

Abstract
We sought to identify factors that are predictive of liver transplantation or death in patients with primary sclerosing cholangitis (PSC), and to develop and validate a contemporaneous risk score for use in a real‐world clinical setting. Analyzing data from 1,001 patients recruited to the UK‐PSC research cohort, we evaluated clinical variables for their association with 2‐year and 10‐year outcome through Cox‐proportional hazards and C‐statistic analyses. We generated risk scores for short‐term and long‐term outcome prediction, validating their use in two independent cohorts totaling 451 patients. Thirty‐six percent of the derivation cohort were transplanted or died over a cumulative follow‐up of 7,904 years. Serum alkaline phosphatase of at least 2.4 × upper limit of normal at 1 year after diagnosis was predictive of 10‐year outcome (hazard ratio [HR] = 3.05; C = 0.63; median transplant‐free survival 63 versus 108 months; P < 0.0001), as was the presence of extrahepatic biliary disease (HR = 1.45; P = 0.01). We developed two risk scoring systems based on age, values of bilirubin, alkaline phosphatase, albumin, platelets, presence of extrahepatic biliary disease, and variceal hemorrhage, which predicted 2‐year and 10‐year outcomes with good discrimination (C statistic = 0.81 and 0.80, respectively). Both UK‐PSC risk scores were well‐validated in our external cohort and outperformed the Mayo Clinic and aspartate aminotransferase‐to‐platelet ratio index (APRI) scores (C statistic = 0.75 and 0.63, respectively). Although heterozygosity for the previously validated human leukocyte antigen (HLA)‐DR*03:01 risk allele predicted increased risk of adverse outcome (HR = 1.33; P = 0.001), its addition did not improve the predictive accuracy of the UK‐PSC risk scores. Conclusion: Our analyses, based on a detailed clinical evaluation of a large representative cohort of participants with PSC, furthers our understanding of clinical risk markers and reports the development and validation of a real‐world scoring system to identify those patients most likely to die or require liver transplantation.
Abbreviations
- Alb
albumin
- ALP
alkaline phosphatase
- APRI
AST‐to‐platelet ratio index
- AST
aspartate aminotransferase
- Bili
bilirubin
- CI
confidence interval
- Hb
hemoglobin
- HLA
human leukocyte antigen
- HR
hazard ratio
- IBD
inflammatory bowel disease
- IgG
immunoglobulin G
- MELD
Model for End‐Stage Liver Disease
- PBC
primary biliary cholangitis
- Plts
platelets
- PSC
primary sclerosing cholangitis
- UDCA
ursodeoxycholic acid
- and ULN
upper limit of normal
Primary sclerosing cholangitis (PSC) is a chronic fibrosing cholestatic liver disease that is frequently associated with inflammatory bowel disease (IBD).1 Disease progression culminates in end‐stage liver disease and a high likelihood of death without liver transplantation.2 Patients with PSC have up to a 15% lifetime risk of developing cholangiocarcinoma, which parallels their risk of colorectal cancer.1, 3, 4, 5 Insights into disease pathogenesis are limited, but genetic studies highlight the importance of the adaptive immune system, with the strongest genetic association found within the human leukocyte antigen (HLA) locus.6, 7, 8
Clinical course is variable, and efforts to individualize risk prediction are important for patients, clinicians, and trials of experimental agents.9 Existing studies suggest that various clinical factors may predict the risk of adverse outcome. For example, elevated immunoglobulin G (IgG) 4 concentration is reportedly associated (although not robustly validated) with an increased risk of progression to cirrhosis.10, 11 Conversely, small‐duct PSC confers an improved survival and lower risk of cholangiocarcinoma,12 as does a reduction in serum alkaline phosphatase (ALP) 1‐2 years following diagnosis.13, 14, 15 Using cutoffs previously defined as stratifiers of risk in small bile duct disease primary biliary cholangitis (PBC), two studies have confirmed the independent prognostic value of ALP in PSC cohorts.13, 14 However, many studies evaluating risk prediction models in PSC have been limited by sample size, tertiary center recruitment bias, failure to control for the interaction of variables with one another, and lack of validation.16 With the exception of the revised Mayo Clinic model (Mayo), previous prognostic models include histological staging.17, 18, 19, 20, 21 Although it is not unexpected that histology is a predictor of outcome, as surrogates of liver fibrosis such as enhanced liver fibrosis score and transient elastography perform equally well22, 23, a simpler prognostic scoring is warranted. The revised Mayo Clinic model that was published in 2000 was designed to predict short‐term survival within the proceeding 4 years and does not predict the need for transplantation.21 Updated scoring systems such as the Amsterdam‐Oxford model are designed to predict PSC‐related death and liver transplant, but demonstrate only moderate predictive power (C statistic = 0.68), likely attributable to its limited study‐cohort size.24
Given our ability to capture the clinical characteristics of a large, clinically representative cohort of patients with PSC through the UK National Institute for Health Research PSC (UK‐PSC) Rare Disease Translational Research Cohort, we sought to describe the clinical course of PSC and to identify clinical and genetic features early in the disease course that are associated with increased risk of transplantation or death. In doing so, our subsequent internationally validated findings provide clinically meaningful approaches to individualized risk prediction.
Materials and Methods
Study Design
Using data from patients recruited to the UK‐PSC research cohort (www.uk-psc.com), we evaluated participants with PSC who were at least 18 years of age with PSC incident or prevalent between August 1, 2008, and March 31, 2015, including liver transplant recipients who had undergone transplantation for PSC at any point before March 31, 2015. Participants were recruited from throughout the United Kingdom across a research network of 155 collaborating National Health Service (NHS) Trusts, including nearly every hospital providing general or specialist hepatology services in the United Kingdom, excluding Northern Ireland.
Inclusion criteria were based on accepted diagnostic criteria for PSC25 and included the presence of cholestatic liver biochemistry tests with characteristic bile duct changes on either endoscopic retrograde cholangio‐pancreatography, magnetic resonance cholangio‐pancreatography and/or liver histology. To address the challenges faced outside of clinical trials with comparing magnetic resonance images, the distinction between intrahepatic and extrahepatic biliary disease was determined by a team review of local radiographic reports of cholangiographic imaging, as opposed to a single expert review. Involvement of first‐order bile ducts (right or left main hepatic duct) and/or common bile duct at cholangiography were classified as extrahepatic biliary disease, as opposed to their absence being classified as intrahepatic biliary disease. Exclusion criteria included congenital abnormalities of the biliary tree, previous biliary surgery likely to cause secondary sclerosing cholangitis, primary bile duct carcinoma, human immunodeficiency virus cholangiopathy, PBC, positive antimitochondrial antibody, hepatic sarcoidosis, and drug‐induced liver injury.
The study was conducted in accordance with the guidelines of the Declaration of Helsinki and the principles of good clinical practice. All participants provided written informed consent. Multiregional Ethics Committee approval was granted by the Cambridgeshire 4 National Ethics Committee (No. 08/45/008) and by the research and development department of each collaborating hospital.
Data Capture
Data were collected using prespecified questionnaires through a systematic review of case notes between March 31, 2013, and March 31, 2015. Data included patient demographics, diagnostic cholangiography/histology reports, serial biochemistry at diagnosis (t0), 1‐year following diagnosis (t1) and 2‐years following diagnosis (t2), IBD status, concomitant autoimmune disease, use of ursodeoxycholic acid (UDCA), development of malignancy or liver decompensation, and progression to transplantation or death. Seventy‐four percent of all data was collected by the lead clinician researcher (E.C.G.) during site visits to each hospital, and completed questionnaires were reviewed by a second clinician researcher for accuracy and missing data. The remaining 26% of data was collected by the responsible clinician or research nurse at each hospital site, and reviewed by the lead clinician researcher (E.C.G.) following return. In cases in which patients were under the care of more than one hospital (e.g., a transplant center and a general hospital), the questionnaire was sent sequentially to each hospital to ensure complete data capture. Missing or inaccurate data were systematically queried with the clinician who had completed the questionnaire to ensure complete and accurate data capture. Data that passed quality control were uploaded into a bespoke secure database.
