

Summary
Congenital dyserythropoietic anaemia type I (CDA‐I) is one of a heterogeneous group of inherited anaemias characterised by ineffective erythropoiesis. CDA‐I is caused by bi‐allelic mutations in either CDAN1 or C15orf41 and, to date, 56 causative mutations have been documented. The diagnostic pathway is reviewed and the utility of genetic testing in reducing the time taken to reach an accurate molecular diagnosis and avoiding bone marrow aspiration, where possible, is described. The management of CDA‐I patients is discussed, highlighting both general and specific measures which impact on disease progression. The use of interferon alpha and careful management of iron overload are reviewed and suggest the most favourable outcomes are achieved when CDA‐I patients are managed with a holistic and multidisciplinary approach. Finally, the current understanding of the molecular and cellular pathogenesis of CDA‐I is presented, highlighting critical questions likely to lead to improved therapy for this disease.
Keywords: congenital dyserythropoetic anaemia, anaemia, dyserythropoiesis, erythropoiesis, clinical haematology
Congenital dyserythropoietic anaemia type I (CDA‐I) (Online Mendelian Inheritance in Man [OMIM] entry: 224120; Orphanet: D64.4 and DCS‐10) is one of a heterogeneous group of disorders termed the congenital dyserythropoietic anaemias (CDAs). Unlike other rare anaemias, the CDAs are characterised by ineffective erythropoiesis and morphological abnormalities of erythroblasts. Other haematopoietic lineages are unaffected and there is a haemolytic component. Dyserythropoiesis is defined as the presence of erythroblast abnormalities indicative of aberrant proliferation or differentiation (Crookston et al, 1966). The World Health Organization recognizes nine types of erythroid dysplasia (Brunning et al, 2008). Although there are further categories and subcategories of abnormalities described on dysplastic bone marrow smears, concordance amongst expert haematologists reviewing identical dyserythropoietic slides is poor (Goasguen et al, 2018). Crookston's original description allows for a minority of dyserythropoiesis in normal bone marrow (Crookston et al, 1966).
There are three main types of CDA (CDA‐I, CDA‐II and CDA‐III) and although each has specific morphological and clinical features, blood films show overlapping abnormalities. All subtypes show anisocytosis and poikilocytosis and CDA‐I has macrocytic red cells while types II and III are usually normocytic (Bain et al, 2010). The major CDA subgroups were originally proposed based on specific erythroblast morphological abnormalities on bone marrow light microscopy of aspirates (Heimpel & Wendt, 1968): CDA‐I is indicated by binucleate macrocytic erythroblasts and internuclear bridging, CDA‐II by the presence of 10–35% binucleate late erythroblasts and CDA‐III by the presence of giant multinucleate erythroblasts with up to 12 nuclei per cell. Electron microscopy of erythroblasts reveals a characteristic pattern of chromatin abnormalities in CDA‐I (see below) and a double cellular membrane in CDA‐II. This classification system has facilitated the systematic collection of CDA cases, allowed the best available treatment to be delivered and led to the identification of causative genes. However, the degree to which these disorders share a molecular basis is unclear.
Since the discovery of CDAN1 as a causative gene for CDA‐I in 2002, the genes for the three main types of CDA have been identified. Approximately 90% of CDA‐I cases are caused by bi‐allelic mutations in CDAN1 or C15orf41 (Dgany et al, 2002; Babbs et al, 2013), CDA‐II is caused by biallelic mutations in SEC23B (Schwarz et al, 2009) and CDA‐III is a dominant disorder caused by the P916R mutation of KIF23 (Liljeholm et al, 2013). Identification of the causative genes has shed light on the pathogenesis of these disorders, opened new avenues for research, allowed accurate molecular diagnosis and carrier testing of family members and impacted disease management. Variants of CDA have also been described, for example the dominantly inherited E325K mutation of KLF1 causing an anaemia termed CDA‐IV and X‐linked forms caused by GATA1 mutations (Nichols et al, 2000; Singleton et al, 2009; Arnaud et al, 2010). Further CDA subtypes have been suggested, however, the extent to which these are distinct entities will become clearer as our understanding of their molecular pathogenesis improves.
The abnormal cellular morphology and phenotypic abnormalities in CDA‐I are reviewed elsewhere (Wickramasinghe, 1997, 1998; Shalev et al, 2002; Heimpel, 2004; Tamary et al, 2005; Wickramasinghe & Wood, 2005; Heimpel et al, 2006, 2010; Iolascon & Delaunay, 2009; Renella & Wood, 2009; Iolascon et al, 2011, 2012, 2013; Gambale et al, 2016). Therefore, this review will focus on recent advances in the understanding of the diagnosis, management and pathogenesis of CDA‐I.
As with many rare disorders, establishing an accurate estimate of incidence and prevalence of CDA‐I is difficult. Over 300 cases have been reported (Iolascon et al, 2013). Most are sporadic cases from diverse regions such as Western Europe, North Africa and Asia, (Iolascon & Delaunay, 2009) while some series are accounted for by a founder effect, particularly in the Middle East (Tamary et al, 2005). The lack of reported cases from Sub‐Saharan Africa or South America may reflect ascertainment bias (Heimpel et al, 2010).
Clinical presentation
Diagnosis of CDA‐I is often predicated on a high index of suspicion. With rare disorders, awareness of the condition is necessary before appropriate investigations are instigated. Traditional pathways for the investigation of rare inherited anaemias follow the usual sequence of history and examination, standard haematological and biochemical assays, possibly radiological imaging, a bone marrow aspirate and trephine and genetic analysis. However, this approach is quickly changing with the advent of clinical‐grade next‐generation sequencing (Fig 1). Nevertheless, all investigations will be guided by a thorough initial consultation with the patient.
Figure 1.
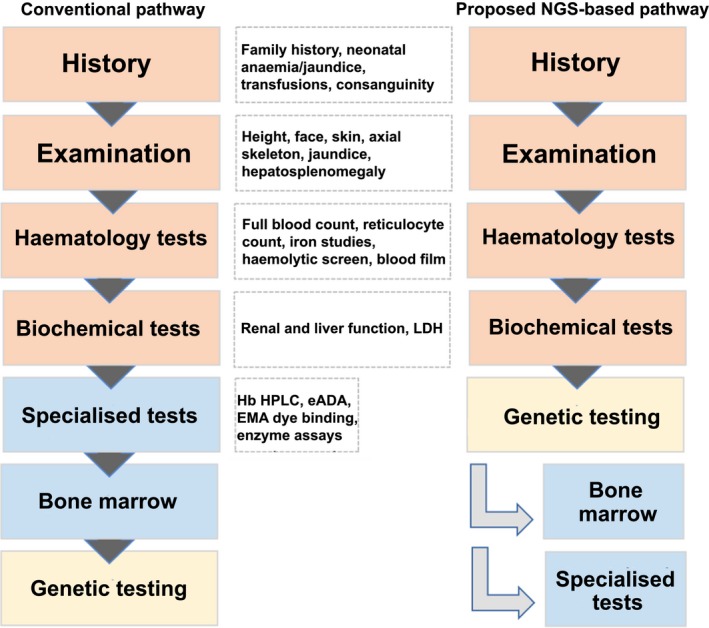
Comparison between the conventional pathway and an alternative NGS‐based pathway for the investigation of inherited anaemias. In the conventional pathway, patients are investigated sequentially where a differential diagnosis is devised based on history, examination and standard blood investigations. Specialised tests are then requested according to the suspected diagnosis and a bone marrow investigation frequently performed. Genetic testing is then reserved as a confirmatory test. In the alternative pathway, NGS is employed early, obviating the need for bone marrow biopsies in some patients where clear molecular diagnoses can be made by NGS. Where variants of uncertain significance are identified functional tests are required to confirm or refute the variant's pathogenicity. This alternative pathway can cut the time to diagnosis, remove the need for some bone marrow biopsies, provide accurate diagnosis of cases and allow genetic counselling. eADA, erythrocyte adenine deaminase; EMA, eosin‐5‐maleimide; Hb HPLC, haemoglobin high performance liquid chromatography; LDH, lactate dehydrogenase; NGS, next‐generation sequencing.
History and examination
While some cases have been identified in utero (Parez et al, 2000; Kato et al, 2001; Lin et al, 2014), most present in childhood or young adulthood (Iolascon et al, 2011; Shalev et al, 2017). Cases detected in utero have led to fetal demise if untreated, but intra‐uterine transfusions support survival to term, followed by lifelong transfusion dependence. Later presentation can be due to intermittent jaundice and fatigue or the requirement for occasional blood transfusion for severe anaemia, while some have presented with secondary iron overload (Kawabata et al, 2012) or pigment gallstones (Fujino et al, 2013). Retrieving a neonatal history can be useful as some neonates with CDA‐I experience prolonged jaundice and/or the requirement for a blood transfusion perinatally, with no further transfusions thereafter. As a recessive disorder, CDA‐I is more likely to occur in consanguineous families and this should be directly questioned. Intriguingly, there is marked heterogeneity in phenotype expression, not only between unrelated patients with different mutations, but even in siblings with identical mutations, where, for example, one sibling presents in the neonatal period and the other at 2 years of age (al‐Fawaz & al‐Mashhadani, 1995; Heimpel et al, 2010).
Examination may reveal pallor, mild jaundice and splenomegaly, which is found in all cases, at least radiologically (Gambale et al, 2016; Shalev et al, 2017). The finding of frontal bossing can be observed in the more severe untreated cases, and this may prompt the need for imaging to assess for extramedullary haematopoiesis (Heimpel et al, 2003). The finding of limb abnormalities in the presence of anaemia should suggest CDA‐I.
Haematology and biochemical assays
A full blood count typically reveals moderate macrocytic anaemia, with a haemoglobin (Hb) of 66–116 g/l (mean 92 g/l), mean corpuscular volume (MCV) 100–120 and reticulocytopenia, although 30% of cases have a normal MCV (Wickramasinghe, 1998). There are reports of transient neutropenia/thrombocytopenia, but these lineages are generally unaffected (Meznarich et al, 2018). The hallmark of CDA‐I is an absolute or relative reticulocytopenia, indicative of ineffective erythropoiesis (Heimpel, 2004). Ineffective erythropoiesis was originally demonstrated using ferrokinetic studies, where the fraction of 59Fe present in peripheral red blood cells is calculated 2 weeks post‐intravenous infusion. Where erythropoiesis is effective, e.g. when anaemia occurs due to bleeding, iron deficiency or haemolysis, the fraction of red cells containing 59Fe is ~75–80%, but in ineffective erythropoiesis, it may be as low as 25–30% (Lewis, 2001). Recently, a new clinical index, termed the bone marrow responsiveness index (BMRI), has been developed to discriminate haemolytic anaemia from ineffective erythropoiesis. This is defined as: [(absolute reticulocyte count) × (patient Hb/normal Hb)] and was shown to be a highly sensitive parameter (90·4%) to achieve a clinical diagnosis of CDA‐II. This metric is likely to be useful in CDA‐I but is yet to be formally validated in this disease.
