

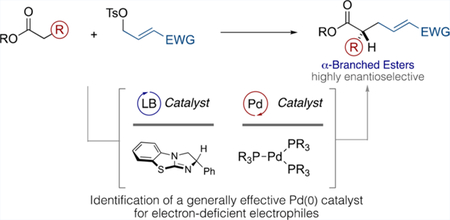
Abstract
We have identified a generally effective Pd catalyst for the highly enantioselective cooperative Lewis base/Pd-catalyzed α-allylation of aryl acetic esters using electron-deficient electrophiles. Changing between aldehyde, ketone, ester, and amide substituents at the terminus of intermediate cationic π-(allyl)Pd species affects both the efficiency of the reaction and, in the case of amides, control over the stereochemistry of the product alkene, as a function of the ligand. Tris[tri(2-thienyl)phosphino]Pd(0) serves as a broadly effective catalyst and overcomes these challenges to provide a general, high-yielding, and operationally simple C(sp3)–C(sp3) bond-forming method that gives products with high levels of enantioselectivity.
Keywords: cooperative catalysis, Lewis base, palladium, allylic alkylation, enantioselective, synergistic
Graphical Abstract
1. INTRODUCTION
Enantioselective Pd-catalyzed allylic alkylation has emerged as one of the most robust and versatile methods for the construction of C(sp3)−C(sp3) bonds.1 During such reactions, a range of carbon-based nucleophiles engage intermediary cationic π-(allyl)Pd(II) electrophiles and, by effectively tailoring the steric and electronic parameters of the supporting chiral nonracemic P(III) ligands on palladium,2 excellent control over both reactivity and enantiofacial selectivity is possible.
Our laboratory has a long-standing interest in enantioselective allylic alkylation, and we have recently described a cooperative Lewis base/Pd dual catalysis platform that efficiently addresses the stereocontrol challenges engendered by acyclic ester pronucleophiles.3 In this regime, the Lewis base serves to control and direct intermediate enolate geometry, as well as enantiofacial selectivity, whereas the Pd catalyst is primarily responsible for regulation of electrophile reactivity (see Figure 1a and Scheme 1 (left)). This process has proven to be general, as a consequence of the disparate roles played by these two catalysts, which permits the reactivity of the Pd center to be tuned via the ancillary ligands without compromising the nature or efficiency of enantiocontrol, which is governed by the Lewis base. We have exploited this feature to overcome unexpected reactivity and stereocontrol challenges posed by differentially functionalized electrophiles.3 Herein, we further demonstrate the utility of this by identifying a broadly effective Pd(0) catalyst that allows for the enantioselective α-alkylation of aryl acetic acid esters by a wide range of carbonyl-substituted electrophiles.
Figure 1.
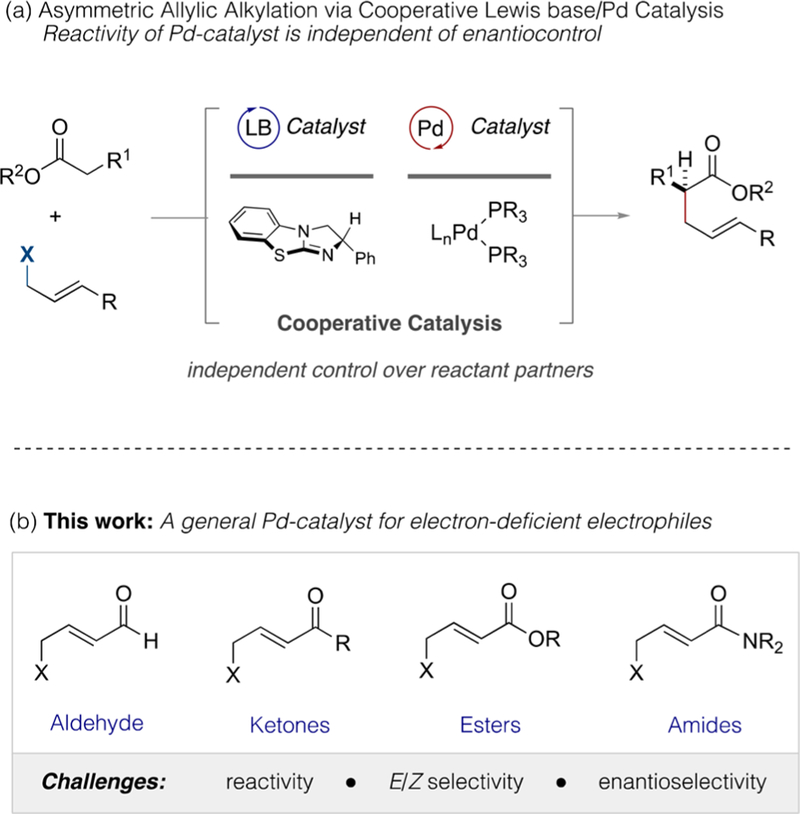
(a) Cooperative Lewis base/Pd-catalyzed enantioselective allylic alkylation using acyclic ester pro-nucleophiles. (b) Can a Pd catalyst be identified to overcome the varying influence of common electron-withdrawing groups on reaction yield, E/Z selectivity, and enantioselectivity?
Scheme 1.
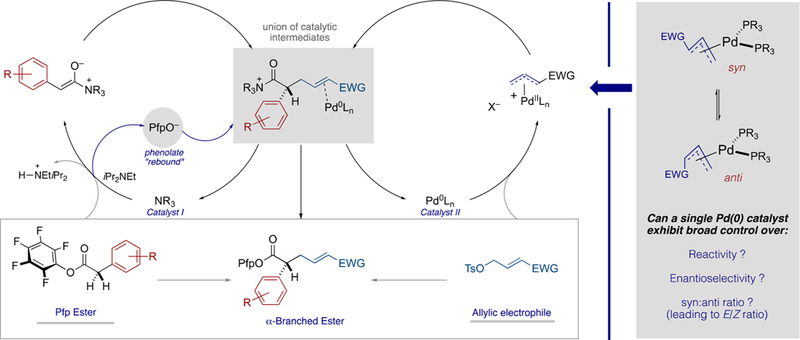
Postulated Mechanism (Left); Challenges for Pd(0) Catalysis (Right)
The efficiency of Pd-catalyzed allylic alkylation is significantly affected by the precise electronic and steric composition of intermediate π-(allyl)Pd(II) species.1 Although their reactivity can be fine-tuned by the supporting ligands, it is nonetheless challenging to identify single-catalyst systems that are broadly effective. For example, the presence of strongly electron-withdrawing carbonyl functional groups at the termini of the organopalladium intermediates directs attack by the incoming nucleophile to the distal carbon by virtue of orbital control,4 thus reinforcing the regiochemical preference in favor of linear products as is common in Pd-catalyzed allylic alkylation. However, the degree of electron-withdrawing character, dipole, and steric demand varies significantly across the carbonyl series5 and can drastically affect the behavior of the electrophilic intermediates. For example, isomeric syn and anti π-(allyl)Pd(II) complexes can differ greatly in their relative electrophilicities and, as such, their ratio and the rate of their interconversion via π−σ−π isomerization can affect both the product alkene stereochemistry and the enantioselectivity of bond formation (see Scheme 1 (right)).6,7 Thus, identification of a broadly effective and general Pd/ligand system is far less trivial than it might first appear (see Figure 1b and Scheme 1 (right)).