Study Entry and Outcome
We calculated the time from PSC diagnosis to outcome event. The value of t0 was defined as the date of first cholangiographic imaging or liver biopsy demonstrating PSC. The first primary endpoint was liver transplantation, chosen as an important hard outcome for which a definitive time point is easily available. In the context of liver disease, it can be difficult to accurately define deaths solely related to liver disease; therefore, the second primary endpoint of all‐cause mortality was chosen as the most encompassing term that would include all liver‐related deaths. Participants who did not reach an endpoint were censored at the date of their most recent blood tests or follow‐up. To be sure we captured all outcome events, we made use of the fact that every UK patient has a unique NHS number, which ensures that clinical coding is linked across primary, secondary, and tertiary care. This practice, in place throughout the 40‐year study follow‐up duration, ensures that the risk of missed events was minimal, and did not bias the analysis.
Explanatory Variables
We considered variables for their association with outcome and inclusion in the risk score, based on clinical relevance or pre‐existing evidence. To account for variability in the measurement of laboratory investigations, alkaline phosphatase (ALP) was taken as a ratio of the upper limit of normal range (ULN) for the reporting laboratory. Other laboratory measures used the following standard units of measurement: hemoglobin (g/L), platelet count × 109/L, albumin (g/L), and bilirubin (μmol/L).
Clinical Data Analysis
We calculated and reported descriptive statistics as numbers or percentages. Variables with more than 40% missing data were excluded from further analysis. For this reason, the following variables were omitted from the analysis: international normalized ratio, aspartate aminotransferase (AST), IgG subclasses, and date of first hepatic decompensation (e.g., ascites, hepatic encephalopathy, jaundice). Time‐to‐event analysis was conducted using Cox's proportional hazards model, ensuring at least 10 events per risk factor would be included in the model.26 To facilitate accurate risk prediction, events were truncated at 10 years of follow‐up. To ensure sufficient variation within the data set, categorical variables were only considered if the categories had more than 5% of the cohort in each category. Variables that were present in less than 5% of the cohort, and thus excluded from the analysis, were smoking status and variceal hemorrhage at t0.
We performed unadjusted/univariate analysis on the raw data set to demonstrate associations between risk factors and outcome. To account for missing data, we performed multivariable imputation using iteratively chained equations, combined the results of 10 imputed data sets using Rubin's equations, and estimated the adjusted/multivariable model using this imputed data set. We selected variables for the final risk score using backward elimination, with removal of risk factors not significant at the 10% level.27 Continuous variables were assessed for nonlinear association using cubic splines. Variables demonstrating a linear association were included in a standard, continuous fashion. Variables demonstrating a nonlinear association were categorized using cubic splines and clinical judgment to allow for ease of interpretation.
ALP
We analyzed the association between ALP at t1 and t2 with outcome, to determine the optimal threshold for predicting 10‐year hazard of outcome. ALP was divided into categorical variables from less than or greater than or equal to 0.5 to 4 × ULN, in increments of 0.1. We plotted each ALP cutoff against the hazard of reaching an endpoint. The optimal threshold for ALP was determined using Harrell's C statistic.
Derivation of the UK‐PSC Risk Scores
We derived three separate risk scores to determine the model with the best discrimination. The first was a score using t0 data to predict 10‐year risk of outcome, the second a short‐term risk score using t0 data to predict 2‐year risk of outcome (RSST), and the third, a long‐term risk score using t0 and t2 data to predict 10‐year risk of outcome (RSLT). The RSLT included only those patients not reaching a primary endpoint within 2 years of diagnosis. The discrimination of each score was compared using Harrell's C statistic. Calibration curves for RSST and RSLT were generated by creating deciles of data and comparing the model's predicted rates with the observed rates in the cohort, estimated by the Kaplan‐Meier curve.
Validation of the UK‐PSC Risk Scores
We used data from two external PSC patient cohorts that were not included in the original analysis, to validate the UK‐PSC risk scores: the first a national validation cohort (n = 352) from two UK hospitals (Transplant Center at Queen Elizabeth Hospital Birmingham and John Radcliffe Hospital, Oxford [a nontransplant center])24 and the second an international validation cohort from Norway (n = 99). Methods of validation cohort data collection were identical to the derivation cohort, and retrospective data from individual auditing of electronic and paper case notes by clinician researchers were followed by quality control. Validation of the scoring system was performed by fitting a Cox model to the validation cohort using the scoring system derived from the derivation cohort.28 Further visual validation was performed by displaying Kaplan‐Meier survival curves for risk groups in both cohorts.28 Risk groups were defined by dividing the derivation cohort into four equally sized groups with increasing RSLT, and the validation cohort divided into four groups according to the same RSLT categories.
Comparison of the UK‐PSC Score With Existing Scores
We analyzed the predictive ability of the modified Mayo risk score and AST‐to‐platelet ratio index (APRI) scores in both the derivation and validation cohorts, comparing them to the UK‐PSC risk scores using Harrell's C statistic. Both the Mayo and APRI score algorithms include AST. However, in most UK hospitals, AST is not measured as part of the standard liver biochemical tests; therefore, AST was not available for calculation of the Mayo risk or APRI scores. Other studies have demonstrated some equivalence of AST and ALT.29 Using a subset of patients for which both AST and ALT data were available at the same time points, we demonstrated the correlation and concordance between the two variables. We then used ALT in place of AST in the calculation of the Mayo risk score and APRI scores.
Genetic Data Analysis
Previous genotyping was conducted using the Illumina Immunochip,7 a targeted genotyping array with dense marker coverage across 186 known disease loci from 12 immune‐mediated diseases. We considered the following HLA risk alleles that are known to be associated with PSC disease risk from genome‐wide association studies6, 7: HLA‐B*08:01 and HLA‐DRB1*03:01, 04:01, 07:01, 13:01, and 15:01. The association between HLA risk alleles and outcome as analyzed using a test for trend across zero, one, and two copies of each allele. We also tested for association between significant risk alleles and important clinical variables. After applying a Bonferroni correction, our threshold for statistical significance was P < 0.008.
All analyses were performed using Stata software (version 14.0/SE; StataCorp, College Station, TX). This study was conducted and reported in accordance with TRIPOD (transparent reporting of a multivariate prediction model for individual prognosis or diagnosis).30
Results
Cohort Characteristics
A total of 1,749 patients were recruited to the UK‐PSC cohort; 1,252 questionnaires were distributed and 1,131 returned; 130 were excluded following quality control (Fig. 1), leaving 1,001 patients for analysis who were recruited from 108 hospitals, including 7 transplant centers. Fifty‐seven percent were recruited from non–transplant centers. The cohort (Table 1) included 64% males who were diagnosed at a median age of 46.8 years with median follow‐up of 14.8 years (range 0.2‐40.4), censored at the time of transplant. Forty‐four percent had extrahepatic biliary disease and 72.5% had concomitant IBD, most commonly ulcerative colitis (80.4%), and 14.3% of the cohort had another autoimmune disease. UDCA was prescribed for 58% of the cohort within the first 2 years following diagnosis.
Figure 1.