The blood film, as illustrated in Fig 2, is markedly abnormal with anisocytosis and various poikilocytes, including teardrops, ovalocytes, elliptocytes, microspherocytes, irregularly contracted red cells (Heimpel et al, 2010) and occasional nucleated red cells (Tamary & Dgany, 2009), although these are not a typical feature. Red cell distribution width, as a quantitative assessment of the aniosopoikilocytosis seen on films, is elevated in CDA‐I (Wickramasinghe, 1998; Kamiya & Manabe, 2010). Renal and liver function are normal unless perturbed secondary to severe iron overload, but unconjugated bilirubin and lactate dehydrogenase will be elevated from the haemolysis and ineffective erythropoiesis. Haptoglobin levels are reduced secondary to intra‐ and extra‐vascular haemolysis (Heimpel, 2004).
Figure 2.
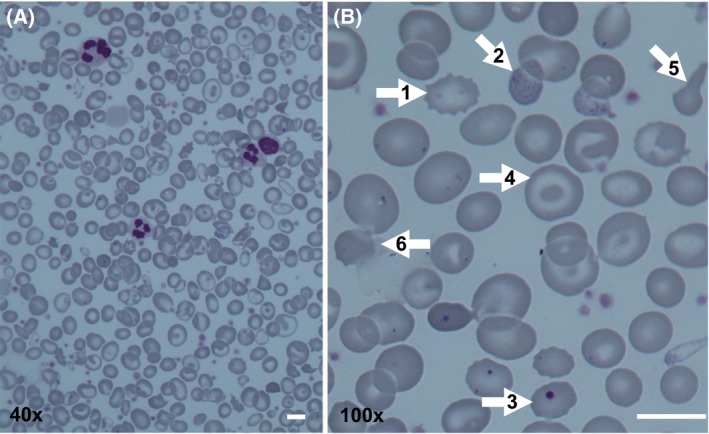
Peripheral blood film of a splenectomised patient with CDA‐I. (A) 40 × magnification, showing marked red cell anisopoikilocytosis. (B) 100 × magnification showing (1) echinocyte; (2) basophilic stippling; (3) Howell‐Jolly body; (4) target cell; (5) teardrop cell; (6) irregularly contracted red cell. Scale bars are 10 μm.
Key alternative diagnoses, which must be ruled out, include vitamin B12 and folate deficiency, autoimmune haemolytic anaemias, haemoglobinopathies or infections, such as human immunodeficiency virus or leishmaniasis. Ferritin and transferrin saturations should be assessed to evaluate for secondary iron overload. A high level of soluble transferrin receptor and low/unrecordable serum hepcidin in the absence of iron deficiency has also been noted in some CDA‐I patients (Cazzola et al, 1999), but neither test is readily available in the clinic.
Bone marrow examination
A bone marrow aspirate and trephine are usually carried out next, allowing rapid differentiation between, for example, Diamond Blackfan Anaemia (DBA) and CDA, both of which present with a reticulocytopenic anaemia. Findings on bone marrow examination were originally described by Heimpel and Wendt (1968); a hallmark of the condition is marked erythroid hyperplasia, with an excess of erythroid precursors compared to myeloid precursors (Heimpel, 2004). Between 2·4% and 10% of late polychromatic erythroblasts precursors are binucleate (Heimpel et al, 2010), where nuclei can be unequal sizes (Gambale et al, 2016). Intermediate erythroblasts exhibit internuclear bridges in 1–8% of cells examined and 30–60% of late erythroblasts exhibit a range of abnormalities, including polychromasia, megaloblastic changes, multinuclearity, karyorrhexis and basophilic stippling (Iolascon et al, 2012). At least 20% of erythroblasts must have an abnormality on light microscopy for a diagnosis of CDA‐I (Heimpel et al, 2010). The diagnosis is based on a constellation of abnormalities but the absence of internuclear bridges calls a diagnosis of CDA‐I into question. A recent study examining concordance in the morphological identification of dyserythropoiesis in two CDA‐I cases found agreement between only 4/7 expert haematologists (Goasguen et al, 2018). Diagnostic certainty relies on genetic confirmation or the gold standard of scanning electron microscopy (SEM) to identify nuclear abnormalities. This often requires a second bone marrow aspirate, as SEM samples require specific preparation for best results and CDA‐I is usually unsuspected prior to the light microscopy findings. SEM findings include widening of nuclear pores, darkening of heterochromatin with electron lucent areas within abnormally electron dense heterochromatin (Fig 3) described as ‘spongy heterochromatin’ (Wickramasinghe, 1998) or ‘Swiss cheese appearance’ (Heimpel et al, 2006). Invagination of the cytoplasm into the nucleus has also been described (Iolascon et al, 2013).
Figure 3.
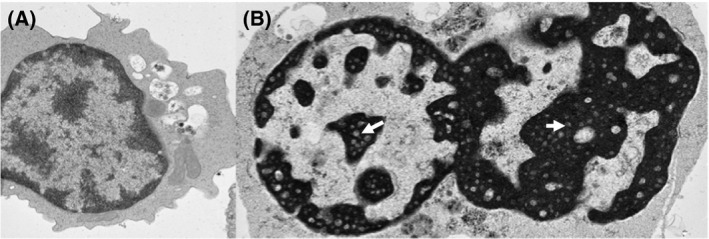
(A) Electron microscopy of a normal cultured erythroblast (day 7 of culture) showing the expected pattern of euchromatin and heterochromatin (magnification × 4000). (B) A CDA‐I cultured erythroblast showing binuclearity and the ‘Swiss cheese’ or ‘spongy’ heterochromatin with electron‐lucent areas (arrows) that is pathognomonic of this condition.
Other features
Non‐haematological manifestations of CDA‐I are described in 10–20% of cases (Wickramasinghe, 1998), mostly involving the axial skeleton, such as missing distal phalanges, syndactyly (especially of the toes), and complete lack of nail formation (Brichard et al, 1994). Brown skin pigmentation (Heimpel & Wendt, 1968) and neurological deficits (Wickramasinghe, 1998) have occasionally been reported. However, as some families in which CDA‐I is diagnosed have a high degree of consanguinity, other features may represent epiphenotypes arising from different recessive conditions (Renella & Wood, 2009).
The presence of angioid streaks has been described in CDA‐I. These represent a break in Bruch's membrane, a collagenous layer underneath the pigment epithelium of the retina, which has been described in a small number of CDA‐I patients and is associated with loss of visual acuity (Frimmel & Kniestedt, 2016).
Imaging
Abdominal ultrasound reveals universal splenomegaly (Heimpel et al, 2006) and gallstones in 50–60% of adults with CDA‐I (Shalev et al, 2002). Imaging with plain radiographs or magnetic resonance imaging (MRI) to investigate and characterise paraspinal masses secondary to extramedullary haematopoiesis may be necessary (Heimpel et al, 2009).
Imaging for organ iron overload by T2* MRI or Ferriscan® is guided by biochemical iron studies. However, it has been reported that 38% of CDAs (type not specified) have cardiac iron on MRI, and 25% acquire cardiac iron loading by 10 years of age. Raised iron in the pancreas was described in six CDA cases and median liver iron concentration from T2* was 5·9 mg/g dry weight (upper limit of normal = 1·8 mg/g dry weight). While the role of imaging for iron loading is yet to be systematically evaluated in CDA‐I patients, this study suggests that this patient cohort warrants routine assessment of iron status (Berdoukas et al, 2013).
Definitive diagnosis
Genetic analysis
CDA‐I is a recessive disease caused by biallelic mutations in either CDAN1 or C15orf41 (Dgany et al, 2002; Babbs et al, 2013). There are 51 causative mutations documented in CDAN1 and 5 in C15orf41 (Fig 4). Currently no patients have been identified with compound mutations in these genes. Approximately 10% of CDA‐I cases remain unexplained by mutations in either of the known genes and there may be cis‐acting regulatory mutations affecting these genes or further, yet to be identified, loci causative of CDA‐I.
Figure 4.
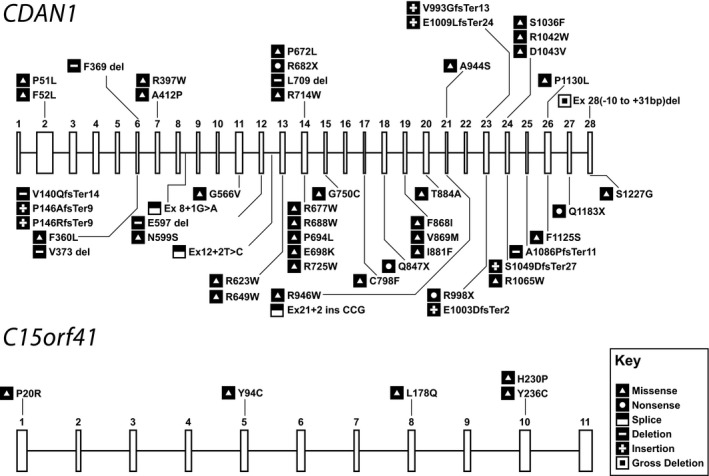
Representation of all known pathogenic variants associated with CDAN1 and C15orf41. The exons in each gene are numbered to show the location of each mutation. Coding mutations are shown according to the amino acid change while splicing changes are shown according to their location relative to exons.
A recent review of the role of SEM in the diagnosis of CDA‐I concluded that genetic analysis for CDAN1 and C15orf41 should only be done once SEM has confirmed the presence of the pathognomonic ‘spongy heterochromatin’ abnormality (Resnitzky et al, 2017). We feel that genetic testing may obviate the need for SEM in some cases, thus avoiding an additional bone marrow aspirate with associated risks, which would be performed under general anaesthetic in the paediatric population. Genetic testing can be performed from a small amount of peripheral blood early in the diagnostic process (Fig 1). While knowledge of the precise underlying mutation does not currently carry prognostic information, it guides discussion, confirms the diagnosis and allows preimplantation genetic diagnosis.