2. RESULTS AND DISCUSSION
2.1. Initial Substituent Assessment.
Beginning with our previously developed conditions employing Birman’s benzotetramisole (BTM),8,9 in conjunction with Buchwald’s thirdgeneration Xantphos-ligated Pd precatalyst,10 we evaluated the influence of four common carbonyl-containing functional groups on the reactivity, alkene stereoselectivity, and enantioselectivity of our cooperative catalysis process (see Scheme 2). Thus, the direct α-allylation of phenyl acetic acid pentafluorophenyl ester with allylic tosylates11 1a–d revealed stark differences in the challenges facing the Pd catalyst and control elements that are inherent to the putative cationic π-(allyl)Pd(II) intermediates. The ester-substituted electrophile 1a provided the expected product in good yield, as a single alkene isomer and with good levels of enantiocontrol.12 However, decreasing the carbonyl oxidation level in electrophiles 1c and 1d provided lower yields of products, which also exhibited slightly lower levels of enantiocontrol, albeit as a single alkene E-isomer. Finally, most challenging was the amide-appended electrophile 1b, which also gave lower chemical yields as well as poor levels of control over alkene stereochemistry. Noteworthy is the high enantioselectivity of the major E-isomer. While these results demonstrate the flexibility of this cooperative catalysis protocol and further validate the broad capability of the Lewis base catalyst to impart useful levels of enantioselectivity, they also reveal disparate challenges facing the Pd catalyst that must be overcome in order to develop a fully general protocol for the direct asymmetric α-allylation of aryl acetic acid esters by electron-deficient electrophiles.
Scheme 2.
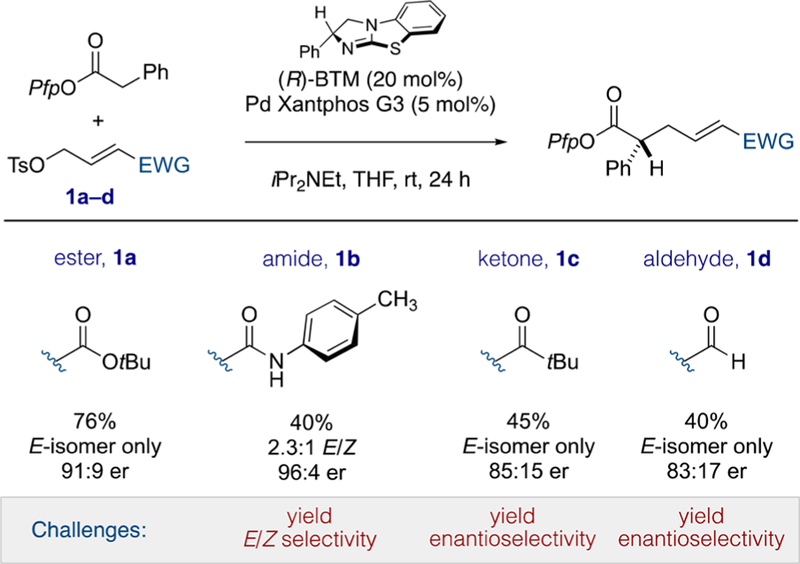
Influence of Commonly Encountered Electron-Withdrawing Groups on Reaction Yield, E/Z Selectivity, and Enantioselectivity
We have previously described the inherent modularity of this mechanistic construct, where the supporting ligands on Pd can be tuned to (i) overcome poor steric-derived reactivity, (ii) control chemoselectivity, and (iii) control alkene stereochemistry, in a manner that is independent of enantioselectivity, which continues to be administered by the Lewis base catalyst.3
In considering these results, and other notable contributions by the groups of Smith,13 Hartwig,14 and Gong,15 we expected that a Pd catalyst could be identified that would address the distinct challenges posed by each of the functional groups presented in Figure 1. As the amide-substituted electrophile 1b possessed the added complication of control over the alkene stereochemistry, we began our investigations with this subclass.
2.2. Amide-Substituted Electrophiles.
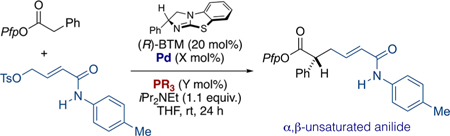
Informed by our earlier studies, we surveyed a representative range of bidentate and monodentate phosphine ligands and assessed their capacity to influence both chemical yield and alkene stereoselectivity (see Table 1). Comparison of Xantphos-ligated Pd(0) derived from Buchwald’s G3 precatalyst,10 or formed in situ by stirring with Pd2dba3 gave similar levels of E/ Z selectivity and enantioselectivity, although product yield using the latter method was slightly improved (Table 1, entries 1 and 2). Using in situ catalyst preparation, other bidentate phosphines with varying bite angle and backbone flexibility offered no improvement (Table 1, entries 3–5). Standard monophosphines (Table 1, entries 6–10) exhibited a range of activity and E/Z selectivity, with PCy3 and P(o-tolyl)3 providing no reaction (Table 1, entries 6 and 7) and P(4-OMePh)3 affording comparable levels of reactivity and selectivity to the parent PdXantphos G3 system (see Table 1, entry 8 vs entry 1). We have previously described the enhanced reactivity engendered by P(2-furyl)3 and P(2-thienyl)3 ligands in related cooperative catalysis processes via C1-ammonium enolate nucleophiles.3c,d In the case of the former, the product was obtained exclusively as the E-isomer, albeit in prohibitively low yield and with a slight reduction in enantioselectivity (Table 1, entry 9). The latter provided the product in useful 6.6:1 E/Z ratio, with excellent levels of enantioselectivity and more useful chemical yield (entry 10). A brief assessment of Pd:ligand stoichiometry (entries 11 and 12) resulted in further yield enhancement (90%) and E/Z stereoselectivity (7.9:1) without compromising the level of enantiocontrol (96:4 er). At this juncture, we considered our optimization complete and moved forward to assess the scope of the appended amide and performed a direct comparison between our previous conditions employing PdXantphos G3 and our newly identified in situ Pd2dba3/P(2-thienyl)3 conditions (see Scheme 3, columns 1 and 2).
Table 1.