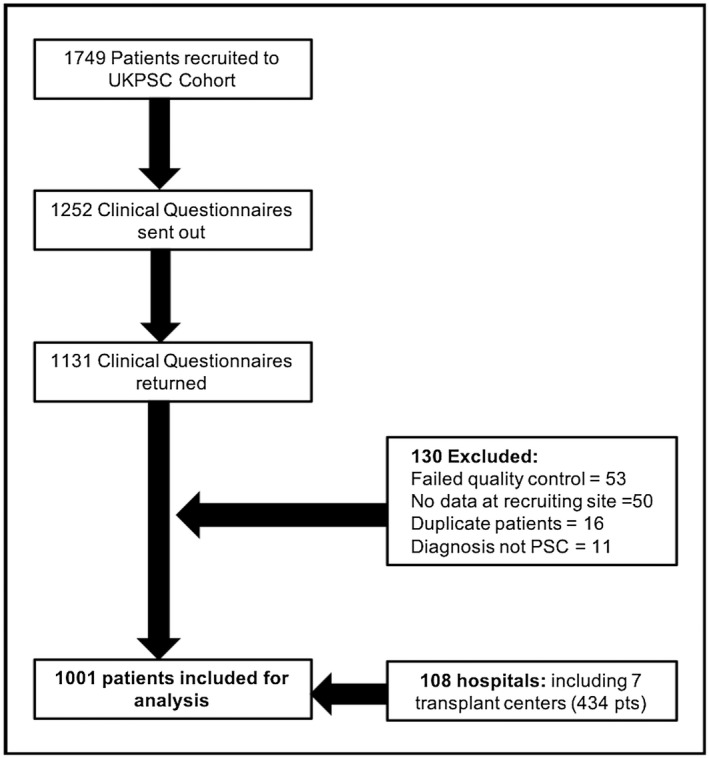
Study flow diagram.
Table 1.
Demographics of the UK‐PSC Derivation Cohort (n = 1,001), National Validation Cohort (n = 352), and International Validation Cohort (n = 99)
| Variable | Derivation Cohort | Validation Cohorts | ||
|---|---|---|---|---|
| n = 1,001 (%) | National n = 352 (%) | International n = 99 (%) | ||
| Demographics | Male | 63.8 | 62.4 | 75.7 |
| Mean age at diagnosis (years) | 46.8 | 45.0 | 35.0 | |
| Median age at transplant | 47 | 47.0 | 39.0 | |
| Median follow‐up (years) | 14.8 | 6.0 | 8.0 | |
| Disease distribution | Extrahepatic biliary disease present | 44.1 | 47.8 | 33.3 |
| IBD | IBD | 72.5 | 71.0 | 86.0 |
| Ulcerative colitis | 80.4 | 73.6 | 77.6 | |
| Crohn's colitis | 14.2 | 10.7 | 15.3 | |
| Indeterminate colitis | 5.4 | 3.2 | 7.1 | |
| Autoimmune disease | Autoimmune disease | 14.3 | — | — |
| Thyroid disease | 6.9 | — | — | |
| Rheumatoid arthritis | 2.3 | — | — | |
| Celiac disease | 2.0 | — | — | |
| Other | 6.2 | — | — | |
| Smoking status | Never smoked | 53.2 | — | — |
| Ex‐smoker | 26.5 | — | — | |
| Current smoker | 3.7 | — | — | |
| Events | Total events | 35.7 | 39.2 | 32.3 |
| Transplants | 27.8 | 13.9 | 11.1 | |
| Deaths (all causes) | 7.9 | 25.3 | 21.2 | |
| Cancers | Gastrointestinal cancer | 10.7 | — | — |
| Colorectal | 5.4 | — | — | |
| Cholangiocarcinoma | 3.3 | — | — | |
| Gall bladder | 1.3 | — | — | |
| Hepatocellular carcinoma | 0.6 | — | — | |
| Pancreatic | 0.1 | — | — | |
| UDCA use | Taking UDCA at year 2 | 57.8 | — | — |
| Median dose (mg/kg) | 11.4 | — | — | |
| Range (mg/kg) | 2.2‐46.8 | — | — | |
A total of 35.7% of patients reached a primary endpoint over a cumulative follow‐up period of 7,904 years; 27.8% underwent liver transplantation at a median age of 47.0 years; 7.9% died without a transplant; and 47.8% of all deaths were PSC‐related. The overall proportion of the cohort who were event‐free at 2, 5, and 10 years was 92%, 82% and 64%, respectively. Thirty‐nine percent of men reached an outcome, compared with 29% of females (χ2 = 10.07, P = 0.002), and 43% of those with extrahepatic biliary disease reached an outcome compared with 23% of those without (χ2 = 40.6, P = 0.00). Patients with extrahepatic biliary disease had a reduced median transplant‐free survival compared with those without extrahepatic biliary disease (11.7 years versus 23 years). UDCA use in the first 2 years following diagnosis was not associated with outcome. Eleven percent of patients developed a gastrointestinal cancer, most commonly colorectal (5.4%), followed by cholangiocarcinoma (3.3%).
Serum ALP is associated with PSC outcome
ALP data at t1 and t2 were available for 72% and 70% of the cohort, respectively. At both time points, elevated ALP was associated with an increased 10‐year hazard of reaching an outcome (P ≤ 0.001) (Fig. 2A,B). There was a log‐linear association between serum ALP and outcome; however, for ease of interpretation we chose to categorize ALP using cubic splines (Supporting Fig. S1). At t1, the optimal threshold for predicting 10‐year outcome was ALP ≥ 2.4 × ULN (HR = 3.05, C = 0.63) (Fig. 2C), where median transplant‐free survival was 63 versus 108 months for those with ALP < 2.4 × ULN (P < 0.0001 [log‐rank test]) (Fig. 2E). At t2, the optimal threshold for predicting 10‐year outcome was ALP ≥ 2.2 × ULN (HR = 3.05, C = 0.66) (Fig. 2D), where median survival was 44 versus more than 96 months for those with a t2 ALP < 2.2 × ULN (P < 0.0001 [log‐rank test]) (Fig. 2F).
Figure 2.
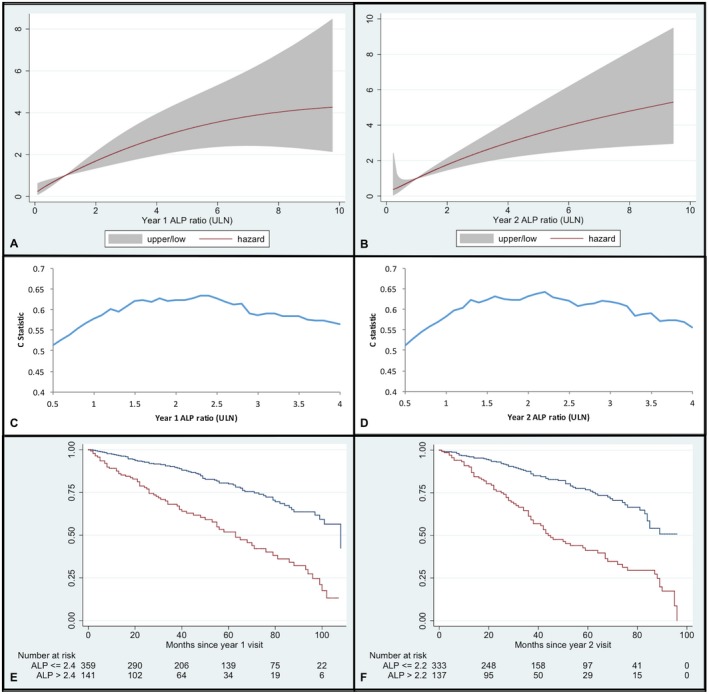
Predictive value of ALP and outcome. (A) Association between ALP (as ratio of ULN) at year 1 and hazard of reaching a clinical endpoint within 10 years, with 95% CI. (B) Association between ALP (as ratio of ULN) at year 2 and hazard of reaching a clinical endpoint within 10 years, with 95% CI. (C) Harrell's C statistic per ALP cutoff point at year 1 for 10‐year hazard of outcome. (D) Harrell's C statistic per ALP cutoff point at year 2 for 10‐year hazard of outcome. (E) Kaplan‐Meier survival curve for transplant‐free survival in patients with ALP 2.4 × ULN (blue line) versus ALP > 2.4 × ULN (red line) at 1 year following diagnosis (0 = 12 months after diagnosis). (F) Kaplan‐Meier survival curve for transplant‐free survival in patients with ALP 2.2 × ULN (blue line) versus ALP > 2.2 × ULN (red line) at 2 years following diagnosis (0 = 24 months after diagnosis).