Current approaches to genetic analysis include targeted panels (containing 50–200 genes), whole exome sequencing (WES) and whole genome sequencing (WGS). Where WES and WGS are undertaken for clinical diagnostics rather than research, e.g. as part of the National Health Service (NHS) England 100 000 genome project, the whole exome/genome is sequenced but only data from a pre‐determined set of genes is analysed. Targeted panels have been shown to be clinically useful in the diagnosis of rare inherited anaemias, including CDA‐I (Gerrard et al, 2013; Roy et al, 2016; Russo et al, 2018; Shefer Averbuch et al, 2018). Reported diagnostic rates vary from ~38% to ~65%, depending on the number and types of genes included and the depth of phenotypic assessment undertaken. In ~10% of cases, this approach reveals an unsuspected diagnosis (Roy et al, 2016). Making an accurate diagnosis is paramount to instituting correct therapy, for example steroids for DBA, splenectomy for pyruvate kinase deficiency and administration of interferon alpha (IFNa) for CDA‐I. In our recent study of a series of 20 cases with a presumed diagnosis of CDA‐I (from blood results and light microscopy and/or SEM), 55% received a molecular diagnosis from the targeted panel. The majority of these confirmed CDA‐I with mutations in CDAN1 or C15orf41, but 20% had an alternative diagnosis, such as DBA with an unusual marrow in early childhood, and PK deficiency (Roy et al, 2016).
Extrapolating from these findings, up to 55% of patients may be able to avoid an unnecessary bone marrow aspiration when genetic analysis is performed earlier in the diagnostic pathway. In addition, time to diagnosis is notoriously long for patients with rare disorders and the delay between onset of symptoms and a formal diagnosis has been reported to be 12 years in some cases of CDA‐I (Fujino et al, 2013). A German CDA Registry reported the age of the 21 patients at the time of initial diagnosis of CDA‐I ranged between 0·1 and 45 years (median 17·3 years) and that 11 of 21 cases were previously misdiagnosed as congenital haemolytic anaemia (Heimpel et al, 2006). This underscores the utility of genetic testing to provide an accurate molecular diagnosis.
Management
Holistic approach
Akin to the management of thalassaemia patients, whose clinical course CDA‐I patients often closely resemble, the most favourable outcomes are achieved when CDA‐I patients are managed with a holistic and multidisciplinary approach. The diagnosis is usually a surprise to patients and their families and, as with rare disorders, patients suffer from a sense of isolation and that their physicians have inadequate knowledge about their condition (Budych et al, 2012). There is often the need for genetic counselling of the parents for future pregnancies. For patients receiving life‐long transfusions, issues such as long‐term intravenous access, logistics of regular transfusion and transfusion complications, are as important as they are in thalassaemia patients. Irrespective of transfusion status, CDA‐I is associated with iron overload and compliance with chelation is particularly important, especially around adolescence. Transition from paediatric to adult services should be carefully managed, and in the UK, National Institute for Health and Care Excellence (NICE) guidelines recommend the use of the “Ready, Steady, Go” approach (https://www.nice.org.uk/sharedlearning/implementing-transition-care-locally-and-nationally-using-the-ready-steady-go-programme). Complex patients may need to be managed by a team of psychologists, dietician, endocrinologists and bone specialists, while others will have a very mild disorder and require only a yearly review with a haematologist.
Management of the anaemia
This depends on severity and patient characteristics and may change according to the patient's lifestyle. Some patients require transfusions only perinatally whilst others require them during additional marrow stress, e.g. intercurrent infections or pregnancy. True transfusion dependence occurs in 3–4% of cases (Heimpel et al, 2006), although genotype‐phenotype correlations have not yet been convincingly described. Transfusions increase the prevalence and severity of iron overload. Transfusion practice should follow guidelines for chronically transfused patients, and managing transfusion in CDA‐I patients according to practices developed for haemoglobinopathies is entirely appropriate. As such, blood units should be as fresh as possible, preferably <10 days since sampling from the donor (Milkins et al, 2013; Davis et al, 2017) and patients should receive the Hepatitis B vaccine and be offered lifelong folic acid replacement due to the haemolytic component. The only difference with haemoglobinopathy patients concerns the requirement for extended phenotyping to reduce the likelihood of developing antibodies. This is less critical than for the haemoglobinopathies where there is commonly a mismatch between the donor and recipient ethnicity (Davis et al, 2017), but is still considered best practice for CDA‐I patients.
Interferon alpha
The only specific treatment available for CDA‐I is interferon alpha (IFNa). Its discovery was fortuitous when IFNa was given to a French patient with transfusion‐dependent CDA‐I who had contracted Hepatitis C through contaminated blood (Lavabre‐Bertrand et al, 1995) and became transfusion independent with reduction in ineffective erythropoiesis and amelioration of SEM features on bone marrow aspirate. Intriguingly, while 4 weeks of treatment ameliorated the erythroid:myeloid ratio and the SEM features reduced from 57% to 15·6%, the percentage of erythroblasts exhibiting internuclear bridges and binuclearity was unchanged. Prolonged treatment with IFNa leads to a stable Hb with ongoing normalisation in SEM features (Heimpel et al, 2006). Bilirubin falls in parallel as intramedullary haemolysis, characteristic of ineffective erythropoiesis, improves. Improvement in ineffective erythropoiesis in response to IFNa is supported by ferrokinetic studies (Lavabre‐Bertrand et al, 1995). Responses to IFNa are rapid, occurring within 4 weeks, but are not universal (Marwaha et al, 2005) and patients have required cessation of treatment due to side effects (Bader‐Meunier et al, 2005). These include gastrointestinal symptoms, flu‐like symptoms and depression. Reported effective doses vary between 4–10 million units per week (Heimpel, 2004; Lavabre‐Bertrand et al, 2004) but individual dose titration is required. Pegylated interferon at a dose of 30–50 μg/week has also been used effectively.
The mechanism of action of IFNa is not understood. In vitro, IFN production by Epstein–Barr virus‐transformed lymphocytes from CDA‐I patients is reduced, (Wickramasinghe et al, 1997) suggesting a deficit. It remains unclear whether CDA‐I patients respond to IFNa due to subnormal production of this in vivo, or whether the molecular defect leading to CDA‐I can be overcome by the over‐expression of IFN‐responsive genes. Further insights into the mechanism of action of IFNa may allow for the development of other, targeted therapies.
Erythropoietin (Epo)
Epo levels are mildly elevated in CDA‐I patients, but remain inappropriately low for the degree of anaemia. Efforts to correct the anaemia using recombinant human Epo did not result in any rise in Hb, increase in reticulocyte count or fall in iron overload (Tamary et al, 1999). As such, Epo is not considered a treatment for CDA‐I.
Splenectomy
Splenectomy in CDA‐I has not been evaluated systematically, but evidence from small case series suggests exercising caution. In one series, six patients were splenectomised for severe anaemia, five of whom had a rise in Hb (Shalev et al, 2017). However, this benefit was offset by a rise in mortality, with 3/6 patients dying between the ages of 40–60 from pulmonary hypertension (n = 1) and sepsis (n = 3). In another series, 7/21 patients underwent splenectomy with no response in Hb (Heimpel et al, 2006). Recent European Haematology Association recommendations highlight the high rate of complications in CDA‐I and suggest splenectomy should be reserved for patients with painful splenomegaly and/or significant thrombocytopenia/leucopenia (Iolascon et al, 2017).
Management of iron overload
As detailed above, the aetiology of iron overload in non‐transfused CDA‐I patients is directly related to ineffective erythropoiesis. CDA‐I patients have unrecordable levels of hepcidin, leading to unopposed gastrointestinal iron absorption and deposition in target organs (Tamary et al, 2008; Kawabata et al, 2012). Suppression of hepcidin is hypothesized to result from excess erythroferrone production by the greatly expanded pool of erythroblasts. This mechanism has been successfully demonstrated in thalassaemia (Kautz et al, 2015) and CDA‐II (Russo et al, 2016). Serum growth differentiation factor 15 (GDF15) levels (a marker of ineffective erythropoiesis) were found to be elevated in CDA‐I patients, with a correlation between GDF15 and ferritin and anti‐correlation with serum hepcidin levels (Tamary et al, 2008).
There is no clinical consensus on the frequency of monitoring for tissue iron overload. In our centre we perform yearly T2* MRI in patients from the age of 10 years (Tamary & Dgany, 2009), although others investigate every 5 years once serum ferritin exceeds 600 μg/l from the age of 20 years (Gambale et al, 2016).
The management of iron overload in CDA‐I patients should be the same as for thalassaemia patients, namely chelators, be that sub‐cutaneous (deferrioxamine) or oral (deferiprone, deferasirox). However, a unique approach in these patients is to titrate the dose of IFNa such that the patient's Hb rises above that needed to avert the symptoms of anaemia so that regular venesections can be carried out. This has proven a very useful technique in some CDA‐I patients in clinical practice in several UK centres of which the authors are aware.
Management of extramedullary haematopoieis
Extramedullary haematopoiesis (EMH), ranging in severity from minor to bulky, is a recognised complication of CDA‐I, although the exact prevalence is unclear (Heimpel et al, 2006, 2009). Management is in line with the EMH in thalassaemia with therapeutic options including surgical debulking, low dose irradiation and commencing regular transfusions to suppress EMH.
Management of osteoporosis
Osteoporosis is present in 89% of CDA‐I cases (Shalev et al, 2017). The aetiology is probably multifactorial due to marrow expansion, diabetes and hypothyroidism, parathyroid gland dysfunction and the toxic effects of iron and chelators on osteoblasts (Voskaridou & Terpos, 2008). Active management entails treatment of calcium and vitamin D deficiency, bone densitometry scanning and regular review by bone specialists (Shalev et al, 2017).
Management of pregnancy
Shalev et al (2004) reviewed the outcomes in 28 spontaneous pregnancies in 18 women over a 15‐year period in a Bedouin tribe. The complication rate was high (64%) and included one first trimester spontaneous abortion, one stillbirth and 42% low birth weight infants. The rate of infants requiring caesarean sections was statistically significantly higher than a control group of Bedouin women (35·7% vs. 11%) for reasons including fetal distress and pre‐eclampsia. Not infrequently, previously transfusion independent women with CDA‐I develop a transfusion requirement during pregnancy (Roy & Pavord, 2018).
Management of endocrinopathies
Endocrinopathies have been described in 10–40% of CDA‐I patients and include diabetes mellitus and hypothyroidism (Heimpel et al, 2006; Shalev et al, 2017), as well as pituitary failure leading to growth retardation (Facon et al, 1990). These are thought to be secondary complications from iron overload and poor compliance with chelation. Their management requires close collaboration with endocrinologists as well as psychologists to aid medication compliance.
