

Abstract
CEACAM1, a homotypic transmembrane receptor with 12 or 72 amino acid cytosolic domain isoforms, is converted from inactive cis-dimers to active trans-dimers by calcium-calmodulin (Ca2+/CaM). Previously, the weak binding of Ca2+/CaM to the human 12 AA cytosolic domain was studied using C-terminal anchored peptides. We now show the binding of 15N labeled Phe-454 cytosolic domain peptides in solution or membrane anchored using NMR demonstrates a significant role for the lipid bilayer. Although binding is increased by the mutation Phe454Ala, this mutation was previously shown to abrogate actin binding. On the other hand, Ca2+/CaM binding is abrogated by phosphorylation of nearby Thr-457, a post-translation modification required for actin binding and subsequent in vitro lumen formation. Binding of Ca2+/CaM to a membrane proximal peptide from the long 72 AA cytosolic domain anchored to lipid nanodiscs was very weak compared to lipid free conditions, suggesting membrane specific effects between the two isoforms. NMR analysis of 15N labeled Ca2+/CaM with unlabeled peptides showed the C-lobe of Ca2+/CaM is involved in peptide interactions, and hydrophobic residues such as Met-109, Val-142 and Met-144 play important roles in binding peptide. This information was incorporated into transmembrane models of CEACAM1 binding to Ca2+/CaM. The lack of Ca2+/CaM binding to phosphorylated Thr-457, a residue we have previously shown to be phosphorylated by CaMK2D, also dependent on Ca2+/CaM, suggests stepwise binding of the cytosolic domain first to Ca2+/CaM and then to actin.
Keywords: CEACAM1, Calmodulin, Phosphorylation, NMR, Lipid nanodisc
1. Introduction
CEACAM1 is an evolutionarily conserved homotypic cell adhesion molecule that is highly expressed in lymphoid and epithelial cells [1]. Among the eleven mRNA splice variants of human CEACAM1 that show a variable number of extracellular IgC-like domains, all have either a short (12 aa) or long (72 aa) cytoplasmic domain [2]. CEACAM1-LF (long form) variants have immunoreceptor tyrosine based motifs (ITIM and ITSM), mainly associated with an inhibitory function in lymphocytes, while CEACAM1-SF (short form) variants lack these motifs and are mainly expressed on epithelial cells [2]. Interestingly the short form variants are able to signal, possessing actin and calmodulin binding amino acid sequences [3,4]. These findings suggest not only a division of labor between the short and long form variants in humans, but also a means of modulating membrane-cytoskeleton interactions [2].
CEACAM1 cis-dimers on the cell surface are thought to represent an inactive state, while trans-dimers lead to cell-cell adhesion and signal activation [5]. In the case of the ITIM containing long isoforms, Src phosphorylation of the ITIMs leads to the recruitment of Shp1/2, that in turn, can terminate signaling of phosphorylated receptors [6]. In the case of the short form isoforms, phosphorylation of a key Thr residue by PKC in the rodent [3] and by CaMK2D in human [7] can affect cytoskeletal rearrangement and cell activation including lumen formation in human breast epithelial cells [8].
Elevated intracellular Ca2+ levels induce calmodulin binding to the cytoplasmic domains of both the short and long variants of CEACAM1, thus, promoting the trans-dimer over the cis-dimer state [5]. For example, Edlund et al. [3] showed that Ca2+ loaded calmodulin (Ca2+/CaM) regulates the monomer to dimer ratio of CEACAM1. More recently, Gray-Owen and coworkers [9] found basal levels of CEACAM1 cis-homodimerization depended on the CEACAM1 transmembrane domain sequence 432GXXXG436. In that study, Gray-Owen and coworkers also showed that Ca2+/CaM bound to the cytoplasmic-domain of CEACAM1, thus, driving the monomer to dimer equilibrium towards a monomeric state. This, in turn, influenced intra- and intercellular cellular signaling and binding, recruitment of effector molecules and cellular responses. Calmodulin binding sites in both isoforms overlap serine/threonine residues that are sites of phosphorylation by protein kinases that further regulate the cis- vs the trans-dimeric states of CEACAM1 [10]. Thus, Ca2+/CaM initiates the formation of the activated state of CEACAM1 in terms of cell signaling, marking this association as a starting point for our study.