Disease distribution is associated with outcome in PSC
Cholangiographic data at t0 were available in 87.2% of the cohort. Presence of extrahepatic biliary disease was associated with adverse outcome (HR = 1.45 (confidence interval [CI] 1.09, 1.92), P = 0.010). Patients without extrahepatic biliary disease had improved 10‐year event‐free survival, although more than 50% of both groups were event‐free at 10 years; thus, the median survival was not reached (Supporting Fig. S2).
Derivation of the UK‐PSC Risk Scores
Our first UK‐PSC risk score used variables available at t0 to predict 10‐year risk of outcome. Following multivariable analysis, seven factors were included in the score: age at t0, bilirubin, ALP, albumin, hemoglobin, platelets, and presence of extrahepatic biliary disease a t0 (Supporting Table S1) (C = 0.78, shrinkage = 0.94). Our cohort demonstrated a high event rate (8%) within the first 2 years of diagnosis. Therefore, to determine whether variables predicting short‐term and long‐term risk differed, we derived a short‐term risk score (RSST) using variables at t0 to predict the risk of outcome within 2 years following diagnosis, and a long‐term risk score (RSLT) using variables from t2 to predict 10‐year risk of outcome (see Box). Mean serum ALT, platelet count, and ALP ratio were all significantly reduced from t0 to t2 (Supporting Table S2). Both RSST and RSLT demonstrated improved predictive ability over the original model.
BOX 1
Short‐Term UK‐PSC Risk Score (RST) = 0.745(Bili_t0 Group 1 [0/1] + 1.613(Bili_t0 Group 2 [0/1]) – 0.061(Alb_t0 [g/l]) – 0.012(Hb_t0 [g/l]) – 0.476(Plts_t0 Group 1 [0/1]) – 0.698(Plts_t0 Group 2 [0/1]) – 0.962(Plts_t0 Group 3 [0/1]).
Long‐Term UK‐PSC Risk Score (RSLT) = 0.026(Age_t0[yrs]) + 1.197(Bili_t2 Group 1 [0/1]) + 1.38(Bili_t2 Group 2 [0/1]) + 0.4(ALP_t2 Group 1 [0/1]) + 0.45(ALP_t2 Group 2 [0/1]) –0.07(Alb_t2[g/l]) –0.543(Plts_t2 Group 1) – 0.503(Plts_t2 group 2) – 0.768(Plts_t2 Group 3 [0/1]) + 0.524(disease type_t0 [0/1]) + 1.014(variceal bleed_t2 [0/1]).
Bili_t0/t2 group 1: 0 = Bili_t0 < 35 μmol/L or > 50 μmol/L, 1 = 35 to ≤ 50 μmol/L
Bili_t0/t2 group 2: 0 = Bili_t0 < 50 μmol/L, 1 = Bili_t0 ≥ 50 μmol/L
Plts_t0/t2 group 1: 0 = Plts_t0 < 150 × 109/L, or ≥ 200 × 109/L, 1 = Plts_t0 150 to < 200 × 109/L
Plts t0/t2 group 2: 0 = < 200 or ≥ 400 × 109/L, 1 = 200 to < 400 × 109/L
Plts_t0/t2 group 3: 0 = < 400 × 109/L, 1 = ≥ 400 × 109/L
ALP_t2 group 1: 0 = ALP_t2 < 1.5 × ULN or ≥ 2.5 × ULN, 1 = 1.5 to < 2.5
ALP_t2 group 2: 0 = ALP_t2 < 2.5 × ULN, 1 = ≥ 2.5 × ULN
Disease type_t0: 0 = no extrahepatic biliary disease; 1 = presence of extrahepatic biliary disease
Variceal bleed_t2: 0 = no bleed by t2; 1 = bleed by t2
Predicted survival rate at time t: (baseline survival at time t) ^ exp (RSST or RSLT)
RSST baseline survival at time t: 1 year: 0.0096612; 2 years: 0.0001109
RSLT baseline survival at time t: 1 year: 0.9218476; 2 years: 0.8227174; 5 years: 0.7070919; 8 years: 0.2771266
Example
An individual aged 47 and with no evidence of extrahepatic disease at diagnosis presented with the following biochemistry at t0: bilirubin 37 μmol/L, albumin 34 g/L, hemoglobin 130 g/L, and platelets 245 × 109/L; and the following biochemistry at t2: bilirubin 24 μmol/L, ALP 2 × ULN, albumin 30 g/L, platelets 152 × 109/L, and no variceal bleed by t2. This would score RSST = (0.745 × 1) – (0.061 × 34) – (0.012 × 130) – (0.698 × 1) = –3.587. Predicted event‐free survival rate at 2 years = (0.0001109)^exp (–3.587) = 0.78 = 78%. Long‐term risk score would be RSLT = (0.026 × 47) + 0.403 + (–0.07 × 30) – 0.543 = –1.018. Predicted event‐free survival rate at 5 years = 0.707^exp (–1.047) = 0.885 = 88.5%.
The UK‐PSC risk scores are available at http://www.uk-psc.com/riskscores .
Short‐Term UK‐PSC Risk Score (RSST)
Four variables at t0 were associated with 2‐year outcome: bilirubin, albumin, hemoglobin, and platelet count (Table 2). Based on these coefficients, a prognostic model was developed to predict the risk of death or liver transplantation by year 2 (C = 0.81, shrinkage = 0.92) (Box 1).
Table 2.
Univariate Analysis Using Unimputed Data, and Multivariate Analysis Using Imputed Data, of Factors at Diagnosis Associated With 2‐Year Risk of Transplantation or Death
| Factor | Univariate Analysis | Multivariate Analysis | ||
|---|---|---|---|---|
| HR (95% CI) | P Value | HHR (95% CI) | P Value | |
| Female | 0.88 (0.54, 1.42) | 0.596 | ||
| Age at diagnosis | 1.01 (1.00, 1.03) | 0.126 | ||
| Extrahepatic biliary disease | 1.30 (0.77, 2.21) | 0.332 | ||
| IBD presence | 1.09 (0.49, 2.44) | 0.832 | ||
| Ulcerative colitis | 1.12 (0.67, 1.89) | 0.665 | ||
| Crohn's disease | 0.39 (0.12, 1.31) | 0.127 | ||
| Indeterminate colitis | 1.38 (0.47, 4.03) | 0.560 | ||
| Autoimmune disease | 0.90 (0.46, 1.75) | 0.757 | ||
| Smoker | 1.22 (0.74, 2.02) | 0.426 | ||
| Bilirubin (μmol/L) | ||||
| 35‐49 | 4.03 (1.36, 11.98) | 0.012 | 2.11 (0.74, 5.96) | 0.159 |
| 50+ | 14.12 (7.89, 25.3) | < 0.001 | 5.02 (2.76, 9.13) | < 0.001 |
| ALP (ratio of ULN) | ||||
| 1.5 to < 2.5 | 1.25 (0.49, 3.17) | 0.634 | ||
| 2.5+ | 2.64 (1.35, 5.17) | 0.005 | ||
| ALT (IU/L)* | 1.02 (0.98, 1.05) | 0.331 | ||
| Albumin (g/L) | 0.87 (0.84, 0.90) | < 0.001 | 0.94 (0.90, 0.99) | 0.011 |
| Hemoglobin (g/L)† | 0.98 (0.97, 0.99) | < 0.001 | 0.99 (0.97, 1.00) | 0.095 |
| Platelets group (×109/L) | ||||
| 150‐199 | 0.23 (0.08, 0.72) | 0.011 | 0.62 (0.26, 1.48) | 0.283 |
| 200‐399 | 0.22 (0.11, 0.45) | < 0.001 | 0.50 (0.25, 0.98) | 0.045 |
| 400+ | 0.32 (0.13, 0.78) | 0.012 | 0.38 (0.15, 0.98) | 0.046 |
| Eosinophils (×109/L) | 1.10 (0.89, 1.36) | 0.368 | ||
| Sodium (mmol/L) | 0.89 (0.82, 0.98) | 0.015 | ||
| Creatinine > 120 (µmol/L) | 4.21 (1.66, 10.68) | 0.002 | ||
| IgG (g/L)* | 1.08 (0.93, 1.25) | 0.313 | ||
Denotes HR for a 10‐unit change.