Optimization of 4-Me-Anilide-Substituted Electrophile: Effect of Ligand
 | |||||
|---|---|---|---|---|---|
| Entrya | Pd(mol%) | PR3(mol%) | Yield[%]b | E:Zc | erd |
| 1 | PdXantphos G3 | -- | 40 | 2.3:1 | 96.4 |
| 2 | Pd2dba3(5) | Xantphos (10) | 56 | 2.5:1 | 95.5 |
| 3 | Pd2dba3(5) | DPEphos (10) | 40 | 3:1 | 96.4 |
| 4 | Pd2dba3(5) | dppf (10) | 37 | 2.1:1 | 95.5 |
| 5 | Pd2dba3(5) | dppe (10) | 26 | 1.9:1 | 96.4 |
| 6 | Pd2dba3(5) | PCy3 (20) | 0 | -- | -- |
| 7 | Pd2dba3(5) | P(o-tolyl)3 (20) | 0 | -- | -- |
| 8 | Pd2dba3(5) | P(4-OMePh)3 (20) | 28 | 2.5:1 | 94.6 |
| 9 | Pd2dba3(5) | P(2-furyl)3 (20) | 15 | 100:1 | 92.8 |
| 10 | Pd2dba3(5) | P(2-thienyl)3 (20) | 43 | 6.6:1 | 96.4 |
| 11 | Pd2dba3(5) | P(2-thienyl)3 (10) | 85 | 7.2:1 | 94.6 |
| 12 | Pd2dba3 (10) | P(2-thienyl)3 (25) | 90 | 7.9:1 | 96.4 |
Reactions performed on a 0.1 mmol scale.
Yields determined by 1H NMR by comparison with an internal standard (1,2,4,5-tetramethylbenzene).
E/Z ratio calculated from crude 1H NMR.
Determined by chiral HPLC analysis.
Scheme 3.
Scope and Comparison of Ligands/Conditions for Amide-Substituted Electrophiles
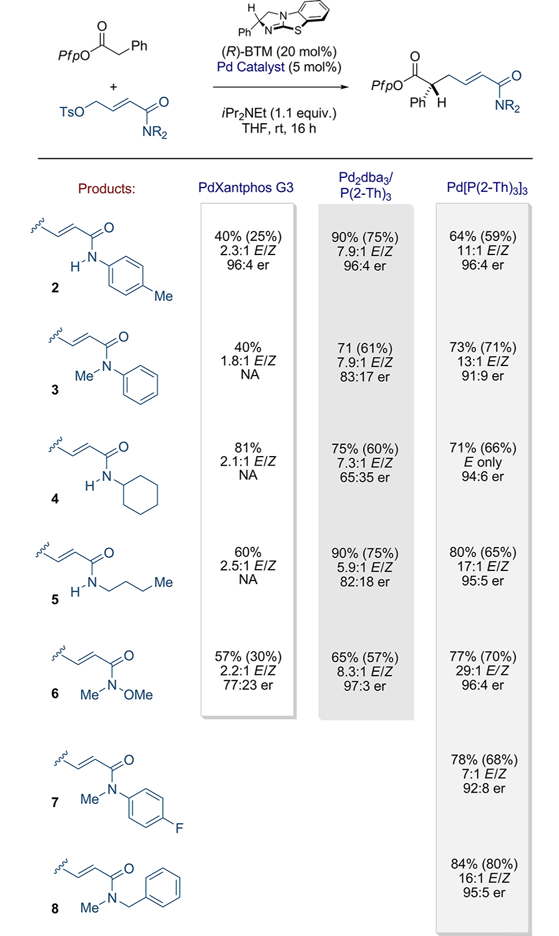
aYields determined by 1H NMR by comparison with an internal standard (1,2,4,5-tetramethylbenzene). Isolated yields in parentheses. Enantiomeric ratio determined by chiral HPLC in comparison to the racemate. P(2-Th)3 = tri(2-thienyl)phosphine.
Comparison of secondary and tertiary anilides (2 and 3), branched secondary amide (4), acyclic secondary amide (5), and Weinreb amide (6) revealed the broad superiority of the Pd2dba3/P(2-thienyl)3 protocol in terms of yield and E/Z selectivity; however, enantioselectivity was largely variable and was attributed to product epimerization during the prolonged reactions times.16 Accordingly, and because of the induction time necessary for in situ catalyst formation, we expected that a preformed Pd(0) catalyst would exhibit greater reactivity and enable a reduction in reaction time (see Supporting Information for full time study results). Employing Pd[P(2-thienyl)3]3 revealed its generally superior control over the alkene E/Z ratio and preservation of the high levels of enantioselectivity (see Scheme 3, column 3).17
At this juncture, it is appropriate to offer some comments concerning the alkene isomer ratio. In Pd-catalyzed allylic alkylation the E/Z selectivity originates from the relative stability (and reactivity) of syn and anti π-(allyl)Pd(II) intermediates,6 which, in turn, can derive from electronic and/or steric bias imparted by both the substrate and the supporting ligands. On a case -by-case basis, some combination of these influence not only the relative energies of syn and anti but also the relative facility of π–σ−π isomerization en route.18 The data presented in Table 1 and Scheme 3 indicate clear ligand dependence, with respect to the obtained alkene stereochemistry and led us to consider those factors responsible. Although neither in situ spectroscopic interrogation nor single-crystal analysis of species relevant to catalysis have offered any insight, available Xantphos ligand analogues provide a toolbox for potential qualitative assessment via modulation of the steric demand or electronic parameters of the P(III) donor atom substituents (see Scheme 4, Question).19 Consequently, formation of the parent α,β-unsaturated anilide 2 (E/Z 2.3:1) was re-evaluated using commercially available Xantphos ligand analogues 9–12 that present an indicative range of steric and electronic features. The gem-dimethyl → NH ligand backbone modification in NiXantphos 9 constitutes a remote electronic modification that does not significantly affect the donor properties of the P(III) donor atom.20 Consistent with this, 9 gave the product in almost-identical alkene ratio (2.5:1 E/Z) to the parent ligand (see Scheme 3, column 1) suggesting little substantive influence over the reactivity of the associated π-(allyl)Pd(II) fragment. More direct influence can be expected by altering the substituents on the P(III) donor atom, and the corresponding bis-phosphonous diamide 10 or Reetz’s (R)-BINOL-derived diphosphonite ligand 11 only further favored the E alkene isomer relative to Xantphos itself. Given the enhanced π-accepting character of these substituents via the classical backdonation from the appropriate metal d-orbital to the lowlying σ*P–N and σ*P–O orbitals,21 respectfully, this might well indicate why 2-(thienyl)phosphine, which is a ligand with low steric demand and strong π-accepting character,22 is so generally effective. Finally, poorer acceptor properties coupled with increased steric demand via di-t-butyl substitution (12) shut down the reaction completely. While this last result might suggest inhibition of catalysis due to deleterious steric effects, the poor reactivity of PCy3 (see Table 1, entry 6) more likely suggest that electron-rich alkyl-substituted phosphines lack the electronic properties necessary for catalysis at the Pd(0) center. While elucidation of the factors responsible for E/Z control must await further study, these data further illustrate the unexpected breadth of effect that relatively minor modifications to a given ligand scaffold can have on product selectivity.
Scheme 4.