With over 300 known binding partners, CaM plays a central role in numerous biological processes from immune response, inflammation, smooth muscle contraction, apoptosis, and metabolism [11]. The Ca2+/CaM interaction with the rat homologs of CEACAM1 short and long isoforms was first demonstrated by Obrink and coworkers [12,13] who demonstrated that short and long cytoplasmic membrane proximal peptides of rat, mouse and human CEACAM1 homologs all bound 125I labeled CaM [3]. In addition, it was demonstrated that the Arginine (Arg) and Lysine (Lys) residues of rat CEACAM1 were key residues involved in CaM binding [3]. However, in that study CEACAM1 peptides were synthesized on cellulose membranes with the C-terminus of the peptide bound to the cellulose membrane, an orientation that does not mimic the natural extension of the cytoplasmic domain peptides from the lipid bilayer. More recently, our group has demonstrated that Phe-454 is a key residue involved in actin binding by the short cytoplasmic domain of human CEACAM1 [8]. Moreover, our group has shown that Thr-457 of the short cytoplasmic domain of CEACAM1 is phosphorylated during lumen formation, and the kinase responsible for this phosphorylation is CaMK2D [7,8]. Based on these studies we performed measurements on null mutations of Phe-454 in the short form and Arg-459 in the long form to determine their effect on binding to CaM. In addition, we studied the effect of phosphorylation of Thr-457 on CaM binding to the short form, since null mutations of both Phe-454 and Thr-457 affected lumen formation [8]. Previously, the short length of the short isoform of CEACAM1 and its close proximity to the lipid bilayer prompted us to perform molecular dynamic simulations of its interaction with actin in the presence or absence of Ca2+, suggesting that the phenyl ring of Phe-454 could flip out of the membrane in the presence of Ca2+ allowing it to associate with actin [14].
Since the lipid bilayer is an integral part of the cell-cell signaling platform, the use of peptides embedded in biomimetic lipid membrane systems that respond to stimuli such as changes in Ca2+ levels is important [15–17]. Several models have been developed to address the needs of biomimetic lipid membranes including planar bilayers, micelles, bicelles, liposomes, and more recently, lipid nanodiscs [15–26]. In particular, insertion of transmembrane proteins into lipid nanodiscs allows physical biochemical studies of their interactions with receptor ligands on the one hand, or signaling partners on the other [27]. In this study, we compared the interaction of CEACAM1 membrane proximal peptides with Ca2+/CaM in both free solution and anchored to lipid nanodiscs to determine the potential contribution of the lipid bilayer to this interaction. In addition, we asked if phosphorylation of the key residue Thr-457 in the short form, essential for lumen formation, affected this binding.
In order to interrogate these interactions, we performed nuclear magnetic resonance (NMR) studies using 15N-labeled CaM and CEACAM1 peptides in lipid bilayer free and in a lipid bilayer environment in the form of lipid nanodiscs. We were able to identify Phe454 and Thr457 as key residues in the CEACAM1 interaction with Ca2+/ CaM, and based on chemical shift studies, identified the C-lobe of CaM that mediates their binding. Based on the differences between lipid free versus the lipid environment, we conclude their interaction strongly depends on the lipid membrane environment.
2. Materials and methods
2.1. Peptide synthesis
Twelve amino acid (aa) long synthetic peptides representing the cytoplasmic membrane proximal region of CEACAM1-SF and CEACAM1-LF were synthesized by the City of Hope Peptide Synthesis Core Facility as described in detail in Chen et al., 2007 [8]. Briefly, peptides were synthesized with N-terminal methylated-mercaptounde-canoic acid (CH3-MUA) (Sigma Aldrich) extension using Fmoc aa and 15N-Fmoc-Phe (Cambridge Isotope Labs). Five CEACAM1 peptides were examined in this work (Table 1): short form wild type (SFWT), short form Phe-454-Ala mutant (SFFA), Thr-457 phosphorylated short form wild type (pSFWT), long form wild type (LFWT) and long form Arg-459-Ala mutant (LFRA).