Bone marrow transplantation
Paediatric bone marrow transplants have been carried out in a few patients with CDA‐I. In a small series, three children underwent matched sibling allografts. All were severe and diagnosed before 6 months of age, had hepatosplenomegaly and two required chelation. Conditioning was with cyclophosphamide 50 mg/kg/day for 4 days, busulfan 4 mg/kg/day for 4 days and antithymocyte globulin 30 mg/kg for 4 doses pre‐stem cell transplantation. All three engrafted and became transfusion independent (Ayas et al, 2002). One of the barriers to transplantation is poor prognosis conferred by pre‐transplant iron overload, yet the most severe patients who would most benefit from transplant are the ones whose response to chelation is limited (Buchbinder et al, 2012).
Pulmonary hypertension
CDA‐I may present with pulmonary hypertension in the neonatal period, in association with other congenital anomalies (El‐Sheikh et al, 2014; Landau et al, 2015). Treatment includes inhaled nitric oxide and high frequency oscillation ventilation. While pulmonary hypertension may be a late complication of CDA‐I, the extent of this is unknown. There are currently no guidelines on whether and how pulmonary hypertension should be assessed and treated in CDA‐I patients. For patients with sickle cell disease, this is screened by tricuspid jet velocity followed by right heart catheterization in patients with tricuspid jet velocity >2·5 m/s. A recent meta‐analysis (Wang et al, 2018) compared different types of treatment for all types of idiopathic pulmonary hypertension including endothelin receptor antagonists, phosphodiesterase type‐5 inhibitors, prostaglandin I2, soluble guanylate cyclase stimulator and selective non‐prostanoid prostacyclin receptor agonists or combination treatment. Results differed depending on the outcome used to measure response but the best responses appeared to be for vardenafil (taken orally) and iloprost + bosantan (inhaled). Whether these would be the best treatment for CDA‐I patients with pulmonary hypertension is not known.
Potential for gene editing
Gene editing could provide hope for a cure. In CDA‐I, gene editing would require homology‐directed repair to integrate a donor template and so each targeted mutation would require individually designed editing, which remains currently out of reach. However, in the longer term, elucidation of the pathway affected in CDA‐I may identify a therapeutic target gene, which would allow a universal therapy to be developed.
Pathogenesis
Rare diseases (defined as those affecting <1:2000 of the population) have been estimated to number between 6000–8000, collectively affecting some 30 million European Union citizens (http://www.eurordis.org/). However, the study of rare diseases has an impact reaching far beyond affected individuals alone: for example, research into the molecular basis behind Fanconi anaemia, a congenital bone marrow failure syndrome affecting ~1000 individuals worldwide, has provided critical insights into the link between genomic instability and malignancy, has significantly advanced our understanding of DNA repair mechanisms (Schindler & Hoehn, 2007 ) and has led directly to the development of therapeutic agents for patients with BRCA1/2 mutations (Fong et al, 2009).
CDA‐I is an example of a rare disease which has the potential to inform about general cellular processes. Codanin‐1 (CDAN1) and C15orf41 expression levels are extremely low in all cell types, yet are widely expressed, and loss of either protein is incompatible with life. Both proteins are likely to play a critical role in DNA repair and/or chromatin assembly following DNA replication and understanding their function will elucidate universal cellular processes. A number of fundamental questions remain unanswered about the biology and pathology of CDA‐I (Fig 5). The main hurdle to advancing our understanding of the function of CDA‐I proteins is the lack of molecular reagents, including antibodies, appropriate cell lines and access to primary material, although attempts have been made to address this by generating erythroid cell lines with engineered tags at the endogenous loci (Moir‐Meyer et al, 2018).
Figure 5.
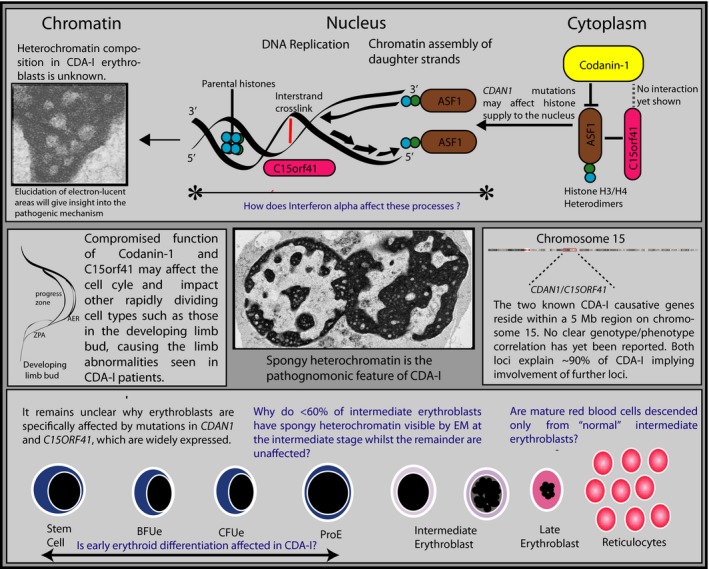
Summary of current knowledge of pathophysiology of CDA‐I (black type) and key questions (blue type). The top panel focuses on different sub‐cellular compartments. While spongy heterochromatin is the pathognomonic feature of CDA‐I, the composition of the electron‐lucent areas is unknown. Whether they are true euchromatin or abnormally packaged heterochromatin would indicate the function(s) of the key proteins. The known interactions between Codanin‐1, C15orf41 and the histone chaperone ASF1 are shown and the abnormal shuttling of these proteins into the nucleus in the context of mutated CDAN1 in non‐erythroid cells suggests a possible problem in delivery of histones in the rechromatinisation of replicating DNA. Because of its predicted role as a nuclease, C15orf41 may play a role in clearing blocks to replication fork progression (such as interstrand cross‐links) or replication intermediaries. Determining whether interferon alpha acts at this level would narrow down the key cellular processes in which Codanin‐1/C15orf41 are involved. In the middle left panel, the possibility that Codanin‐1 has a direct function in the developing limb bud by affecting the coordinated interaction between the signalling centres is suggested. In the right middle panel the chromosomal location of CDAN1 and C15orf41 is shown and the existence of further loci is strongly suggested by the lack of molecular diagnoses in 10% of EM‐proven CDA‐I cases. The bottom panel illustrates erythroid differentiation and the conundrums of the predominantly erythroid abnormalities in CDA‐I despite broad expression of CDAN1 and C15orf41 and the fact that only subset of erythroid progenitors are morphologically affected in the bone marrow. AER, apical ectodermal ridge; Stem cell, haematopoietic stem cell; BFUe, burst forming unit (erythroid); CFUe, colony forming unit (erythroid); ProE, pro‐erythroblast; ZPA, zone of polarising activity.
Gene function
Much of our knowledge about Codanin‐1 and C15orf41 derives from studies in osteosarcoma (U‐2‐OS) cells and cervical cancer (HeLa) cells, both of which are cytogenetically abnormal cell lines that may not reflect biology in primary erythroid cells. For example, Codanin‐1 knock‐down in U‐2‐OS cells results in a faster cell cycle (Ask et al, 2012), whereas deletion of the endogenous Cdan1 in mice results in early embryonic lethality (Renella et al, 2011). Both CDA‐I proteins are widely expressed and yet, when mutated, affect only the erythroid lineage, suggesting the pathological mechanism must be investigated in this cell type. However, insights into potential functions of these proteins gained from studies in non‐erythroid cells will be presented.
Codanin‐1
Bi‐allelic mutations of CDAN1 account for ~80% of CDA‐I cases. The gene comprises 28 exons and encodes the protein Codanin‐1, which is relatively large (~134 kD) and highly evolutionarily conserved in fish, frogs and flies with no human orthologs and no apparent homologue in worms and yeast (Dgany et al, 2002). The Drosophila homologue, discs lost (Dlt), is required for cell survival and cell‐cycle progression (Pielage et al, 2003). There are no functionally conserved domains in Codanin‐1 to facilitate functional predictions, however, a putative peptide binding site has been identified through which Codanin‐1 may interact with the well‐described histone chaperone Asf1 (Tamary et al, 2010; Ask et al, 2012).
Regulation of CDAN1 expression appears to depend on the cell cycle as the promoter contains several binding sites for the cell‐cycle regulated transcription factor E2F that increases expression of Codanin‐1 in S‐phase in HeLa cells (Noy‐Lotan et al, 2009). Codanin‐1 is reported to be enriched in the nucleus in the K562 erythroleukaemia line, U‐2‐OS and HeLa cells. Codanin‐1 has been found to associate with DNA during interphase in HeLa cells and excluded from mitotic condensing chromosomes (Noy‐Lotan et al, 2009). However, other reports suggest that Codanin‐1 is mainly localised to the cytoplasm in U‐2‐OS cells (Ask et al, 2012). This discrepancy needs to be resolved by determining the localisation in primary erythroid cells using independent antibodies when the protein is expressed at native levels.
C15orf41
Bi‐allelic mutations in C15orf41 cause ~10% of CDA‐I cases (Babbs et al, 2013). Similar to Codanin‐1, C15orf41 protein is widely conserved with orthologs broadly distributed in eukaryotes and in members of the archaea and viruses, indicating a highly conserved function. C15orf41 is present in all species where CDAN1 is found, and none where it is not, suggesting the proteins function in concert. Additionally, the pathognomonic CDA‐I heterochromatin defects that arise when either gene is mutated strongly suggest a common pathway, although a direct interaction between the two proteins has not been shown (Fig 5). Sequence conservation shows C15orf41 protein belongs to the PD‐(D/E)XK family of restriction endonucleases, a diverse group of phosphodiesterases involved in genome maintenance processes, such as DNA damage repair, Holliday junction resolution and RNA processing (Laganeckas et al, 2011). Notable members of this family include the DNA repair nucleases Mus81 and XPF (ERCC4/FANCQ) that play key roles in DNA lesion resolution and maintenance of genome stability (Steczkiewicz et al, 2012). This points to a defect in DNA repair in CDA‐I, however, the nature of the lesions that result from impaired C15orf41 function remain unknown.
