
Figure 1.
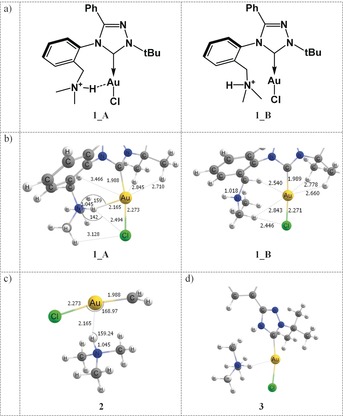
a) Molecule 1 in conformation A with a Au⋅⋅⋅H+−N contact, and conformation B with the N−H+ group pointing away from the AuI ion (further denoted 1_A, 1_B). b) Equilibrium geometries of 1_A and 1_B calculated at PBE0‐D3/def2‐TZVPP. c) Model system 2 and d) model system 3, used to evaluate the magnitude of the Au⋅⋅⋅H+−N interaction.