Denotes HR for a 1‐unit change.
Long‐Term UK‐PSC Risk Score (RSLT)
Seven variables at t2 were associated with 10‐year risk of outcome: age at diagnosis, bilirubin at t2, ALP at t2, albumin at t2, platelets at t2, presence of extrahepatic biliary disease at t0, and variceal hemorrhage by t2 (C = 0.80, shrinkage = 0.96) (Table 3 and Box 1). Calibration of RSST and RSLT using predicted versus observed survival rates estimated by Kaplan‐Meier curve demonstrated good correlation. The scores are available at http://www.uk-psc.com/riskscores.
Table 3.
Univariate Analysis Using UnImputed Data, and Multivariate Analysis Using Imputed Data, of Factors at Year 2 Associated With 10‐Year Risk of Transplantation or Death
| Factor | Univariate Analysis | Multivariate Analysis | ||
|---|---|---|---|---|
| HR (95% CI) | P Value | HHR (95% CI) | P Value | |
| Female | 0.81 (0.60, 1.10) | 0.181 | ||
| Age at diagnosis | 1.01 (1.00, 1.03) | 0.005 | 1.03 (1.01, 1.04) | < 0.001 |
| Extrahepatic biliary disease | 1.95 (1.42, 2.69) | < 0.001 | 1.70 (1.15, 2.48) | 0.008 |
| IBD | 0.91 (0.59, 1.38) | 0.646 | ||
| Ulcerative colitis | 0.92 (0.70, 1.22) | 0.558 | ||
| Crohn's disease | 0.68 (0.41, 1.11) | 0.122 | ||
| Indeterminate colitis | 1.28 (0.71, 2.31) | 0.416 | ||
| Autoimmune disease | 1.27 (0.88, 1.83) | 0.200 | ||
| Smoker | 0.96 (0.70, 1.32) | 0.790 | ||
| Bilirubin (μmol/L) | ||||
| 35‐49 | 6.77 (3.87, 11.85) | < 0.001 | 3.31 (1.65, 6.62) | 0.001 |
| 50+ | 7.92 (5.62, 11.18) | < 0.001 | 3.96 (2.37, 6.62) | < 0.001 |
| ALP (ratio of ULN) | ||||
| 1.5‐2.4 | 1.75 (0.98, 3.15) | 0.061 | 1.50 (1.09, 2.30) | 0.015 |
| 2.5+ | 1.40 (1.04, 1.88) | 0.025 | 1.57 (1.12, 2.52) | 0.011 |
| ALT (IU/L)* | 1.05 (1.03, 1.08) | < 0.001 | ||
| Albumin (g/L) | 0.88 (0.85, 0.90) | < 0.001 | 0.93 (0.90, 0.96) | < 0.001 |
| Hemoglobin (g/L)* | 0.75 (0.69, 0.81) | < 0.001 | ||
| Platelets group (×109/L) | ||||
| 150‐199 | 0.35 (0.20, 0.60) | < 0.001 | 0.58 (0.31, 1.10) | 0.092 |
| 200‐399 | 0.29 (0.20, 0.43) | < 0.001 | 0.60 (0.40, 0.91) | 0.016 |
| 400+ | 0.32 (0.17, 0.60) | < 0.001 | 0.46 (0.23, 0.92) | 0.028 |
| Eosinophils (×109/L) | 0.81 (0.52, 1.29) | 0.380 | ||
| Sodium (mmol/L) | 0.90 (0.96, 0.93) | < 0.001 | ||
| Creatinine > 120 (µmol/L) | 0.66 (0.21, 2.07) | 0.474 | ||
| IgG (g/L)* | 1.01 (0.92, 1.12) | 0.774 | ||
| UDCA use | 0.96 (0.72, 1.28) | 0.795 | ||
| Variceal bleed by year 2 | 5.97 (2.93, 12.16) | < 0.001 | 2.76 (1.14, 6.66) | 0.024 |
Denotes HR for a 10‐unit change.
To define low‐risk and high‐risk disease groups according to RSLT, we divided the derivation cohort into four equal quartiles. Event‐free survival, plotted on a Kaplan‐Meir survival curve (Fig. 3A), demonstrated observed event rates of 6.0%, 8.4%, 19.1%, and 55.8% in the four respective risk groups. Curves were generally well separated, although the model was less able to distinguish between the two lowest risk groups. The value of RSLT, which defines the four risk groups, is provided in Supporting Table S3 .
Figure 3.
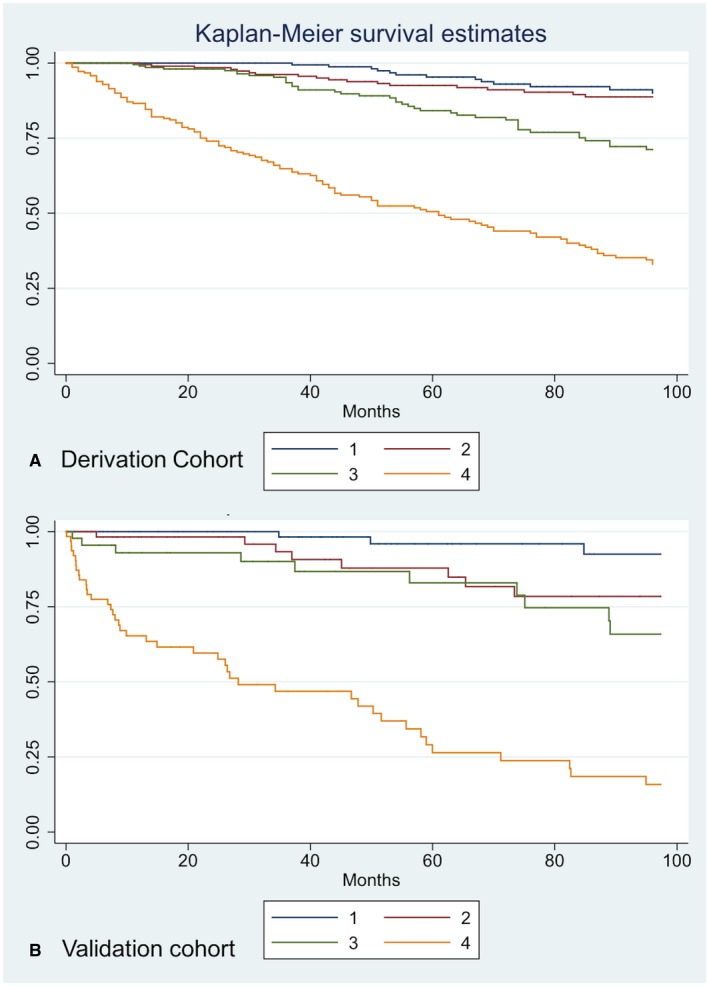
Kaplan‐Meir survival curves for four risk groups. Risk group 1: RSLT > −2.019879 (blue line); risk group 2: −1.463874 < RSLT < −2.019879 (red line); risk group 3: −0.8146346 < RSLT < −1.463874 (green line); risk group 4: 2.737384 < RSLT < −0.8146346 (orange line). (A) Derivation cohort Kaplan‐Meir survival curves for four risk groups. (B) Validation cohort Kaplan‐Meir survival curves for four risk groups.