Role in chromatin assembly
C15orf41 and Codanin‐1 both interact with the histone chaperone Asf1b (anti silencing factor 1b) (Ewing et al, 2007; Ask et al, 2012). Asf1 is essential for chromatin assembly in human cells (Groth et al, 2005, 2007), playing a role donating new histones to chromatin assembly factor 1 (CAF1) (Mello et al, 2002) for incorporation into nucleosomes following DNA replication. Asf1 binds histone H3‐H4 heterodimers in the cytoplasm and chaperones them into the nucleus where they are transferred to downstream chromatin assembly factors (Campos et al, 2010; Jasencakova et al, 2010).
Codanin‐1 sequesters Asf1 in the cytoplasm, thereby negatively regulating the supply of histone bound Asf1 to the nucleus (Ask et al, 2012). Mutations in Codanin‐1, which impair the interaction with Asf1, may allow unregulated Asf1 to access the nucleus thereby disrupting the fine‐tuned delivery of histones known to be critical in correctly rechromatinising newly synthesised DNA (Jasencakova & Groth, 2010). It remains to be shown to what extent abnormal histone delivery at the replication fork leads to the specific abnormalities in chromatin and heterochromatin seen in CDA‐I erythroblasts.
Lineage specificity
CDAN1 and C15orf41 are ubiquitously expressed, albeit at a relatively low level in most tissues, and no individual harbouring two loss of function alleles of either gene has been identified. Additionally, mice embryos homozygous for null Cdan1 alleles die prior to implantation, suggesting that Codanin‐1 is essential prior to the onset of erythropoiesis (Renella et al, 2011). Given that Codanin‐1 and C15orf41 are highly conserved, ubiquitously expressed and appear essential, it is of great interest that abnormalities in CDA‐I are restricted to the erythroid lineage, suggesting that erythroblasts have a specific requirement for Codanin‐1 and C15orf41.
One possibility may be that erythroid progenitors have a uniquely fast cell cycle, although CDA patients do not manifest abnormalities of other tissues containing fast‐dividing cell types, such as gut epithelium or hair follicles. It has been reported that, in mice, a particularly rapid cell cycle is required at the start of terminal erythroid differentiation for erythroid lineage commitment, through a mechanism of passive genome demethylation causing the PU.1 (also termed SPI1) switch and lineage commitment (Pop et al, 2010; Shearstone et al, 2011). However, it remains to be shown whether a similar phenomenon exists in human erythroid differentiation and how this may be impacted by impaired chromatin assembly. Other hypotheses include nuclear extrusion in erythroblasts, which requires the eviction of histones such as H3 and H4 (Zhao et al, 2016) and C15orf41 and Codanin‐1 may play a role in this process. It also may be of significance that the proportion of erythroblasts displaying chromatin abnormalities varies from patient to patient and remaining erythroblasts appear to undergo normal terminal maturation, suggesting a threshold effect.
Whether the non‐haematological manifestations of CDA‐I reflect severe anaemia in utero or are directly due to the effects of a mutated or reduced amount of Codanin‐1, as has been previously suggested (Goede et al, 2006), is difficult to ascertain. Certainly, some features, such as diabetes and growth retardation are more readily ascribed to the iron overload that accompanies CDA‐I. However, the ubiquitous expression of Codanin‐1 and C15orf41 in all tissue types suggests a direct effect in tissues beyond the erythroid lineage. Compromised cell division may affect the rapidly dividing cells of the progress zone and the apical ectodermal ridge in developing limb buds (Tickle, 2015). Limb abnormalities in CDA‐I patients are usually asymmetrical reductions, suggesting a defined signalling pathway is not uniformly affected. However, compromised cell division is likely to be stochastic with a threshold effect, as seen in erythroblasts, and therefore malformations would not be expected to be uniform or bilateral. In addition, if the skeletal defects were due to tissue hypoxia, they could be expected to mirror those seen in Bart's Hydrops Fetalis Syndrome where the absence of HbF creates severe hypoxia. However, in that form of thalassaemia non‐haematological manifestations are more neurological and urogenital, with no descriptions of acral dysostosis (Songdej et al, 2017).
Erythroblast abnormalities
Characterisation of the cellular abnormalities in CDA‐I erythroblasts could shed light on the function of the two genes involved. Analysis of cell cycle distribution in cultured erythroblasts showed an increase in cells in S‐phase in CDA‐I, which are, paradoxically, not actively synthesising DNA when tested, suggesting an S‐phase arrest in CDA‐I (Wickramasinghe & Pippard, 1986). This points to a problem with DNA replication, but needs to be refined using a larger number of patients. Unrepaired DNA lesions act as physical barriers to replication fork progression and nicks, gaps and stretches of ssDNA can be both sources and symptoms of stress (Zeman & Cimprich, 2014). Because C15orf41 is predicted to be an endonuclease it may be that there are more unrepaired lesions in CDA‐I patients and therefore more stalled replication forks, underlying the proposed S‐phase arrest. Alternatively, replication intermediaries that are usually cleared by C15orf41 may underlie some of the nuclear abnormalities seen in CDA‐I erythroblasts. Given that these events are stochastic and would also be likely to be affected by genetic background, this may go some way to explaining the variability between patients. It would be very informative to distinguish these possibilities (blocked replication vs. unresolved replication intermediates).
Elucidating the nature of the electron‐lucent areas in the heterochromatin seen by SEM (Fig 3) would shed light on the underlying pathology of CDA‐I, especially as resolving this abnormality is associated with improved Hb levels in patients treated with IFNa (Lavabre‐Bertrand et al, 1995, 2004). The differentiating stain used in SEM is osmium tetroxide, however, why this differentially binds the heterochromatin in affected nuclei is unclear. It may be that the “holes” contain protein, lipids or improperly packaged heterochromatin affecting its transcriptional status.
Do the mutations in either CDAN1 or C15orf41 represent different entities? It has been suggested that CDA‐I arising from mutations of C15orf41 may be more severe (Gambale et al, 2016). In our clinical and laboratory experience, at the time of writing there are insufficient data to draw this distinction with certainty. Given that CDA‐I with specific chromatin abnormalities arises from mutation of either gene, we propose that this disease simply be termed CDA‐I until new insights on genotype/phenotype correlations are obtained.
Conclusions
Patient welfare
CDA‐I remains a rare disorder and shares some of the hurdles and obstacles borne by patients with other rare conditions. Delays in diagnosis will hopefully be reduced by the implementation of gene panels as part of routine testing. Earlier diagnosis should allow treatment prior to the development of iron overload and associated organ damage. Due to the rarity of the condition, CDA‐I patients should be reviewed at least annually in a centre with a specialist interest in rare anaemias and access to specialized monitoring and multidisciplinary meetings. Such clinicians belong to national and international networks of experts which collaborate to provide optimal care and access to research developments. EuroBloodNet, a European rare disease network, promotes trans‐national working and sharing of expertise. Finally, patients with rare conditions benefit from support offered by national patient networks. A recent James Lind Alliance Priority Setting Partnership identified the number one (of ten) research questions important to these patients and their families as “Would a national formal network of clinicians with expertise and/or a national MDT (multidisciplinary team meeting) improve care for patients with rare inherited anaemias?” (http://www.jla.nihr.ac.uk/priority-setting-partnerships/rare-inherited-anaemias/top-10-priorities.htm). Therefore, collaboration between clinicians, patients and research partners is critical to guiding and securing optimal clinical care for CDA‐I.
Author Contributions
NR and CB conducted the literature review and wrote the manuscript.
Acknowledgements
The authors thank Charlotte Brierley, Jill Brown and Veronica Buckle for critically reading the manuscript and Josh Simmonds for making Fig 4. CB is funded by the Medical Research Council (MC_uu_12009). This work was supported by the National Institute for Health Research (NIHR) Oxford Biomedical Research Centre Haematology Theme at Oxford University Hospitals NHS Trust and University of Oxford and the charities Blood Buddies, Action Medical Research for Children and the Henry Smith Charity.
References
- Arnaud, L. , Saison, C. , Helias, V. , Lucien, N. , Steschenko, D. , Giarratana, M.C. , Prehu, C. , Foliguet, B. , Montout, L. , de Brevern, A.G. , Francina, A. , Ripoche, P. , Fenneteau, O. , Da Costa, L. , Peyrard, T. , Coghlan, G. , Illum, N. , Birgens, H. , Tamary, H. , Iolascon, A. , Delaunay, J. , Tchernia, G. & Cartron, J.P.A. (2010) Dominant mutation in the gene encoding the erythroid transcription factor KLF1 causes a congenital dyserythropoietic anemia. American Journal of Human Genetics, 87, 721–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ask, K. , Jasencakova, Z. , Menard, P. , Feng, Y.P. , Almouzni, G. & Groth, A. (2012) Codanin‐1, mutated in the anaemic disease CDAI, regulates Asf1 function in S‐phase histone supply. Embo Journal, 31, 3229–3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayas, M. , al‐Jefri, A. , Baothman, A. , al‐Mahr, M. , Mustafa, M.M. , Khalil, S. , Karaoui, M. & Solh, H. (2002) Transfusion‐dependent congenital dyserythropoietic anemia type I successfully treated with allogeneic stem cell transplantation. Bone Marrow Transplantation, 29, 681–68. [DOI] [PubMed] [Google Scholar]
- Babbs, C. , Roberts, N.A. , Sanchez‐Pulido, L. , McGowan, S.J. , Ahmed, M.R. , Brown, J.M. , Sabry, M.A. , Bentley, D.R. , McVean, G.A. , Donnelly, P. , Gileadi, O. , Ponting, C.P. , Higgs, D.R. & Buckle, V.J. (2013) Homozygous mutations in a predicted endonuclease are a novel cause of congenital dyserythropoietic anemia type I. Haematologica, 98, 1383–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bader‐Meunier, B. , Leverger, G. , Tchernia, G. , Schischmanoff, O. , Cynober, T. , Bernaudin, F. , Leblanc, T. , Munzer, M. , Roda, L. , Soler, C. , Thuret, I. & Delaunay, J. (2005) Clinical and laboratory manifestations of congenital dyserythropoietic anemia type I in a cohort of French children. Journal of Pediatric Hematology/Oncology, 27, 416–419. [DOI] [PubMed] [Google Scholar]
- Bain, B.J. , Clark, D.M. & Wilkins, B.S. (2010) Bone Marrow Pathology, pp. 461–499. Wiley‐Blackwell, Chichester, UK. [Google Scholar]
- Berdoukas, V. , Nord, A. , Carson, S. , Puliyel, M. , Hofstra, T. , Wood, J. & Coates, T.D. (2013) Tissue iron evaluation in chronically transfused children shows significant levels of iron loading at a very young age. American Journal of Hematology, 88, E283–E285. [DOI] [PubMed] [Google Scholar]
- Brichard, B. , Vermylen, C. , Scheiff, J.M. , Michaux, J.L. , Ninane, J. & Cornu, G. (1994) Two cases of congenital dyserythropoietic anaemia type I associated with unusual skeletal abnormalities of the limbs. British Journal of Haematology, 86, 201–202. [DOI] [PubMed] [Google Scholar]
- Brunning, R.D. , Orazi, A. , Germing, U. , Le Beau, M.M. , Porwit, A. , Baumann, I. , Vardiman, J.W. & Hellstrom‐Lindberg, E. (2008) Myelodysplastic syndromes/neoplasms, overview In: World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues (eds. by Swerdlow S.H., Campo E., Harris N.L., Jaffe E.S., Pileri S.A., Stein H., Thiele J. & Vardiman J.W.). IARC Press, Lyon. [Google Scholar]
- Buchbinder, D. , Nugent, D. , Vu, D. , Soni, A. , Stites, J. , Hsieh, L. & Puthenveetil, G. (2012) Unrelated hematopoietic stem cell transplantation in a patient with congenital dyserythropoietic anemia and iron overload. Pediatric Transplantation, 16, E69–E73. [DOI] [PubMed] [Google Scholar]
- Budych, K. , Helms, T.M. & Schultz, C. (2012) How do patients with rare diseases experience the medical encounter? Exploring role behavior and its impact on patient‐physician interaction. Health Policy, 105, 154–164. [DOI] [PubMed] [Google Scholar]
- Campos, E.I. , Fillingham, J. , Li, G. , Zheng, H. , Voigt, P. , Kuo, W.H. , Seepany, H. , Gao, Z. , Day, L.A. , Greenblatt, J.F. & Reinberg, D. (2010) The program for processing newly synthesized histones H3.1 and H4. Nature Structural & Molecular Biology, 17, 1343–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazzola, M. , Beguin, Y. , Bergamaschi, G. , Guarnone, R. , Cerani, P. , Barella, S. , Cao, A. & Galanello, R. (1999) Soluble transferrin receptor as a potential determinant of iron loading in congenital anaemias due to ineffective erythropoiesis. British Journal of Haematology, 106, 752–755. [DOI] [PubMed] [Google Scholar]
- Crookston, J.H. , Godwin, T.F. , Wightman, K.J.R. , Dacie, J.V. , Davis, J.A. , Lewis, S.M. & Patterson, M.J.L. (1966) Congenital Dyserythropoietic Anaemia. Abstracts from XIth Congress of the International Society of Haematology, Sydney, pp. 18.
