less conspicuous primitive spindle cells. Mitotic activity is absent to rare, cytologic atypia is not seen, and necrosis is usually absent [2]. The main differential diagnosis is rhabdomyosarcoma, embryonal type. Rhabdomyosarcoma typically presents as a rapidly growing mass with aggressive clinical presentation. Microscopically, a dense cambium layer under the epithelium is typically present. Mitotic activity is easy to identify and is typically more than 10/10 high power field. Necrosis and severe cytologic atypia are common findings [2]. Another differential diagnosis is harmatomas with skeletal muscle component, e.g., Triton tumor and rhabdomyomatous mesenchymal hamartoma of the skin. Both are very rare lesions seen in young children. The presence of neural and/or adipose tissue distinguishes hamartoma from rhabdomyoma [23].
In summary, FTR is a rare benign tumor that predominantly occurs in the head and neck region of young children. It is very rare in adult patients, with less than 30 cases reported in the literature. We report a case of myxoid FTR in a 37 year old female and reviewed the clinical and pathologic features of this rare tumor. Proper recognition of this rare entity is essential to avoid misdiagnosis of rhabdomyosarcoma.
Conflict of interest
None of the authors have any conflicts of interest to disclose.
Research Involving Human and Animal Participants
This article does not contain any studies with human participants or animals performed by any of the authors.
References
- 1.Mills SE. Histology for pathologist. 4. Philadelphia: Lippincott Williams & Wilkins; 2012. Musculoskeletal system. [Google Scholar]
- 2.Goldblum JR, Folpe AL, Weiss SW. Soft tissue tumors. 6. Amsterdam: Elservier; 2014. Rhabdomyoma. [Google Scholar]
- 3.Thompson LD. Ear fetal rhabdomyoma. Ear Nose Throat J. 2017;96(9):358. doi: 10.1177/014556131709600904. [DOI] [PubMed] [Google Scholar]
- 4.Weeks DA, Chase DR, Malott RL, Chase RL, Zuppan CW, Beckwith JB, Mierau GW. HMB-45 staining in angiomyolipoma, cardiac rhabdomyoma, other mesenchymal processes, and tuberous sclerosis-associated brain lesions. Int J Surg Pathol. 1994;1:191. doi: 10.1177/106689699400100307. [DOI] [Google Scholar]
- 5.Vinaitheerthan M, Wei J, Mizuguchi M, Greco A, Barness EG. Tuberous sclerosis: immunohistochemistry expression of tuberin and hamartin in a 31-week gestational fetus. Fetal Pediatr Pathol. 2004;23(4):241–249. doi: 10.1080/15227950490923606. [DOI] [PubMed] [Google Scholar]
- 6.Diociaiuti A, Inserra A, De Vega IF, Rota C, Surrenti T, Giraldi L, Piemontese MR, Giovannoni I, Callea F, El Hachem M. Naevoid basal cell carcinoma syndrome in a 22-month-old child presenting with multiple basalcell carcinomas and a fetal rhabdomyoma. Acta Derm Venereol. 2015;95(2):243–244. doi: 10.2340/00015555-1892. [DOI] [PubMed] [Google Scholar]
- 7.de Trey LA, Schmid S, Huber GF. Multifocal adult rhabdomyoma of the head and neck manifestation in 7 locations and review of the literature. Case Rep Otolaryngol. 2013;2013:758416. doi: 10.1155/2013/758416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dehner LP, Enzinger FM, Font RL. Fetal rhabdomyoma. An analysis of nine cases. Cancer. 1972;30(1):160–166. doi: 10.1002/1097-0142(197207)30:1<160::AID-CNCR2820300123>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 9.Kapadia SB, Meis JM, Frisman DM, Ellis GL, Heffner DK. Fetal rhabdomyoma of the head and neck: a clinicopathologic and immunophenotypic study of24 cases. Hum Pathol. 1993;24(7):754–765. doi: 10.1016/0046-8177(93)90013-7. [DOI] [PubMed] [Google Scholar]