Comparison of Xantphos Ligands, Showing the Effect of Electronic and Steric Modification on E/Z Selectivitya
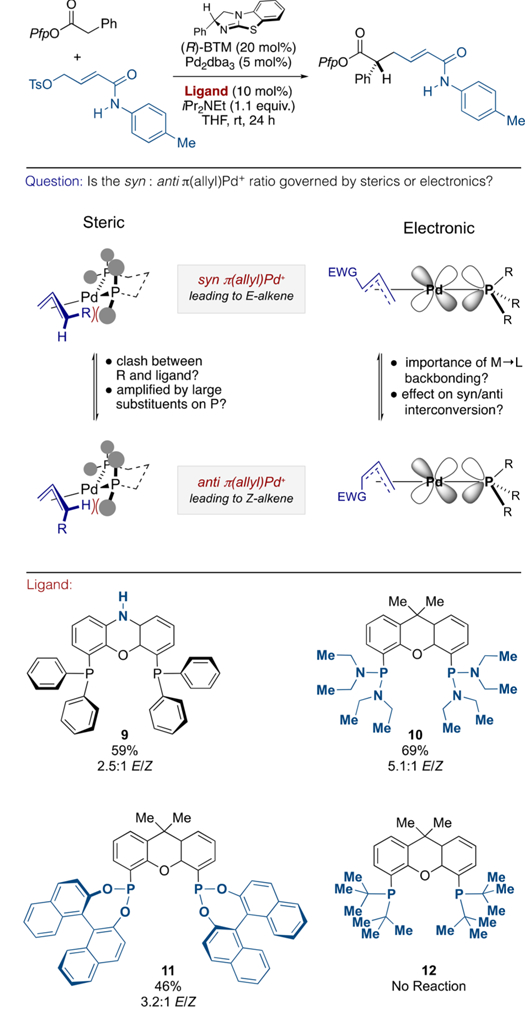
aStructural variations from Xantphos are highlighted in blue.
2.3. Ketone-Substituted Electrophiles.
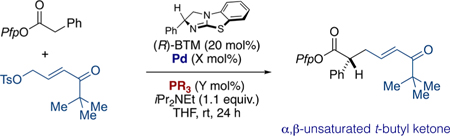
As depicted in Scheme 2, using the PdXantphos G3 catalyst,10 ketone, and aldehyde-substituted electrophiles provided products in low yield, although they did not suffer from alkene isomer distributions. As indicated by the results of our amide assessment, we reasonably expected that the chemical yield could be improved upon identification of an appropriate Pd catalyst, and that any decrease in enantioselectivity might also be addressed through the ligand selection or by limiting in situ product racemization. Assessment of the same ligands/ conditions again revealed the critical influence of the supporting phosphine ligand (see Table 2, entries 2–10), and again revealed P(2-thienyl)3 to be superior (Table 2, entries 10–12). Finally, the preformed zerovalent Pd[P(2-thienyl3)]3 again proved optimal (Table 2, entry 13).
Table 2.
Optimization of t-Butylketone-Substituted Electrophile: Effect of Ligand
 | |||||
|---|---|---|---|---|---|
| Entrya | Pd (mol%) | PR3 (mol%) | Yield [%]b | E:Zc | erd |
| 1 | PdXantphos G3 | -- | 45 | E only | 85:15 |
| 2 | Pd2dba3(5) | Xantphos (10) | 41 | E only | -- |
| 3 | Pd2dba3(5) | DPEphos (10) | 0 | -- | -- |
| 4 | Pd2dba3(5) | dppf (10) | 36 | E only | - |
| 5 | Pd2dba3(5) | dppe (10) | 0 | -- | - |
| 6 | Pd2dba3(5) | PCy3 (20) | NA | NA | NA |
| 7 | Pd2dba3(5) | P(o-tolyl)3 (20) | NA | NA | NA |
| 8 | Pd2dba3(5) | P(4-OMePh)3 (20) | 0 | -- | - |
| 9 | Pd2dba3(5) | P(2-furyl)3 (20) | 50 | E only | - |
| 10 | Pd2dba3(5) | P(2-thienyl)3 (20) | 70 | E only | 86:14 |
| 11 | Pd2dba3(5) | P(2-thienyl)3(10) | 70 | E only | 93:7 |
| 12 | Pd2dba3 (10) | P(2-thienyl)3 (25) | 70 | E only | 93:7 |
| 13 | Pd[P(2-thienyl)3]3 (5) | -- | 87 | E only | 96:4 |
Reactions performed on a 0.1 mmol scale.
Yields determined by 1H NMR by comparison with an internal standard (1,2,4,5-tetramethylbenzene).
E/Z ratio calculated from crude 1H NMR.
Determined by chiral HPLC analysis
Using these optimal conditions, we assessed the steric profile of aliphatic ketones (Scheme 5, 13–16), which provided the products in good isolated yields as single E-isomers and with consistently high levels of enantioenrichment. Cyclopropyl ketone 17 and phenyl ketone 18 were similarly effective.
Scheme 5.
Scope of Ketone-Substituted Electrophiles
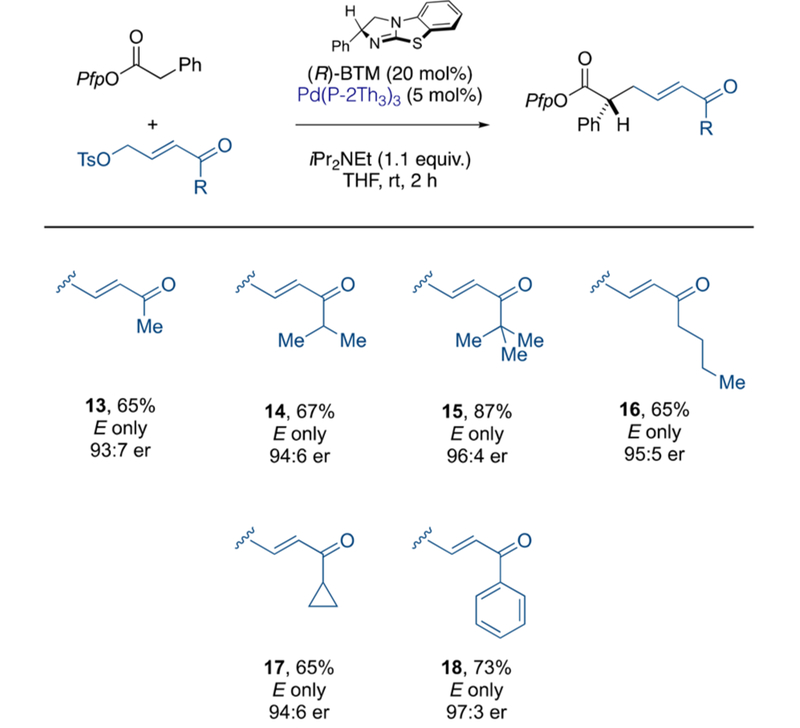
aIsolated yields. Enantiomeric ratio determined by chiral HPLC in comparison to the racemate. P(2-Th)3 = tri(2-thienyl)phosphine.