- Davis, B.A. , Allard, S. , Qureshi, A. , Porter, J.B. , Pancham, S. , Win, N. , Cho, G. & Ryan, K. ; on behalf of the British Committee for Standards in Haematology . (2017) Guidelines on red cell transfusion in sickle cell disease. Part I: principles and laboratory aspects. British Journal of Haematology, 176, 179–191. [DOI] [PubMed] [Google Scholar]
- Dgany, O. , Avidan, N. , Delaunay, J. , Krasnov, T. , Shalmon, L. , Shalev, H. , Eidelitz‐Markus, T. , Kapelushnik, J. , Cattan, D. , Pariente, A. , Tulliez, M. , Cretien, A. , Schischmanoff, P.O. , Iolascon, A. , Fibach, E. , Koren, A. , Rossler, J. , Le Merrer, M. , Yaniv, I. , Zaizov, R. , Ben‐Asher, E. , Olender, T. , Lancet, D. , Beckmann, J.S. & Tamary, H. (2002) Congenital dyserythropoietic anemia type I is caused by mutations in codanin‐1. American Journal of Human Genetics, 71, 1467–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El‐Sheikh, A.A. , Hashem, H. , Holman, C. & Vyas, Y.M. (2014) Congenital dyserythropoietic anemia type I presenting as persistent pulmonary hypertension with pigeon chest deformity. Pediatric Blood & Cancer, 61, 1460–1462. [DOI] [PubMed] [Google Scholar]
- Ewing, R.M. , Chu, P. , Elisma, F. , Li, H. , Taylor, P. , Climie, S. , McBroom‐Cerajewski, L. , Robinson, M.D. , O'Connor, L. , Li, M. , Taylor, R. , Dharsee, M. , Ho, Y. , Heilbut, A. , Moore, L. , Zhang, S. , Ornatsky, O. , Bukhman, Y.V. , Ethier, M. , Sheng, Y. , Vasilescu, J. , Abu‐Farha, M. , Lambert, J.P. , Duewel, H.S. , Stewart, I.I. , Kuehl, B. , Hogue, K. , Colwill, K. , Gladwish, K. , Muskat, B. , Kinach, R. , Adams, S.L. , Moran, M.F. , Morin, G.B. , Topaloglou, T. & Figeys, D. (2007) Large‐scale mapping of human protein‐protein interactions by mass spectrometry. Molecular Systems Biology, 3, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Facon, T. , Mannessier, L. , Lepelley, P. , Weill, J. , Fenaux, P. , Dupriez, B. , Morel, P. & Jouet, J.P. (1990) Congenital diserythropoietic anemia type I. Report on monozygotic twins with associated hemochromatosis and short stature. Blut, 61, 248–250. [DOI] [PubMed] [Google Scholar]
- al‐Fawaz, I.M. & al‐Mashhadani, S.A. (1995) Congenital dyserythropoietic anaemia type I. Report of two siblings from Saudi Arabia. Acta Haematologica, 93, 50–53. [DOI] [PubMed] [Google Scholar]
- Fong, P.C. , Boss, D.S. , Yap, T.A. , Tutt, A. , Wu, P.J. , Mergui‐Roelvink, M. , Mortimer, P. , Swaisland, H. , Lau, A. , O'Connor, M.J. , Ashworth, A. , Carmichael, J. , Kaye, S.B. , Schellens, J.H.M. & de Bono, J.S. (2009) Inhibition of poly(ADP‐ribose) polymerase in tumors from BRCA mutation carriers. New England Journal of Medicine, 361, 123–134. [DOI] [PubMed] [Google Scholar]
- Frimmel, S. & Kniestedt, C. (2016) Angioid streaks in types I and II congenital dyserythropoietic anaemia (CDA). Klin Monbl Augenheilkd, 233, 482–487. [DOI] [PubMed] [Google Scholar]
- Fujino, H. , Doisaki, S. , Park, Y.D. , Hama, A. , Muramatsu, H. , Kojima, S. & Sumimoto, S. (2013) Congenital dyserythropoietic anemia type 1 with a novel mutation in the CDAN1 gene previously diagnosed as congenital hemolytic anemia. International Journal of Hematology, 97, 650–653. [DOI] [PubMed] [Google Scholar]
- Gambale, A. , Andolfo, I. , Russo, R. & Iolascon, A. (2016) Diagnosis and management of congenital dyserythropoietic anemias. Expert Review of Haematology, 9, 283–296. [DOI] [PubMed] [Google Scholar]
- Gerrard, G. , Valganon, M. , Foong, H.E. , Kasperaviuciute, D. , Iskander, D. , Game, L. , Muller, M. , Aitman, T.J. , Roberts, I. , de la Fuente, J. , Foroni, L. & Karadimitris, A. (2013) Target enrichment and high‐throughput sequencing of 80 ribosomal protein genes to identify mutations associated with Diamond‐Blackfan anaemia. British Journal of Haematology, 162, 530–536. [DOI] [PubMed] [Google Scholar]
- Goasguen, J.E. , Bennett, J.M. , Bain, B.J. , Brunning, R. , Vallespi, M.T. , Tomonaga, M. , Zini, G. & Renault, A. (2018) Dyserythropoiesis in the diagnosis of the myelodysplastic syndromes and other myeloid neoplasms: problem areas. British Journal of Haematology, 182, 526–533. [DOI] [PubMed] [Google Scholar]
- Goede, J.S. , Benz, R. , Fehr, J. , Schwarz, K. & Heimpel, H. (2006) Congenital dyserythropoietic anemia type I with bone abnormalities, mutations of the CDAN I gene, and significant responsiveness to alpha‐interferon therapy. Annals of Hematology, 85, 591–595. [DOI] [PubMed] [Google Scholar]
- Groth, A. , Ray‐Gallet, D. , Quivy, J.P. , Lukas, J. , Bartek, J. & Almouzni, G. (2005) Human Asf1 regulates the flow of S phase histones during replicational stress. Molecular Cell, 17, 301–311. [DOI] [PubMed] [Google Scholar]
- Groth, A. , Corpet, A. , Cook, A.J. , Roche, D. , Bartek, J. , Lukas, J. & Almouzni, G. (2007) Regulation of replication fork progression through histone supply and demand. Science, 318, 1928–1931. [DOI] [PubMed] [Google Scholar]
- Heimpel, H. (2004) Congenital dyserythropoietic anemias: epidemiology, clinical significance, and progress in understanding their pathogenesis. Annals of Hematology, 83, 613–621. 10.1007/s00277-004-0892-5 [DOI] [PubMed] [Google Scholar]
- Heimpel, H. & Wendt, F. (1968) Congenital dyserythropoietic anemia with karyorrhexis and multinuclearity of erythroblasts. Helvetica Medica Acta, 34, 103–115. [PubMed] [Google Scholar]
- Heimpel, H. , Anselstetter, V. , Chrobak, L. , Denecke, J. , Einsiedler, B. , Gallmeier, K. , Griesshammer, A. , Marquardt, T. , Janka‐Schaub, G. , Kron, M. & Kohne, E. (2003) Congenital dyserythropoietic anemia type II: epidemiology, clinical appearance, and prognosis based on long‐term observation. Blood, 102, 4576–4581. [DOI] [PubMed] [Google Scholar]
- Heimpel, H. , Schwarz, K. , Ebnother, M. , Goede, J.S. , Heydrich, D. , Kamp, T. , Plaumann, L. , Rath, B. , Roessler, J. , Schildknecht, O. , Schmid, M. , Wuillemin, W. , Einsiedler, B. , Leichtle, R. , Tamary, H. & Kohne, E. (2006) Congenital dyserythropoietic anemia type I (CDA I): molecular genetics, clinical appearance, and prognosis based on long‐term observation. Blood, 107, 334–340. [DOI] [PubMed] [Google Scholar]
- Heimpel, H. , Dührsen, U. , Hofbauer, P. , Rigamonti‐Wermlinger, V. , Kreuser, E.D. , Schwarz, K. , Solenthaler, M. & Pauls, S. (2009) Bulky extramedullary hematopoiesis is not a rare complication of congenital dyserythropoietic anemia. Annals of Hematology, 88, 937–941. [DOI] [PubMed] [Google Scholar]
- Heimpel, H. , Kellermann, K. , Neuschwander, N. , Hogel, J. & Schwarz, K. (2010) The morphological diagnosis of congenital dyserythropoietic anemia: results of a quantitative analysis of peripheral blood and bone marrow cells. Haematologica, 95, 1034–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iolascon, A. & Delaunay, J. (2009) Close to unraveling the secrets of congenital dyserythropoietic anemia types I and II. Haematologica, 94, 599–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iolascon, A. , Russo, R. & Delaunay, J. (2011) Congenital dyserythropoietic anemias. Current Opinion in Hematology, 18, 146–151. [DOI] [PubMed] [Google Scholar]
- Iolascon, A. , Esposito, M.R. & Russo, R. (2012) Clinical aspects and pathogenesis of congenital dyserythropoietic anemias: from morphology to molecular approach. Haematologica, 97, 1786–1794. 10.3324/haematol.2012.072207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iolascon, A. , Heimpel, H. , Wahlin, A. & Tamary, H. (2013) Congenital dyserythropoietic anemias: molecular insights and diagnostic approach. Blood, 122, 2162–2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iolascon, A. , Andolfo, I. , Barcellini, W. , Corcione, F. , Garçon, L. , De Franceschi, L. , Pignata, C. , Graziadei, G. , Pospisilova, D. , Rees, D.C. , de Montalembert, M. , Rivella, S. , Gambale, A. , Russo, R. , Ribeiro, L. , Vives‐Corrons, J. , Martinez, P.A. , Kattamis, A. , Gulbis, B. , Cappellini, M.D. , Roberts, I. & Tamary, H. (2017) Recommendations regarding splenectomy in hereditary hemolytic anemias. Haematologica, 102, 1304–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasencakova, Z. & Groth, A. (2010) Restoring chromatin after replication: how new and old histone marks come together. Seminars in Cell & Developmental Biology, 21, 231–237. [DOI] [PubMed] [Google Scholar]