Validation of the UK‐PSC Scores
We analyzed the predictive ability of both risk scores in a separate national and international patient cohort. In the respective national and international validation cohorts (Table 1), 62.4% and 75.7% of the cohort were male, diagnosed at a median age of 47 and 39 years, with 71% and 86% diagnosed with concomitant IBD. The most notable differences between the derivation and validation cohorts were the shorter median follow‐up (6 years and 8 years in the national and international cohorts, versus 14.8 years), higher death rate (25.3% and 21.2% versus 7.9%), and lower transplant rate (13.9% and 11.1% versus 27.8%).
Both the RSST and RSLT were associated with outcome in the national validation cohort (P < 0.001). The slope of the Cox model for the RSST in the validation cohort was 1.09, which is not significantly different from 1, indicating that the discrimination was preserved. The slope for the RSLT was 1.36 (P = 0.0071), which is significantly different from 1, suggesting that the score is more predictive of outcome in the validation than the derivation cohort. In the international validation cohort, the lack of events within the first 2 years meant that only the RSLT could be validated. The RSLT was associated with outcome in the international validation cohort (P < 0.001): The slope was 1.60 and not significantly different from 1 (P = 0.014), indicating preserved discrimination, although this was based on only 37 individuals.
Further visual validation of the RSLT was performed by comparing Kaplan‐Meier survival curves for the validation cohort according to the same four previously defined risk groups as the derivation cohort (Fig. 3A,B). Event rates were similar to the derivation cohort at 2.9%, 10.4%, 20.0%, and 47.9% (Supporting Table S3). Both sets of four curves were quite well separated, confirming that the model had discrimination in both cohorts; however, the model was less able to distinguish between the two intermediate risk groups in the validation cohort.
Comparison of UK‐PSC Score With Existing Scores
We compared the predictive accuracy of the Mayo and APRI scores to the RSST and RSLT in the imputed derivation data set. Based on a subset of 170 patients from the validation cohort, for which both AST and ALT measurement were available for t0 and t2, there was a strong correlation (r = 0.94, P < 0.0001) and strong concordance (C = 0.92, P < 0.0001) between the two variables. ALT was therefore used in place of AST for calculation of the Mayo and APRI scores. In predicting 2‐year outcome, the RSST out‐performed the APRI and Mayo scores with C statistics of 0.81, 0.63, and 0.75, respectively. In predicting 10‐year outcome the RSLT demonstrated an incremental improvement over the APRI and Mayo scores with C statistics of 0.80, 0.59, and 0.79, respectively.
We then compared the predictive accuracy of the Mayo and Model for End‐Stage Liver Disease (MELD) scores to the RSST and RSLT in the validation data set. In predicting 2‐year outcome, the RSST out‐performed the Mayo and MELD scores with C statistics of 0.81, 0.73, and 0.78, respectively. In predicting 10‐year outcome, the RSLT demonstrated a markedly improved predictive accuracy compared with Mayo and MELD, with C statistics of 0.85, 0.69, and 0.70, respectively.
HLA Risk Alleles Are Associated With Outcome
THE HLA genotype was available for 635 patients. Twenty‐seven percent and 9% of the cohort were heterozygous and homozygous for the HLA‐DR*03:01 risk allele, respectively. Presence of this allele was associated with outcome in a dose‐dependent manner (HR = 1.33, CI 1.13, 1.58, P = 0.001) (Supporting Table S4). After testing for association between HLA‐DR*03:01 and clinical characteristics at diagnosis, we found HLA‐DR*03:01 risk alleles to be inversely correlated with mean age at diagnosis (no copies = 47.6 years, heterozygote = 46.6 years, homozygote = 40.8 years, P = 0.007) (Supporting Table S5). The addition of the HLA‐DR*03:01 risk allele to the risk score did not improve the discrimination of the model. No association was observed with HLA‐B*08, HLA‐DR*04:01, 07:01, 13:01, or 15:01.
Discussion
Using a large cohort of 1,001 patients from across the entire United Kingdom, including 57% recruited from non–transplant centers, representing the full spectrum of PSC disease severity, we provide important, externally validated clinical and genetic modeling based on readily available clinical factors for prediction of short‐term and long‐term outcome. Based on the presence of extrahepatic biliary disease at t0, age, bilirubin, ALP, albumin and platelets at t2, and variceal hemorrhage by t2, we present a scoring system that has value both in individual risk evaluation as well as in being a potential mechanism to stratify recruitment to clinical trials.
Our study confirms the importance of ALP as a prognostic indicator, both individually and as part of our RSLT. We demonstrate that ALP < 2.4 × ULN and < 2.2 × ULN at 1 and 2 years following diagnosis respectively, is associated with improved transplant‐free survival. Understanding the behavior of ALP as a biomarker in PSC is of interest, and parallels interest in PBC. In PBC, dichotomous risk scores have C statistics of approximately 0.6, with the dynamic scores reporting C statistics of 0.8 and above.31, 32 Although in PSC, the serum ALP was not associated with short‐term outcome, the prognostic importance as a longer‐term predictor of clinical events is highlighted by its inclusion in our long‐term risk score. This may be explained by fluctuations in ALP in the early stages of diagnosis, which limit its prognostic value, and the rationale that short‐term risk is driven by factors that measure cholestasis and portal hypertension. Thus, when considering ALP in isolation, we chose to consider ALP at t1 and t2 rather than t0, and used this to predict long‐term risk rather than short‐term risk. In addition, our study demonstrated the poor prognostic effect of extrahepatic biliary disease, highlighting the importance of further study into cholangiographic monitoring in PSC. Although simple cholangiographic imaging used at diagnosis does carry meaningful prognostic data, improving cholangiographic evaluation remains important.
We observed a high event rate (8%) within the first 2 years following diagnosis, suggesting that there is a patient cohort who present late in disease course, or who experience rapidly progressive disease. Recognizing this, we developed separate risk scores for short‐term (RSST) and long‐term (RSLT) prediction, the key differences between them being that the former includes only laboratory parameters (bilirubin, albumin, hemoglobin, and platelets), suggesting that intrinsic liver function is most important in predicting immediate outcome. Conversely the RSLT includes laboratory factors (bilirubin, albumin, platelets, and ALP) in addition to variceal bleeding and cholangiographic disease distribution. By using a dichotomous approach to risk stratification, we improved the predictive utility from C = 0.78 with our original score to C = 0.80 and 0.81 for short‐term prediction, and C = 0.80 and 0.85 for long‐term prediction, in the derivation and validation cohorts, respectively. In practice, this would allow clinicians to recalculate risk at 2 years following diagnosis for greater prognostic accuracy.
There remains a risk of better performance of our model, due to data fitting; this is a risk that we acknowledge. We tried to address this by comparing our risk scores versus existing scores, including the Mayo score. Although we reconfirm the Mayo risk score's prognostic value, it was out‐performed by our RSST and RSLT, which also confer several other advantages. Even though the Mayo score is based on the parameters of age, bilirubin, AST, variceal bleeding and albumin, the UK‐PSC risk scores consider more aspects of disease progression, including age, ALP, albumin, platelets, extrahepatic biliary disease, and variceal hemorrhage. The Mayo risk score predicts only 4‐year risk of all‐cause mortality and does not provide a strong long‐term predictor of outcome. It performs best in patients with end‐stage liver disease and does not consider the important outcome of liver transplantation. In comparison, the dichotomous UK‐PSC risk scores predict short‐term (2‐year) and long‐term (10‐year) outcomes, ensuring that the predictive ability is as good for those patients presenting with early‐stage, as well as late‐stage, liver disease, and includes the important outcome of liver transplantation in addition to all‐cause mortality.