2.4. Ester-Substituted and Related Electrophiles.
The identification of Pd[P(2-thienyl)3]3 as an effective Pd catalyst offered some prospect of a general Lewis base/Pd protocol independent of the carbonyl substituent on the electrophile. Therefore, we assessed its competence with electrophiles bearing ester (19–23), thioester (24), and nitrile (25) moieties (see Scheme 6). All products were formed in high chemical yield, with high levels of enantioenrichment and with exclusive control over the alkene stereochemistry. The nitrile product 25 displayed only minimal deviation from this (9:1 E/Z) and slightly lower levels of enantioselectivity. Noteworthy is the performance of the aldehyde-substituted electrophile, which is sensitive and must be used immediately. The product 26 was obtained in 50% yield and with enhanced levels of enantioenrichment (cf Scheme 2).
Scheme 6.
Scope of Ester-Substituted Electrophiles*
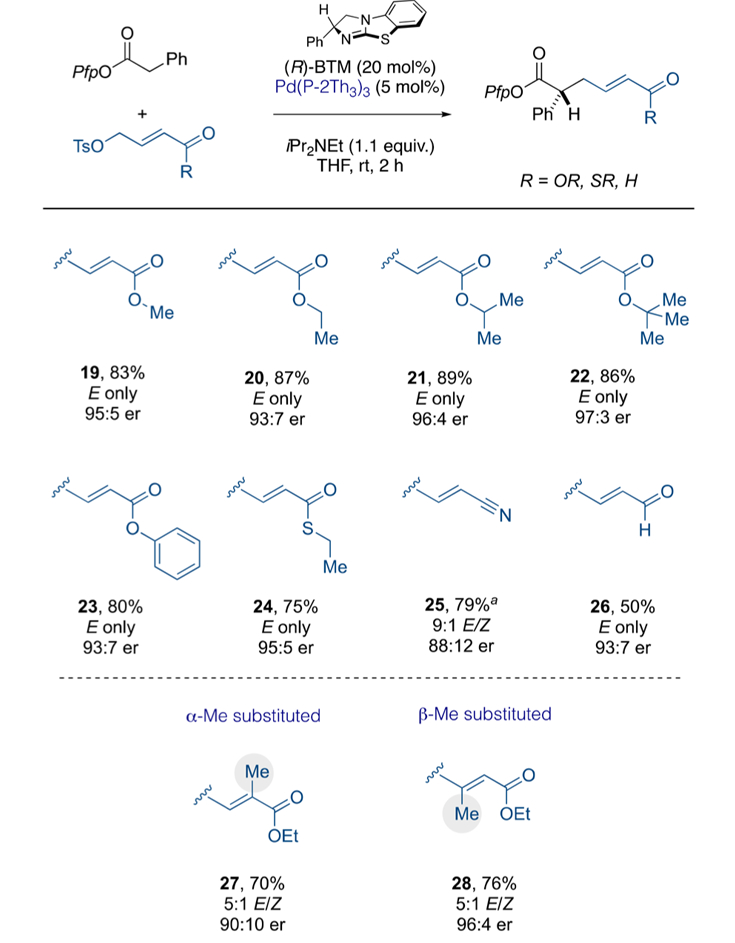
*Isolated yields. Enantiomeric ratio determined by chiral HPLC in comparison to the racemate. aReaction performed at 0 °C. P(2-Th)3 = tri(2-thienyl)phosphine.
Finally, we sought to evaluate control over alkene stereochemistry in trisubstituted α,β-unsaturated esters. In the event, α-Me and β-Me substituents (27 and 28) displayed marked preference for the E-stereoisomer while retaining the levels of enantioselectivity (see Scheme 6, bottom).
2.5. Nucleophile Scope.
Our systematic investigation of carbonyl-based substituent effects on the stereochemical outcome of our direct asymmetric α-allylation of prochiral aryl acetic acid ester nucleophiles23 has led to the identification of a general and effective cooperative Lewis base/Pd-catalyzed protocol. In concert with this broad tolerance of electrophile structure, we evaluated a range of aryl acetic acid ester nucleophiles and further confirmed the effectiveness of this catalytic system (see Scheme 7). We began by assessing the reaction of the ethyl ester-bearing electrophile with three standard nucleophiles, all of which performed as expected (29–31) and provided the products in good yields as single E-isomers and with high levels of enantioselectivity. From here, we explored the efficiency of this catalyst via different nucleophile–electrophile combinations (32–43). Again, these all performed as expected. Noteworthy is the ability to incorporate aryl halides in conjunction with different α,β-unsaturated carbonyl systems. In combination with electro-philic Pfp esters,24 these moieties provide ample opportunity for further orthogonal diversification.
Scheme 7.
Demonstration of Generality–Nucleophile Scope, in Combination with Different Electrophiles*
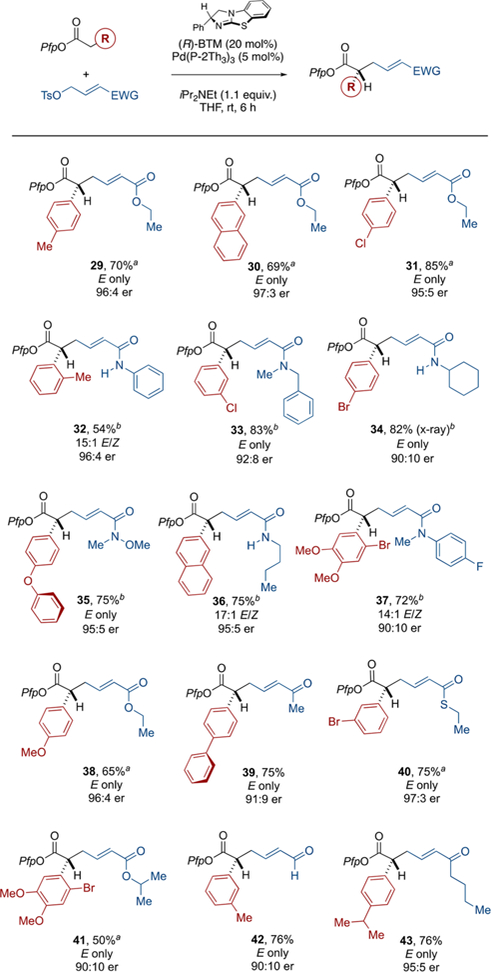
*Isolated yields. Enantiomeric ratio determined by chiral HPLC in comparison to the racemate. aReaction time = 2 h. bReaction performed in 1,4-dioxane. P(2-Th)3 = tri(2-thienyl)phosphine.
3. CONCLUSIONS
In summary, we have identified the compatibility and broadly effective cooperative catalytic effect of BTM/Pd[P(2-thienyl3)]3 for the direct enantioselective α-allylation of prochiral esters. Products are obtained exclusively as the linear regioisomer, with high levels of E-selectivity (or exclusively E) and with high levels of enantiocontrol. Within the context of our own efforts, this cements BTM/Pd as a general reactivity platform for the direct enantioselective alkylation of acyclic prochiral esters. Moreover, this study furthers the notion that Lewis base/transition-metal cooperative catalysis provides unique opportunities for enantioselective reaction design as the properties of the metal can be tuned and modulated independently of the source of enantioselectivity, which resides on the Lewis base catalyst. This can be considered complementary to traditional ligand-centered asymmetric induction.