- Jasencakova, Z. , Scharf, A.N. , Ask, K. , Corpet, A. , Imhof, A. , Almouzni, G. & Groth, A. (2010) Replication stress interferes with histone recycling and predeposition marking of new histones. Molecular Cell, 37, 736–743. [DOI] [PubMed] [Google Scholar]
- Kamiya, T. & Manabe, A. (2010) Congenital dyserythropoietic anemia. International Journal of Hematology, 92, 432–438. [DOI] [PubMed] [Google Scholar]
- Kato, K. , Sugitani, M. , Kawataki, M. , Ohyama, M. , Aida, N. , Koga, N. , Ijiri, R. , Imaizumi, K. , Kigasawa, H. , Tanaka, Y. & Itani, Y. (2001) Congenital dyserythropoietic anemia type 1 with fetal onset of severe anemia. Journal of Pediatric Hematology/Oncology, 23, 63–66. [DOI] [PubMed] [Google Scholar]
- Kautz, L. , Jung, G. , Du, X. , Gabayan, V. , Chapman, J. , Nasoff, M. , Nemeth, E. & Ganz, T. (2015) Erythroferrone contributes to hepcidin suppression and iron overload in a mouse model of β‐thalassemia. Blood, 126, 2031–2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawabata, H. , Doisaki, S. , Okamoto, A. , Uchiyama, T. , Sakamoto, S. , Hama, A. , Hosoda, K. , Fujikura, J. , Kanno, H. , Fujii, H. , Tomosugi, N. , Nakao, K. , Kojima, S. & Takaori‐Kondo, A. (2012) A case of congenital dyserythropoietic anemia type 1 in a Japanese adult with a CDAN1 gene mutation and an inappropriately low serum hepcidin‐25 level. Internal Medicine, 51, 917–920. [DOI] [PubMed] [Google Scholar]
- Laganeckas, M. , Margelevicius, M. & Venclovas, C. (2011) Identification of new homologs of PD‐(D/E)XK nucleases by support vector machines trained on data derived from profile‐profile alignments. Nucleic Acids Research, 39, 1187–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landau, D. , Kapelushnik, J. , Harush, M.B. , Marks, K. & Shalev, H. (2015) Persistent pulmonary hypertension of the newborn associated with severe congenital anemia of various etiologies. Journal of Pediatric Hematology/Oncology, 37, 60–62. [DOI] [PubMed] [Google Scholar]
- Lavabre‐Bertrand, T. , Blanc, P. , Navarro, R. , Saghroun, M. , Vannereau, H. , Braun, M. , Wagner, A. , Taïb, J. , Lavabre‐Bertrand, C. & Navarro, M. (1995) alpha‐Interferon therapy for congenital dyserythropoiesis type I. British Journal of Haematology, 89, 929–932. [DOI] [PubMed] [Google Scholar]
- Lavabre‐Bertrand, T. , Ramos, J. , Delfour, C. , Henry, L. , Guiraud, I. , Carillo, S. , Wagner, A. , Bureau, J.P. & Blanc, P. (2004) Long‐term alpha interferon treatment is effective on anaemia and significantly reduces iron overload in congenital dyserythropoiesis type I. European Journal of Haematology, 73, 380–383. [DOI] [PubMed] [Google Scholar]
- Lewis, S.M. (2001) Diagnostic radionuclides in haematology In: Dacie and Lewis Practical Haematology (eds. by Lewis S.M., Bain B.J. & Bates I.), pp. 324–326. Elsevier Ltd, London, UK. [Google Scholar]
- Liljeholm, M. , Irvine, A.F. , Vikberg, A.L. , Norberg, A. , Month, S. , Sandstrom, H. , Wahlin, A. , Mishima, M. & Golovleva, I. (2013) Congenital dyserythropoietic anemia type III (CDA III) is caused by a mutation in kinesin family member, KIF23. Blood, 121, 4791–4799. [DOI] [PubMed] [Google Scholar]
- Lin, S.M. , Chen, M. , Ma, E.S. , Lam, Y.H. , Wong, K.Y. & Tang, M.H. (2014) Intrauterine therapy in a fetus with congenital dyserythropoietic anaemia type I. Journal of Obstetrics and Gynaecology, 34, 352–353. [DOI] [PubMed] [Google Scholar]
- Marwaha, R.K. , Bansal, D. , Trehan, A. & Garewal, G. (2005) Interferon therapy in congenital dyserythropoietic anemia type I/II. Pediatric Hematology and Oncology, 22, 133–138. [DOI] [PubMed] [Google Scholar]
- Mello, J.A. , Sillje, H.H. , Roche, D.M. , Kirschner, D.B. , Nigg, E.A. & Almouzni, G. (2002) Human Asf1 and CAF‐1 interact and synergize in a repair‐coupled nucleosome assembly pathway. EMBO Reports, 3, 329–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meznarich, J.A. , Draper, L. , Christensen, R.D. , Yaish, H.M. , Luem, N.D. , Pysher, T.J. , Jung, G. , Nemeth, E. , Ganz, T. & Ward, D.M. (2018) Fetal presentation of congenital dyserythropoietic anemia type 1 with novel compound heterozygous CDAN1 mutations. Blood Cells, Molecules, & Diseases, 71, 63–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milkins, C. , Berryman, J. , Cantwell, C. , Elliott, C. , Haggas, R. , Jones, J. , Rowley, M. , Williams, M. , Win, N. ; for the British Committee for Standards in Haematology . (2013) Guidelines for pre‐transfusion compatibility procedures in blood transfusion laboratories. Transfusion Medicine, 23, 3–35. [DOI] [PubMed] [Google Scholar]
- Moir‐Meyer, G. , Cheong, P.L. , Olijnik, A.A. , Brown, J.M. , Knight, S. , King, A. , Kurita, R. , Nakamura, Y. , Gibbons, R.J. , Higgs, D.R. , Buckle, V.J. & Babbs, C. (2018) Robust CRISPR/Cas9 genome editing of the HUDEP‐2 erythroid precursor line using plasmids and single‐stranded oligonucleotide donors. Methods and Protocols, 1, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols, K.E. , Crispino, J.D. , Poncz, M. , White, J.G. , Orkin, S.H. , Maris, J.M. & Weiss, M.J. (2000) Familial dyserythropoietic anaemia and thrombocytopenia due to an inherited mutation in GATA1. Nature Genetics, 24, 266–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noy‐Lotan, S. , Dgany, O. , Lahmi, R. , Marcoux, N. , Krasnov, T. , Yissachar, N. , Ginsberg, D. , Motro, B. , Resnitzky, P. , Yaniv, I. , Kupfer, G.M. & Tamary, H. (2009) Codanin‐1, the protein encoded by the gene mutated in congenital dyserythropoietic anemia type I (CDAN1), is cell cycle‐regulated. Haematologica, 94, 629–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parez, N. , Dommergues, M. , Zupan, V. , Chambost, H. , Fieschi, J.B. , Delaunay, J. , Miélot, F. , Cramer, E.M. , Dommergues, J.P. , Wickramasinghe, S.N. & Tchernia, G. (2000) Severe congenital dyserythropoietic anaemia type I: prenatal management, transfusion support and alpha‐interferon therapy. British Journal of Haematology, 110, 420–423. [DOI] [PubMed] [Google Scholar]
- Pielage, J. , Stork, T. , Bunse, I. & Klämbt, C. (2003) The Drosophila cell survival gene discs lost encodes a cytoplasmic Codanin‐1‐like protein, not a homolog of tight junction PDZ protein Patj. Developmental Cell, 5, 841–851. [DOI] [PubMed] [Google Scholar]
- Pop, R. , Shearstone, J.R. , Shen, Q. , Liu, Y. , Hallstrom, K. , Koulnis, M. , Gribnau, J. & Socolovsky, M. (2010) A key commitment step in erythropoiesis is synchronized with the cell cycle clock though mutual inhibition between PU.1 and S‐phase progression. Plos Biology, 8, pii: e1000484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renella, R. & Wood, W.G. (2009) The congenital dyserythropoietic anemias. Hematology/Oncology Clinics of North America, 23, 283–306. [DOI] [PubMed] [Google Scholar]
- Renella, R. , Roberts, N.A. , Brown, J.M. , De Gobbi, M. , Bird, L.E. , Hassanali, T. , Sharpe, J.A. , Sloane‐Stanley, J. , Ferguson, D.J. , Cordell, J. , Buckle, V.J. , Higgs, D.R. & Wood, W.G. (2011) Codanin‐1 mutations in congenital dyserythropoietic anemia type 1 affect HP1α localization in erythroblasts. Blood, 117, 6928–6938. [DOI] [PubMed] [Google Scholar]
- Resnitzky, P. , Shaft, D. , Shalev, H. , Kapelushnik, J. , Dgany, O. , Krasnov, T. & Tamary, H. (2017) Morphological features of congenital dyserythropoietic anemia type I: the role of electron microscopy in diagnosis. European Journal of Haematology, 99, 366–371. [DOI] [PubMed] [Google Scholar]
- Roy, N.B. & Pavord, S. (2018) Management of other inherited red cell disorders in pregnancy In: The Obstetric Hematology Manual (eds. by Pavord S. & Hunt B.), pp. 93–103. Cambridge University Press, Cambridge, UK. [Google Scholar]