We found that HLA‐DR*03:01, a previously validated HLA risk allele, was an important predictor of disease outcome, demonstrating a gene‐dose effect; however, the addition of HLA‐DR*03:01 did not improve the predictive ability of our prognostic score. When considering effect size, HLA‐DR*03:01 had an adjusted HR of 1.33 (CI 1.13, 1.58, P = 0.001) for outcome, which is more comparable to that of extrahepatic biliary disease or ALP, in comparison to the strongest associated clinical variable, bilirubin, which had an adjusted odds ratio of 3.96 (CI 2.4, 6.9, P < 0.001).
A strength of our cohort is the representative nature of the participants, notably identifying low‐risk as well as high‐risk patients. In our study, we could define a “low‐risk” disease group (patients with a RSLT of ≤ 2.02 had a less than 10% chance of an event by their 10‐year follow‐up) and a “high‐risk” group (patients with RSLT −0.81 < RSLT < 2.74 had an approximate 50% chance of an event by 10 years). Both UK‐PSC risk scores (http://www.uk-psc.com/riskscores) were well validated in a separate patient cohort. The major differences between the derivation and validation cohorts were a lower death and higher transplant rate in the former. There are some biases in our derivation cohort, reflective of ascertainment processes. Recruitment to the UK‐PSC derivation cohort was retrospective through prevalent case ascertainment. Recruitment was therefore inherently biased toward those patients with or without transplant, who survived to 2008 to be recruited to the study, compared with those patients who died before 2008. Furthermore, there was a low prevalence of cholangiocarcinoma (3.3%) in our cohort. Retrospective cohort recruitment is not well‐suited to capturing data on outcome markers associated with very poor survival; nearly 50% of all PSC‐associated cholangiocarcinomas manifest within 2 years of PSC diagnosis.2 Despite these limitations, the UK‐PSC risk scores were nationally and internationally well‐validated in two external cohorts, lending weight to their importance as robust prognostic models.
Retrospective data collection also carries the inherent drawback of incomplete data collection. Not surprisingly, rates of missing data were higher for patients diagnosed many years previously. Given that it was only related to the year of diagnosis, we considered these data to be missing at random, and used imputation to improve the validity of the results. Although a date of diagnosis before 1990 was associated with an improved transplant‐free survival, removal of these patients from the analysis did not alter the strength of the reported associations with either short‐term or long‐term outcome; thus, they were retained for the purpose of statistical power.
In our study, we did not observe any differences in outcome according to sex or subtype of IBD. The evidence for female sex and Crohn's disease conferring a favorable outcome in patients with PSC has only been robustly supported by evidence from one large study that included more than 7,000 patients with PSC.2 It is therefore likely that with a total sample size of 1,452, our study was underpowered to detect any such effect. Further studies of even larger cohorts, adjusted for multiple factors, are needed to confidently validate this finding. In particular, this necessitates careful, consistent classification of PSC‐associated IBD.
We chose the endpoints of all‐cause mortality and liver transplantation rather than hepatic decompensation (e.g., ascites, hepatic encephalopathy), because they provide a definitive and easily identifiable event and time of event that are necessary for a retrospective observational study. In comparison, hepatic decompensation can remain undiagnosed for several months and the precise date of diagnosis remains subjective; such issues are less relevant in well‐designed clinical trial/prospective cohort settings in which such endpoints are clearly meaningful and can be collected robustly. Arguably, there are challenges with our chosen endpoints due to possible variation in clinical practice and outcomes over time. Indeed, it must be acknowledged that changes in disease course over the time period of a study is a potential confounding factor. Evaluating such changes can be hard: For example, contemporaneous reference literature may not reflect clinical changes to a disease manifestation, but report and investigate changes in clinical practices. Despite PSC being infrequent overall, and the difficulty in recruiting large cohorts for the development of well‐powered evaluations, we have collated a unique data set that captures approximately 15% of the total UK PSC population, with substantial power to evaluate all‐cause mortality and transplantation. Although accepting limitations inherent to our approach, there is no evidence to date that PSC outcomes have varied over the time course of our study, simply on the basis of era, and recent data reporting liver transplant practice in the United Kingdom additionally supports this.33 Our approach is of significance to clinicians, as the risk score analyses best reflect collective real‐world clinical practice, with a focus on a spectrum of patients reflective of the disease. Moreover, the study was designed so that, through a large cohort size, it could accommodate the weaknesses introduced by non‐trial‐based uniform evaluation and data capture.
In conclusion, our analyses, which are based on a detailed clinical evaluation of a large representative cohort of participants with PSC, have furthered our understanding of clinical risk markers for predicting outcome in patients with PSC.
Author Contributions
E.C.G., G.M.H., and S.M.R. were involved in the study concept design and oversight. R.N.S., G.J.A., R.W.C., and M.W. contributed to the overall study inception and setup. E.C.G. and A.B.C. were involved in the statistical analysis. B.S., G.F.M., and W.T.H.G. were involved in the database design and management. G.F.M., P.J.T., K.D.W., M.N.V., E.C., T.H.K., S.G.J., D.T., M. Hudson, M. Heneghan, M.A.A., A.B., G.M.H., S.M.R., and C.A.A. contributed data to the study. E.C.G., A.B.C., G.M.H., and S.M.R. wrote the manuscript and all of its revisions; all authors approved the submission.
Supporting information
Acknowledgment
The authors gratefully acknowledge the work of the UK‐PSC Consortium members. We thank all participants who granted access to their medical records, enabling us to conduct this study. Dr Muhammad F Dawwas was responsible for collation of data pertaining to those patients with PSC who underwent liver transplantation. The UK‐PSC study is a portfolio study of the National Institute for Health Research Comprehensive Research Network. The views expressed are those of the authors and not necessarily those of the National Health Service, the National Institute for Health Research, or the Department of Health. We thank the ongoing support of the UK patient support group PSC Support.
Supported by the National Institute of Health Research (RD‐TRC and Birmingham Biomedical Research Centre); Isaac Newton Trust; Addenbrooke's Charitable Trust; Norwegian PSC Research Center; PSC Support; Lily and Terry Horner Chair in Autoimmune Liver Disease Research (to G.M.H.). E.C.G. is supported by a fellowship from the Wellcome Trust.
Potential conflict of interest: Dr. Bathgate received grants from Gilead. Dr. Hirschfield consults and received grants from Intercept; he consults for GlaxoSmithKline, Novartis, and Cymabay; he received grants from Gilead (to continue subsequent unrestricted support of the UK‐PSC cohort) and Falk. Dr. Rushbrook advises Falk and Intercept. Dr. Sandford received grants from Intercept. Dr. Vesterhus advises Intercept.
References
Author names in bold designate shared co‐first authorship.