Supplementary Material
ACKNOWLEDGMENTS
We gratefully acknowledge Indiana University and the NIH (R01GM121573) for generous financial support. We thank Dr. Maren Pink and Dr. Chun-Hsing Chen (IU) for X-ray crystallography. We are indebted to Dr. James W. B. Fyfe for helpful discussions and assistance with the preparation of this manuscript. This project was partially supported by the IU Vice Provost for Research through the Research Equipment Fund.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscatal.8b03507.
NMR spectra (CIF)
Experimental details and data (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).(a) For reviews, see: Tsuji J. Dawn of organopalladium chemistry in the early 1960s and a retrospective overview of the research on palladium-catalyzed reactions. Tetrahedron 2015, 71, 6330–6348. [Google Scholar]; (b) Trost BM Metal catalyzed allyic alkylation: its development in the Trost laboratories. Tetrahedron 2015, 71, 5708–5733. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Weaver JD; Recio A III; Grenning AJ; Tunge JA Transition Metal-Catalyzed Decarboxylative Allylation and Benzylation Reactions. Chem. Rev 2011, 111, 1846–1913. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Trost BM; Machacek MR; Aponick AP Predicting the Stereochemistry of Diphenylphosphino Benzoic Acid (DPPBA)-based Palladium-Catalyzed Asymmetric Allylic Alkylation Reactions: A Working Model. Acc. Chem. Res 2006, 39, 747–760. [DOI] [PubMed] [Google Scholar]; (e) Trost BM; Crawley ML Asymmetric Transition Metal-Catalyzed Allylic Alkylation: Applications in Total Synthesis. Chem. Rev 2003, 103, 2921–2944. [DOI] [PubMed] [Google Scholar]; (f) Helmchen G; Pfaltz A Phosphinooxazolines – A New Class of Versatile, Modular P,N-Ligands for Asymmetric Catalysis. Acc. Chem. Res 2000, 33, 336–345. [DOI] [PubMed] [Google Scholar]; (g) Helmchen G Enantioselective palladium-catalyzed allylic substitutions with asymmetric chiral ligands. J. Organomet. Chem 1999, 576, 203–214. [Google Scholar]; (h) Trost BM; Van Vranken DL Asymmetric Transition Metal-Catalyzed Allylic Alkylations. Chem. Rev 1996, 96, 395–422. [DOI] [PubMed] [Google Scholar]
- (2).(a) For selected examples using chiral counterions to control the absolute stereochemistry of Pd-catalyzed allylic alkylation with both cyclic and acyclic ester nucleophiles, see: Ohmatsu K; Hara Y; Kusano Y; Ooi T. Anion-Stoichiometry-Dependent Selectivity Enhancement in Ion-Paired Chiral Ligand–Palladium Complex Catalyzed Enantioselective Allylic Alkylation. Synlett 2016, 27, 1047–1050. [Google Scholar]; (b) Ohmatsu K; Ito M; Ooi T Ligand-controlled E/ Z-selectivity and enantioselectivity in palladium-catalyzed allylation of benzofuranones with 1,2-disubstituted allylic carbonates. Chem. Commun 2014, 50, 4554–4557. [DOI] [PubMed] [Google Scholar]; (c) Ohmatsu K; Ito M; Kunieda T; Ooi T Exploiting the Modularity of Ion-Paired Chiral Ligands for Palladium-Catalyzed Enantioselective Allylation of Benzofuran-2(3H)-ones. J. Am. Chem. Soc 2013, 135, 590–593. [DOI] [PubMed] [Google Scholar]; (d) Ohmatsu K; Ito M; Kunieda T; Ooi T Ion-paired chiral ligands for asymmetric palladium catalysis. Nat. Chem 2012, 4, 473–477. [DOI] [PubMed] [Google Scholar]
- (3).(a) Scaggs WR; Snaddon TN Enantioselective α-Allylation of Acyclic Esters using B(pin)-Substituted Electrophiles: Independent Regulation of Stereochemical Control of Elements via Cooperative Pd/Lewis Base Catalysis. Chem. -Eur. J 2018, 24, 14378–14381. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Schwarz KJ; Yang C; Fyfe JWB; Snaddon TN. Enantioselective α-Benzylation of Acyclic Esters Using π-Extended Electrophiles. Angew. Chem., Int. Ed 2018, 57, 12102–12105. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Schwarz KJ; Pearson CM; Cintron-Rosado GA; Liu P; Snaddon TN. Traversing Steric Limitations via Cooperative Lewis Base/Pd Catalysis: An Enantioselective Synthesis of α-Branched Esters Using 2-Substituted Electrophiles. Angew. Chem., Int. Ed 2018, 57, 7800–7803. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Fyfe JWB; Kabia O; Pearson CM; Snaddon TN Si-Directed regiocontrol in asymmetric Pd-catalyzed allylic alkylations using C1-ammonium enolate nucleophiles. Tetrahedron 2018, 74, 5383–5391. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Schwarz KJ; Amos JL; Klein JC; Do DT; Snaddon TN Uniting C1-Ammonium Enolates and Transition Metal Electrophiles via Cooperative Catalysis: The Direct Asymmetric α-Allylation of Aryl Acetic Acid Esters. J. Am. Chem. Soc 2016, 138, 5214–5217. [DOI] [PubMed] [Google Scholar]
- (4).(a) Grant DP; Murrall NW; Welch AJ Asymmetrically Bonded π-Ligands: II. Hinging towards metal of substituted allyls: synthesis of three 1-syn-EtOOCC3H4 complexes, and the molecular structure of [(η–1-EtOOC3H4)Pd(tmeda)]BF4 (tmeda = N,N,N’,N’-tetramethylethylenediamine) at 185 K and of [(η–1-EtOOCC3H4)-PdCl2] at 291 K. J. Organomet. Chem 1987, 333, 403–414. [Google Scholar]; (b) Tanikaga R; Jun T-X; Kaji A Regioselective and stereospecific Pd(0)-catalyzed reactions of 4-chloroacetoxyalk-2-enoic esters with carbon and nitrogen nucleophiles. J. Chem. Soc., Perkin Trans 1 1990, 1, 1185–1191. [Google Scholar]; (c) Ogura K; Shibuya N; Iida H Palladium(II)-assisted conversion of a 2-alkenyl sulfone into a 3-acetoxy(or chloro)-1-alkenyl sulfone. Tetrahedron Lett 1981, 22, 1519–1522. [Google Scholar]; (d) Öhler E; Kanzler S. Regioselective Pd(0) Catalyzed Amination of Carbonates of Allylic α-Hydroxyphosphonates with Hydroxylamine Derivatives: A Convenient Route to Phosphonic Acids Related to the Antibiotic Fosmidomycin. Synthesis 1995, 1995, 539–543.; (e) Principato B; Maffei M; Siv C; Buono G; Peiffer G Palladium catalyzed allylic acetoxylation of dialkyl allyl phosphonates. Tetrahedron 1996, 52, 2087–2096. [Google Scholar]; (f) Godleski SA; Villhauer EB Application of a Transition Metal-Mediated Stereospecific Michael Reaction Equivalent to the Synthesis of Alloyohimbone. J. Org. Chem 1986, 51, 486–491. [Google Scholar]; (g) Yamamoto Y; Al-Masum M; Takeda A Palladium-catalysed γ-addition of pronucleophiles to allenyl sulfides. Chem. Commun 1996, 831–832.; (h) Tsuji J; Ueno H; Kobayashi Y; Okumoto H Palladium-catalyzed regioselective reactions of α-acetoxy-β,γ-unsaturated nitriles and γ-acetoxy-α,β-unsaturated ester with nucleophiles. Tetrahedron Lett 1981, 22, 2573–2574. [Google Scholar]