- Roy, N.B. , Wilson, E.A. , Henderson, S. , Wray, K. , Babbs, C. , Okoli, S. , Atoyebi, W. , Mixon, A. , Cahill, M.R. , Carey, P. , Cullis, J. , Curtin, J. , Dreau, H. , Ferguson, D.J. , Gibson, B. , Hall, G. , Mason, J. , Morgan, M. , Proven, M. , Qureshi, A. , Sanchez Garcia, J. , Sirachainan, N. , Teo, J. , Tedgard, U. , Higgs, D. , Roberts, D. , Roberts, I. & Schuh, A. (2016) A novel 33‐Gene targeted resequencing panel provides accurate, clinical‐grade diagnosis and improves patient management for rare inherited anaemias. British Journal of Haematology, 175, 318–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo, R. , Andolfo, I. , Manna, F. , De Rosa, G. , De Falco, L. , Gambale, A. , Bruno, M. , Mattè, A. , Ricchi, P. , Girelli, D. , De Franceschi, L. & Iolascon, A. (2016) Increased levels of ERFE‐encoding. Blood, 128, 1899–1902. [DOI] [PubMed] [Google Scholar]
- Russo, R. , Andolfo, I. , Manna, F. , Gambale, A. , Marra, R. , Rosato, B.E. , Caforio, P. , Pinto, V. , Pignataro, P. , Radhakrishnan, K. , Unal, S. , Tomaiuolo, G. , Forni, G.L. & Iolascon, A. (2018) Multi‐gene panel testing improves diagnosis and management of patients with hereditary anemias. American Journal of Hematology, 93, 672–682. [DOI] [PubMed] [Google Scholar]
- Schindler, D. & Hoehn, H. (eds) (2007) Fanconi anemia. A paradigmatic disease for the understanding of cancer and aging. Monographs in Human Genetics, 15, 1–8. [Google Scholar]
- Schwarz, K. , Iolascon, A. , Verissimo, F. , Trede, N.S. , Horsley, W. , Chen, W. , Paw, B.H. , Hopfner, K.P. , Holzmann, K. , Russo, R. , Esposito, M.R. , Spano, D. , De Falco, L. , Heinrich, K. , Joggerst, B. , Rojewski, M.T. , Perrotta, S. , Denecke, J. , Pannicke, U. , Delaunay, J. , Pepperkok, R. & Heimpel, H. (2009) Mutations affecting the secretory COPII coat component SEC23B cause congenital dyserythropoietic anemia type II. Nature Genetics, 41, 936–U105. [DOI] [PubMed] [Google Scholar]
- Shalev, H. , Kapleushnik, Y. , Haeskelzon, L. , Degani, O. , Kransnov, T. , Sphilberg, O. , Moser, A. , Yaniv, I. & Tamary, H. (2002) Clinical and laboratory manifestations of congenital dyserythropoietic anemia type I in young adults. European Journal of Haematology, 68, 170–174. [DOI] [PubMed] [Google Scholar]
- Shalev, H. , Kapelushnik, J. , Moser, A. , Dgany, O. , Krasnov, T. & Tamary, H. (2004) A comprehensive study of the neonatal manifestations of congenital dyserythropoietic anemia type I. Journal of Pediatric Hematology/Oncology, 26, 746–748. [DOI] [PubMed] [Google Scholar]
- Shalev, H. , Al‐Athamen, K. , Levi, I. , Levitas, A. & Tamary, H. (2017) Morbidity and mortality of adult patients with congenital dyserythropoietic anemia type I. European Journal of Haematology, 98, 13–18. [DOI] [PubMed] [Google Scholar]
- Shearstone, J.R. , Pop, R. , Bock, C. , Boyle, P. , Meissner, A. & Socolovsky, M. (2011) Global DNA demethylation during mouse erythropoiesis in vivo. Science, 334, 799–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shefer Averbuch, N. , Steinberg‐Shemer, O. , Dgany, O. , Krasnov, T. , Noy‐Lotan, S. , Yacobovich, J. , Kuperman, A.A. , Kattamis, A. , Ben Barak, A. , Roth‐Jelinek, B. , Chubar, E. , Shabad, E. , Dufort, G. , Ellis, M. , Wolach, O. , Pazgal, I. , Abu Quider, A. , Miskin, H. & Tamary, H. (2018) Targeted next generation sequencing for the diagnosis of patients with rare congenital anemias. European Journal of Haematology, 101, 297–304. [DOI] [PubMed] [Google Scholar]
- Singleton, B.K. , Fairweather, V.S.S. , Lau, W. , Parsons, S.F. , Burton, N.M. , Frayne, J. , Brady, R.L. & Anstee, D.J. (2009) A novel EKLF mutation in a patient with dyserythropoietic anemia: the first association of EKLF with disease in man. Blood, 114, 72–72. [Google Scholar]
- Songdej, D. , Babbs, C. & Higgs, D.R. (2017) An international registry of survivors with Hb Bart's hydrops fetalis syndrome. Blood, 129, 1251–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steczkiewicz, K. , Muszewska, A. , Knizewski, L. , Rychlewski, L. & Ginalski, K. (2012) Sequence, structure and functional diversity of PD‐(D/E)XK phosphodiesterase superfamily. Nucleic Acids Research, 40, 7016–7045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamary, H. & Dgany, O. (2009) Congenital dyserythropoietic anemia type I In: GeneReviews® (eds. by Adam M.P., Ardinger H.H., Pagon R.A., Wallace S. E., Bean L.J.H., Stephens K. & Amemiya A.). University of Washington, Seattle, WA, USA; 1993‐2018. Apr 21 [updated 2016 Aug 25]. [PubMed] [Google Scholar]
- Tamary, H. , Shalev, H. , Pinsk, V. , Zoldan, M. & Zaizov, R. (1999) No response to recombinant human erythropoietin therapy in patients with congenital dyserythropoietic anemia type I. Pediatric Hematology and Oncology, 16, 165–168. [DOI] [PubMed] [Google Scholar]
- Tamary, H. , Dgany, O. , Proust, A. , Krasnov, T. , Avidan, N. , Eidelitz‐Markus, T. , Tchernia, G. , Genevieve, D. , Cormier‐Daire, V. , Bader‐Meunier, B. , Ferrero‐Vacher, C. , Munzer, M. , Gruppo, R. , Fibach, E. , Konen, O. , Yaniv, I. & Delaunay, J. (2005) Clinical and molecular variability in congenital dyserythropoietic anaemia type I. British Journal of Haematology, 130, 628–634. [DOI] [PubMed] [Google Scholar]
- Tamary, H. , Shalev, H. , Perez‐Avraham, G. , Zoldan, M. , Levi, I. , Swinkels, D.W. , Tanno, T. & Miller, J.L. (2008) Elevated growth differentiation factor 15 expression in patients with congenital dyserythropoietic anemia type I. Blood, 112, 5241–5244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamary, H. , Marcoux, N. , Noy‐Lotan, S. , Yaniv, I. & Dgany, O. (2010) Codanin‐1, the product of the gene mutated in congenital dyserythropoietic anemia type I (CDA I), binds to histone chaperone Asf1a and inhibits its nucleosome assembly activity. Blood, 116, 442–442. [Google Scholar]
- Tickle, C. (2015) How the embryo makes a limb: determination, polarity and identity. Journal of Anatomy, 227, 418–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voskaridou, E. & Terpos, E. (2008) Pathogenesis and management of osteoporosis in thalassemia. Pediatric Endocrinology Reviews: PER, 6 (Suppl 1), 86–93. [PubMed] [Google Scholar]
- Wang, S. , Yu, M. , Zheng, X. & Dong, S. (2018) A Bayesian network meta‐analysis on the efficacy and safety of eighteen targeted drugs or drug combinations for pulmonary arterial hypertension. Drug Delivery, 25, 1898–1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickramasinghe, S.N. (1997) Dyserythropoiesis and congenital dyserythropoietic anaemias. British Journal of Haematology, 98, 785–797. [DOI] [PubMed] [Google Scholar]
- Wickramasinghe, S.N. (1998) Congenital dyserythropoietic anaemias: clinical features, haematological morphology and new biochemical data. Blood Reviews, 12, 178–200. [DOI] [PubMed] [Google Scholar]
- Wickramasinghe, S.N. & Pippard, M.J. (1986) Studies of erythroblast function in congenital dyserythropoietic anemia, type‐I ‐ evidence of impaired DNA, RNA, and protein‐synthesis and unbalanced globin chain synthesis in ultrastructurally abnormal‐cells. Journal of Clinical Pathology, 39, 881–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickramasinghe, S.N. & Wood, W.G. (2005) Advances in the understanding of the congenital dyserythropoietic anaemias. British Journal of Haematology, 131, 431–446. [DOI] [PubMed] [Google Scholar]
- Wickramasinghe, S.N. , Hasan, R. & Smythe, J. (1997) Reduced interferon‐alpha production by Epstein‐Barr virus transformed B‐lymphoblastoid cell lines and lectin‐stimulated lymphocytes in congenital dyserythropoietic anaemia type I. British Journal of Haematology, 98, 295–298. [DOI] [PubMed] [Google Scholar]
- Zeman, M.K. & Cimprich, K.A. (2014) Causes and consequences of replication stress. Nature Cell Biology, 16, 2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, B.B. , Mei, Y. , Schipma, M.J. , Roth, E.W. , Bleher, R. , Rappoport, J.Z. , Wickrema, A. , Yang, J. & Ji, P. (2016) Nuclear condensation during mouse erythropoiesis requires caspase‐3‐mediated nuclear opening. Developmental Cell, 36, 498–510. [DOI] [PMC free article] [PubMed] [Google Scholar]