- 10.Schoolmeester JK, Xing D, Keeney GL, Sukov WR. Genital rhabdomyoma of the lower female genital tract: a study of 12 cases with molecular cytogenetic findings. Int J Gynecol Pathol. 2017 doi: 10.1097/PGP.0000000000000428. [DOI] [PubMed] [Google Scholar]
- 11.Sharma SJ, Kreisel M, Kroll T, Gattenloehner S, Klussmann JP, Wittekindt C. Extracardiac juvenile rhabdomyoma of the larynx: a rare pathological finding. Eur Arch Otorhinolaryngol. 2013;270(2):773–776. doi: 10.1007/s00405-012-2245-7. [DOI] [PubMed] [Google Scholar]
- 12.Wang CP, Chang YH, Chang YT. Fetal rhabdomyoma of the right tonsil with polyp-like appearance. Case Rep Otolaryngol. 2015;2015:713278. doi: 10.1155/2015/713278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Misch KA. Rhabdomyoma purum: a benign rhabdomyoma of tongue. J Pathol Bacteriol. 1958;75(1):105–108. doi: 10.1002/path.1700750112. [DOI] [PubMed] [Google Scholar]
- 14.Nath K, Nema HV, Hameed S, Shukla BR. Orbital rhabdomyoma. Am J Ophthalmol. 1965;59:1130–1134. doi: 10.1016/0002-9394(65)93438-0. [DOI] [PubMed] [Google Scholar]
- 15.Sobel HJ, Marquet E, Schwarz R. Is schwannoma related to granular cell myoblastoma? Arch Pathol. 1973;95(6):396–401. [PubMed] [Google Scholar]
- 16.Ferlito A, Frugoni P. Rhabdomyoma purum of the larynx. J Laryngol Otol. 1975;89(11):1131–1141. doi: 10.1017/S0022215100081482. [DOI] [PubMed] [Google Scholar]
- 17.Fu YS, Perzin KH. Nonepithelial tumors of the nasal cavity paranasal sinuses, and nasopharynx: a clinicopathologic study. V. Skeletal muscle tumors (rhabdomyoma and rhabdomyosarcoma) Cancer. 1976;37(1):364–376. doi: 10.1002/1097-0142(197601)37:1<364::AID-CNCR2820370147>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 18.Di Sant’Agnese PA, Knowles DM. Extracardiac rhabdomyoma: a clinicopathologic study and review of the literature. Cancer. 1980;46(4):780–789. doi: 10.1002/1097-0142(19800815)46:4<780::AID-CNCR2820460423>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 19.Hansen T, Katenkamp D. Rhabdomyoma of the head and neck: morphology and differential diagnosis. Virchows Arch. 2005;447(5):849–854. doi: 10.1007/s00428-005-0038-8. [DOI] [PubMed] [Google Scholar]
- 20.González-Pérez L, Alvarez-Argüelles H, Ramos Gutiérrez VJ, Hernández SG. Bello AP, Masip TC, Ruiz ES. Bladder fetal rhabdomyoma intermediate type. Urol Int. 10.1159/000485256. [DOI] [PubMed]
- 21.Jacques SM, Lawrence WD, Malviya VK. Uterine mixed embryonal rhabdomyosarcoma and fetal rhabdomyoma. Gynecol Oncol. 1993;48(2):272–276. doi: 10.1006/gyno.1993.1047. [DOI] [PubMed] [Google Scholar]
- 22.Bale PM, Parsons RE, Stevens MM. Diagnosis and behavior of juvenile rhabdomyosarcoma. Hum Pathol. 1983;14(7):596–611. doi: 10.1016/S0046-8177(83)80203-2. [DOI] [PubMed] [Google Scholar]
- 23.Walsh SN, Hurt MA. Cutaneous fetal rhabdomyoma: a case report and historical review of the literature. Am J Surg Pathol. 2008;32(3):485–491. doi: 10.1097/PAS.0b013e318148545c. [DOI] [PubMed] [Google Scholar]