- (5).Modern Carbonyl Chemistry; Otera J, Ed.; Wiley–VCH: Weinheim, Germany, 2000. [Google Scholar]
- (6).(a) For seminal and instructive investigations concerning the mechanism of asymmetric Pd-catalyzed allylic alkylation, including the many factors governing reactivity, control, and selectivity, see: Auburn PR; Mackenzie PB; Bosnich B. Asymmetric synthesis. Asymmetric catalytic allylation using palladium chiral phosphine complexes. J. Am. Chem. Soc 1985, 107, 2033–2046. [Google Scholar]; (b) Mackenzie PB; Whelan J; Bosnich B Asymmetric synthesis. Mechanism of asymmetric catalytic allylation. J. Am. Chem. Soc 1985, 107, 2046–2054. [Google Scholar]
- (7).(a) Faller JW; Thomsen ME; Mattina MJ Organometallic conformational equilibriums. X. Steric Factors and their Mechanistic Implications in π-Allyl(amine)chloropalladium(II) Complexes. J. Am. Chem. Soc 1971, 93, 2642–2653. [Google Scholar]; (b) Faller JW; Tully MT Organometallic conformational equilibriums. XV. Preparation and Resolution of 1,2,3-h3-(1-Acetyl-2,3-dimethylallyl)-[(S)-α-phenethylamine]chloropalladium]. J. Am. Chem. Soc 1972, 94, 2676–2679. [Google Scholar]
- (8).(a) Liu P; Yang X; Birman VB; Houk KN Origin of Enantioselectivity in Benzotetramisole-Catalyzed Dynamic Kinetic Resolution of Azlactones. Org. Lett 2012, 14, 3288–3291. [DOI] [PubMed] [Google Scholar]; (b) Bumbu VD; Birman VB Kinetic Resolution of N-Acyl-β-lactams via Benzotetramisole-Catalyzed Enantioselective Alcoholysis. J. Am. Chem. Soc 2011, 133, 13902–13905. [DOI] [PubMed] [Google Scholar]; (c) Yang X; Lu G; Birman VB Benzotetramisole-Catalyzed Dynamic Kinetic Resolution of Azlactones. Org. Lett 2010, 12, 892–895. [DOI] [PMC free article] [PubMed] [Google Scholar]; d () Birman VB; Li X. Benzotetramisole: A Remarkably Enantioselective Acyl Transfer Catalyst. Org. Lett 2006, 8, 1351–1354. For discussion of isothioureas as enantioselective Lewis base catalysts, see: [DOI] [PubMed] [Google Scholar]; (e) Merad J; Pons J-M; Chuzel O; Bressy C Enantioselective Catalysis by Chiral Isothioureas. Eur. J. Org. Chem 2016, 2016, 5589–5560. [Google Scholar]
- (9).(a) For reviews discussing Lewis base catalysis and C1-ammonium enolates, see: Morrill LC; Smith AD. Organocatalytic Lewis base functionalisation of carboxylic acids, esters and anhydrides via C1-ammonium or azolium enolates. Chem. Soc. Rev 2014, 43, 6214–6226. [DOI] [PubMed] [Google Scholar]; b () Paull DH; Weatherwax A; Lectka T. Catalytic, asymmetric reactions of ketenes and ketene enolates. Tetrahedron 2009, 65, 6771–6803. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) France S; Guerin DJ; Miller SJ; Lectka T Nucleophilic Chiral Amines as Catalysts in Asymmetric Synthesis. Chem. Rev 2003, 103, 2985–3012. [DOI] [PubMed] [Google Scholar]
- (10).Bruno NC; Tudge MT; Buchwald SL Design and preparation of new palladium precatalysts for C–C and C–N cross-coupling reactions. Chem. Sci 2013, 4, 916–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11). We have previously described the critical influence the nucleofuge has on the obtained enantioselectivity, and we have established sulfonates as being the most broadly effective. In previous studies, we have employed mesylates (see refs 3a–3e); however, in the case of the electron-deficient allyl mesylates described in this study, they displayed poor stability when stored for prolonged periods. Accordingly, the corresponding tosylates were employed, which could be stored for prolonged periods (>2 weeks) without noticeable degradation.
- (12). For an example of controlling E/Z alkene stereochemistry in asymmetric Pd-catalyzed allylic alkylation, see ref 2b.
- (13).Spoehrle SS; West TH; Taylor JE; Slawin AMZ; Smith AD Tandem Palladium and Isothiourea Relay Catalysis: Enantioselective Synthesis of a-Amino Acid Derivatives via Allylic Amination and [2,3]-Sigmatropic Rearrangement. J. Am. Chem. Soc 2017, 139, 11895–11902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Jiang Z; Beiger JJ; Hartwig JF Stereodivergent Allylic Substitutions with Aryl Acetic Acid Esters by Synergistic Iridium and Lewis Base Catalysis. J. Am. Chem. Soc 2017, 139, 87–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).(a) Lu X; Ge L; Cheng C; Chen J; Cao W; Wu X Enantioselective Cascade Reaction for Synthesis of Quinolinones through Synergistic Catalysis Using Cu-Pybox and Chiral Benzotetramisole as Catalysts. Chem. -Eur. J 2017, 23, 7689–7693. [DOI] [PubMed] [Google Scholar]; (b) Song J; Zhang Z-J; Gong L-Z Asymmetric [4 + 2] Annulation of C1 Ammonium Enolates with Copper-Allenylidenes. Angew. Chem., Int. Ed 2017, 56, 5212–5216. [DOI] [PubMed] [Google Scholar]; (c) Song J; Zhang Z-J; Chen S-S; Fan T; Gong L-Z Lewis Base/Copper Cooperatively Catalyzed Asymmetric α-Amination of Esters with Diazirinone. J. Am. Chem. Soc 2018, 140, 3177–3180. [DOI] [PubMed] [Google Scholar]
- (16). Epimerization over prolonged reaction times was confirmed by over time analysis of product enantioselectivity. See the Supporting Information (SI) for details.