- 1. Bergquist A, Ekbom A, Olsson R, Kornfeldt D, Loof L, Danielsson A, et al. Hepatic and extrahepatic malignancies in primary sclerosing cholangitis. J Hepatol 2002;36:321‐327. [DOI] [PubMed] [Google Scholar]
- 2. Weismuller TJ, Trivedi PJ, Bergquist A, Imam M, Lenzen H, Ponsioen CY, et al. Patient age, sex, and inflammatory bowel disease phenotype associate with course of primary sclerosing cholangitis. Gastroenterology 2017;152:1975‐1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sano H, Nakazawa T, Ando T, Hayashi K, Naitoh I, Okumura F, et al. Clinical characteristics of inflammatory bowel disease associated with primary sclerosing cholangitis. J Hepatobiliary Pancreat Sci 2011;18:154‐161. [DOI] [PubMed] [Google Scholar]
- 4. Claessen MM, Vleggaar FP, Tytgat KM, Siersema PD, van Buuren HR. High lifetime risk of cancer in primary sclerosing cholangitis. J Hepatol 2009;50:158‐164. [DOI] [PubMed] [Google Scholar]
- 5. Razumilava N, Gores GJ, Lindor KD. Cancer surveillance in patients with primary sclerosing cholangitis. Hepatology 2011;54:1842‐1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Karlsen TH, Franke A, Melum E, Kaser A, Hov JR, Balschun T, et al. Genome‐wide association analysis in primary sclerosing cholangitis. Gastroenterology 2010;138:1102‐1111. [DOI] [PubMed] [Google Scholar]
- 7. Liu JZ, Hov JR, Folseraas T, Ellinghaus E, Rushbrook SM, Doncheva NT, et al. Dense genotyping of immune‐related disease regions identifies nine new risk loci for primary sclerosing cholangitis. Nat Genet 2013;45:670‐675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ji SG, Juran BD, Mucha S, Folseraas T, Jostins L, Melum E, et al. Genome‐wide association study of primary sclerosing cholangitis identifies new risk loci and quantifies the genetic relationship with inflammatory bowel disease. Nat Genet 2017;49:269‐273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Trivedi PJ, Corpechot C, Pares A, Hirschfield GM. Risk stratification in autoimmune cholestatic liver diseases: opportunities for clinicians and trialists. Hepatology 2016;63:644‐659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Broome U, Lofberg R, Veress B, Eriksson LS. Primary sclerosing cholangitis and ulcerative colitis: evidence for increased neoplastic potential. Hepatology 1995;22:1404‐1408. [DOI] [PubMed] [Google Scholar]
- 11. Bjornsson E, Chari S, Silveira M, Gossard A, Takahashi N, Smyrk T, et al. Primary sclerosing cholangitis associated with elevated immunoglobulin G4: clinical characteristics and response to therapy. Am J Ther 2011;18:198‐205. [DOI] [PubMed] [Google Scholar]
- 12. Bjornsson E, Boberg KM, Cullen S, Fleming K, Clausen OP, Fausa O, et al. Patients with small duct primary sclerosing cholangitis have a favourable long term prognosis. Gut 2002;51:731‐735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Al Mamari S, Djordjevic J, Halliday JS, Chapman RW. Improvement of serum alkaline phosphatase to <1.5 upper limit of normal predicts better outcome and reduced risk of cholangiocarcinoma in primary sclerosing cholangitis. J Hepatol 2013;58:329‐334. [DOI] [PubMed] [Google Scholar]
- 14. Lindstrom L, Hultcrantz R, Boberg KM, Friis‐Liby I, Bergquist A. Association between reduced levels of alkaline phosphatase and survival times of patients with primary sclerosing cholangitis. Clin Gastroenterol Hepatol 2013;11:841‐846. [DOI] [PubMed] [Google Scholar]
- 15. Stanich PP, Bjornsson E, Gossard AA, Enders F, Jorgensen R, Lindor KD. Alkaline phosphatase normalization is associated with better prognosis in primary sclerosing cholangitis. Dig Liver Dis 2011;43:309‐313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Boonstra K, Weersma RK, van Erpecum KJ, Rauws EA, Spanier BW, Poen AC, et al. Population‐based epidemiology, malignancy risk, and outcome of primary sclerosing cholangitis. Hepatology 2013;58:2045‐2055. [DOI] [PubMed] [Google Scholar]
- 17. Broome U, Olsson R, Loof L, Bodemar G, Hultcrantz R, Danielsson A, et al. Natural history and prognostic factors in 305 Swedish patients with primary sclerosing cholangitis. Gut 1996;38:610‐615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wiesner RH, Grambsch PM, Dickson ER, Ludwig J, MacCarty RL, Hunter EB, et al. Primary sclerosing cholangitis: natural history, prognostic factors and survival analysis. Hepatology 1989;10:430‐436. [DOI] [PubMed] [Google Scholar]
- 19. Farrant JM, Hayllar KM, Wilkinson ML, Karani J, Portmann BC, Westaby D, et al. Natural history and prognostic variables in primary sclerosing cholangitis. Gastroenterology 1991;100:1710‐1717. [DOI] [PubMed] [Google Scholar]
- 20. Dickson ER, Murtaugh PA, Wiesner RH, Grambsch PM, Fleming TR, Ludwig J, et al. Primary sclerosing cholangitis: refinement and validation of survival models. Gastroenterology 1992;103:1893‐1901. [DOI] [PubMed] [Google Scholar]
- 21. Kim WR, Therneau TM, Wiesner RH, Poterucha JJ, Benson JT, Malinchoc M, et al. A revised natural history model for primary sclerosing cholangitis. Mayo Clin Proc 2000;75:688‐694. [DOI] [PubMed] [Google Scholar]
- 22. Corpechot C, Gaouar F, El Naggar A, Kemgang A, Wendum D, Poupon R, et al. Baseline values and changes in liver stiffness measured by transient elastography are associated with severity of fibrosis and outcomes of patients with primary sclerosing cholangitis. Gastroenterology 2014;146:970‐979. [DOI] [PubMed] [Google Scholar]
- 23. Vesterhus M, Hov JR, Holm A, Schrumpf E, Nygard S, Godang K, et al. Enhanced liver fibrosis score predicts transplant‐free survival in primary sclerosing cholangitis. Hepatology 2015;62:188‐197. [DOI] [PubMed] [Google Scholar]
- 24. de Vries EM, Wang J, Williamson KD, Leeflang MM, Boonstra K, Weersma RK, et al. A novel prognostic model for transplant‐free survival in primary sclerosing cholangitis. Gut 2018;67:1864‐1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chapman R, Fevery J, Kalloo A, Nagorney DM, Boberg KM, Shneider B, et al. Diagnosis and management of primary sclerosing cholangitis. Hepatology 2010;51:660‐678. [DOI] [PubMed] [Google Scholar]
- 26. Vittinghoff E, McCulloch CE. Relaxing the rule of ten events per variable in logistic and Cox regression. Am J Epidemiol 2007;165:710‐718. [DOI] [PubMed] [Google Scholar]
- 27. Mallett S, Royston P, Dutton S, Waters R, Altman DG. Reporting methods in studies developing prognostic models in cancer: a review. BMC Med 2010;8:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Royston P, Altman DG. External validation of a Cox prognostic model: principles and methods. BMC Med Res Methodol 2013;13:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Carbone M, Sharp SJ, Flack S, Paximadas D, Spiess K, Adgey C, et al. The UK‐PBC risk scores: derivation and validation of a scoring system for long‐term prediction of end‐stage liver disease in primary biliary cirrhosis. Hepatology 2016;63:930‐950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Collins GS, Reitsma JB, Altman DG, Moons KG. Transparent Reporting of a multivariable prediction model for Individual Prognosis Or Diagnosis (TRIPOD). Ann Intern Med 2015;162:735‐736. [DOI] [PubMed] [Google Scholar]
- 31. Lammers WJ, Kowdley KV, van Buuren HR. Predicting outcome in primary biliary cirrhosis. Ann Hepatol 2014;13:316‐326. [PubMed] [Google Scholar]
- 32. Lammers WJ, van Buuren HR, Hirschfield GM, Janssen HL, Invernizzi P, Mason AL, et al. Levels of alkaline phosphatase and bilirubin are surrogate end points of outcomes of patients with primary biliary cirrhosis: an international follow‐up study. Gastroenterology 2014;147:1338‐1349. [DOI] [PubMed] [Google Scholar]
- 33. Webb GJ, Rana A, Hodson J, Akhtar MZ, Ferguson JW, Neuberger JM, et al. Twenty‐year comparative analysis of patients with autoimmune liver diseases on transplant waitlists. Clin Gastroenterol Hepatol 2018;16:278‐287. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials