
Supplemental Figure S1.
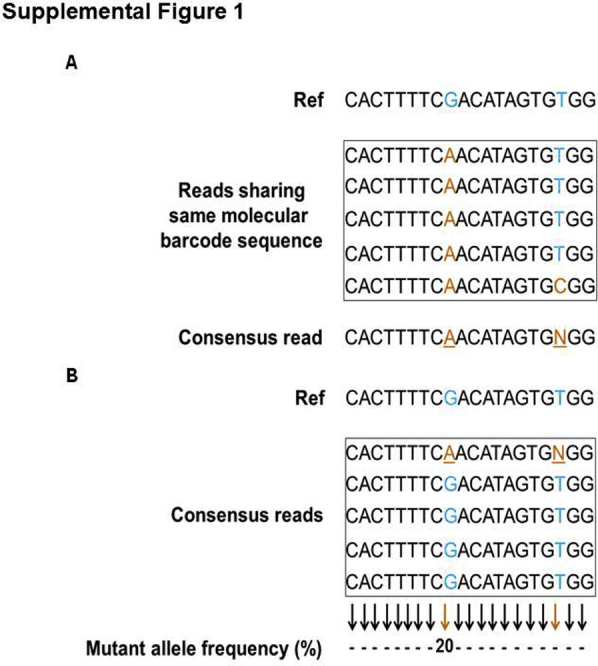
Error elimination approach to removing false-positive variant alleles. A: Derivation of the consensus read sequence. All sequencing reads are grouped into families on the basis of molecular barcode information. All reads within the family share the same molecular barcode sequence; a consensus read sequence is derived from these reads. For any chosen position, if the same nucleotide is present in all of the reads of the family, it is chosen as a consensus nucleotide for that position. If a fraction of reads contains a particular nucleotide and the remaining reads contain a different nucleotide, a consensus base for that position is assigned the letter N. Note that in this example, the underlined nucleotides A and N are the consensus nucleotides, derived from reads that shared the same molecular barcode sequence. N denotes the nucleotide ambiguity of the assigned position. B: Identification of true variant alleles. Variant alleles are determined from the derived consensus read sequences of each molecular barcode family. In this example, a fraction of reads contains the nonreference nucleotide A; it is a true variant allele and accounts for 20% allelic frequency. N was ambiguous and was eliminated during variant allele determination. The positions of the reference sequence, at which variants are shown in the piled sequencing reads, are denoted blue; the variant positions in the original sequencing reads or derived consensus reads are shown in orange, and the corresponding wild-type nucleotide positions are indicated with blue. The orange and black arrows denote the expected mutant allele frequencies at the positions where variants were and were not detected, respectively.