- (17).Li W; Han Y; Li B; Liu C; Bo Z Tris[tri(2-thienyl)phosphine]palladium as the catalyst precursor for thiophenebased Suzuki–Miyaura cross-coupling and polycondensation. J. Polym. Sci., Part A: Polym. Chem 2008, 46, 4556–4563. [Google Scholar]
- (18).(a) The relative population of syn and anti complexes is primarily governed by the substituent on the allyl fragment itself and is often difficult to control by a distal ligand, see:Tsuji J; Shimizu I; Minami I; Ohashi Y; Sugiura T; Takahashi K. Allylic carbonates. Efficient allylating agents of carbonucleophiles in palladium-catalyzed reactions under neutral conditions. J. Org. Chem 1985, 50, 1523–1529. [Google Scholar]; (b) Ferroud D; Gaudin JM; Genet J-P Bis (alrylsulfonyl)-methane: A versatile synthon in pheromone synthesis. Tetrahedron Lett 1986, 27, 845–846. [Google Scholar]; (c) Gamez P; Ariente C; Gore JJ; Cazes B Stereoselectivity of the carbopalladation-functionalization of allenic compounds: A mechanistic study. Tetrahedron 1998, 54, 14835–14844. [Google Scholar]; (d) Commandeur C; Thorimbert S; Malacria M Chemo-and Stereoselective Palladium-Catalyzed Allylic Alkylations Controlled by Silicon. J. Org. Chem 2003, 68, 5588–5592. [DOI] [PubMed] [Google Scholar]; (e) Tsukamoto H; Uchiyama T; Suzuki T; Kondo Y Palladium-(0)-catalyzed direct cross-coupling reaction of allylic alcohols with aryl-and alkenyboronic acids. Org. Biomol. Chem 2008, 6, 3005–3013 For pertinent examples of ligand effects overriding substrate bias to control syn/anti π-(allyl)Pd+ isomer rations during allylic alkylation, see refs 2a and 11. [DOI] [PubMed] [Google Scholar]
- (19).(a) van Haaren RJ; Oevering H; Coussens BB; van Strijdonck GPF; Reek JNH; Kamer PJ; van Leeuwen PWNM On the Influence of the Bite Angle of Bidentate Phosphane Ligands on the Regioselectivity in Allylic Alkylation. Eur. J. Inorg. Chem 1999, 1999, 1237–1241. [Google Scholar]; (b) Sjogren M; Hansson S; Norrby PO; Aakermark B; Cucciolito ME; Vitagliano A Selective Stablilization of the Anti Isomer of (η3-Allyl)palladium and -platinum Complexes. Organometallics 1992, 11, 3954–3964. [Google Scholar]; (c) Aakermark B; Krakenberger B; Hansson S; Vitagliano A Ligand effects and nucleophilic addition to (η3-allyl)palladium complexes. A carbon-13 NMR study. Organometallics 1987, 6, 620–628. [Google Scholar]; (d) Akemark B; Zetterberg K; Hansson S; Krakenberger B; Vitagliano A The mechanism of nucleophilic addition to η3-allylpalladium complexes: the influence of ligands on rates and regiochemistry. J. Organomet. Chem 1987, 335, 133–142. [Google Scholar]
- (20).(a) Zhang J; Bellomo A; Trongsiriwat N; Jia T; Carroll PJ; Dreher SD; Tudge MT; Yin H; Robinson JR; Schelter EJ; Walsh PJ NiXantphos: A Deprotonable Ligand for Room-Temperature Palladium-Catalyzed Cross-Couplings of Aryl Chlorides. J. Am. Chem. Soc 2014, 136, 6276–6287. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhang J; Bellomo A; Creamer AD; Dreher SD; Walsh PJ Palladium-Catalyzed C(sp3)–H Arylation of Diarylmethanes at Room Temperature: Synthesis of Triarylmethanes via Deprotonative-Cross-Coupling Processes. J. Am. Chem. Soc 2012, 134, 13765–13772. [DOI] [PubMed] [Google Scholar]
- (21).(a)For a general discussion, see: Organotransition Metal Chemistry: From Bonding to Catalysis; Hartwig JF, Ed.; University Science Books: Sausalito, CA, 2010. For more-detailed studies concerning the elucidation of hybrid orbital character and the symmetry of key backbonding interactions.see: Xiao SX; Trogler WC; Ellis DE; Berkovitchyellin Z. Nature of the frontier orbitals in phosphine, trimethylphosphine, and trifluorophosphine. J. Am. Chem. Soc 1983, 105, 7033–7037.Marynick DS π-Accepting Abilities of Phosphines in Transition-Metal Complexes. J. Am. Chem. Soc 1984, 106, 4064–4065.Tossell JA; Moore JH; Giordan JC Energies of π-Acceptor Orbitals in SiH4, PH3, H2S, and HCl and Their Permethylated Derivatives. Inorg. Chem 1985, 24, 1100–1103.Giordan JC; Moore JH; Tossell JA Anion States of Organometallic molecules and their ligands. Acc. Chem. Res 1986, 19, 281–286.Dunne BJ; Morris RB; Orpen AG Structural systematics. Part 3. Geometry deformations in triphenylphosphine fragments: a test of bonding theories in phosphine complexes. J. Chem. Soc., Dalton Trans 1991, 653–661.Orpen AG; Connelly NG Structural Systematics: Role of P–A σ* Orbitals in Metal–Phosphorus π-Bonding in Redox-Related Pairs of M–PA3 Complexes (A = R, Ar, OR; R = Alkyl). Organometallics 1990, 9, 1206–1210.
- (22).Andersen NG; Keay BA 2-Furyl Phosphines as Ligands for Transition-Metal-Mediated Organic Synthesis. Chem. Rev 2001, 101, 997–1030. [DOI] [PubMed] [Google Scholar]
- (23).For a discussion of prochiral ester nucleophiles in the context of enantioselective Ir-catalyzed allylic alkylation, see: Hethcox JC; Shockley SE; Stoltz BM. Iridium-Catalyzed Diastereo-, Enantio-, and Regioselective Allylic Alkylation with Prochiral Enolates. ACS Catal 2016, 6, 6207–6213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).(a) Gayo LM; Suto MJ Use of pentafluorophenyl esters for one-pot protection/activation of amino and thiol carboxylic acids. Tetrahedron Lett 1996, 37, 4915–4918. [Google Scholar]; (b) Green M; Berman J Preparation of pentafluorophenyl esters of FMOC protected amino acids with pentafluorphenyl trifluoroacetate. Tetrahedron Lett 1990, 31, 5851–5852. [Google Scholar]; (c) Kisfaludy L; Roberts JE; Johnson RH; Mayers GL; Kovacs J Synthesis of N-carbobenzoxyamino acid and peptide pentafluorophenyl esters as intermediates in peptide synthesis. J. Org. Chem 1970, 35, 3563–3565. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.