Table 1.
CEACAM1 peptidesa.
| CEACAM1 Peptides | Lipid free | Lipid nanodiscs |
|---|---|---|
| Short form wild type (SFWT) | HFGKTGSSGPLQ | CH3MUA-HFGKTGSSGPLQ |
| Short form mutant ( SFFA) | HAGKTGSSGPLQ | |
| Phosphothreonine (pSFWT) | HFGKpTGSSGPLQ | |
| Long form wild type (LFWT) | HFGKTGRASDQR | CH3MUA-HFGKTGRASDQR |
| Long form mutant (LFRA) | HFGKTGAASDQR |
CEACAM1 peptide sequences used in this study for the lipid free and lipid nanodisc experiments. The mutated and phosphorylated residues are shown in red. Methylated-mercaptoundecanoic acid (CH3MUA, in blue) was used as a lipid insertion moiety.
2.2. Buffers
All buffers used here were prepared from a 1 M Tris-d11 (Sigma Aldrich) stock solution. A 15 mM Tris-d11, 6% D2O, pH 7.5 stock solution was prepared (Buffer A). Buffer A was used to prepare our NMR buffer (Buffer B) used in all our NMR experiments. Buffer B had 15 mM Tris-d11 pH 7.5, 6% D2O, 0.05 mM DSS, 2 mM DTT, 5mM CaCl2 (free solution experiments) or 2.5 mM CaCl2 (nanodisc experiments), 0.05% NaN3, 100 mM NaCl and protease inhibitor. Buffer C was also prepared from Buffer A and is identical to Buffer B except Buffer C had 150 mM NaCl.
2.3. Peptide stock solution
CEACAM1 peptide stock solutions were prepared using Buffer B without DSS or CaCl2. The DSS was added to peptide solution aliquots for concentration determination and NMR experiment samples at the time of experimentation at a concentration of 0.05 mM DSS. The calcium concentration of peptide solution aliquots were adjusted to 5 mM CaCl2 or 2.5 mM CaCl2 for the free solution and nanodisc experiments, respectively. All peptide stock solutions were kept at pH 4 with concentrations ranging from 2 to 5 mM. Peptide concentrations were determined by diluting stock peptide solution in DSS and CaCl2 free Buffer B 1:10 to final volume of 0.5 mL. The DSS concentration was adjusted to 0.05 mM and the pH was adjusted to 6.5. Concentrations of peptides were determined by 1H NMR spectroscopy at 25 °C.
2.4. Calmodulin dialysis and concentration
Human recombinant CaM with and without 15N-labeling was purchased from GiottoBiotech at a concentration of 1 mg/mL in 20 mM Tris pH 8, 150 mM NaCl and 2 mM CaCl2. A 5 mL sample of undialyzed CaM was concentrated using an Amicon Ultra Centrifuge Filter of 10 kDa MWCO (Millipore) to 0.5 mL. CaM was dialysed vs three 10 mL volumes of Ca2+ free Buffer C (150 mM NaCl) pH 7.5 followed by three times 10 volumes of Ca2+ free Buffer B (100 mM NaCl) pH 7.5. Dialyzed CaM was concentrated to a final volume of 0.8 mL, and the concentration was determined via UVε280 reading using a Model ND-1000 NanoDrop™ 1000 Spectrophotometer using a coefficient of 2560 M–1 cm–1 for CaM (Thermo Fischer, Inc., Waltham, MA).
2.5. Nanodisc preparation
Nanodiscs were prepared following published protocols [20,22,25,28]. Membrane scaffolding protein MSP1D1, cholate and solubilized dimyristoylphosphatidylcholine (DMPC) (Avanti Polar Lipids) were mixed to maintain a MSP1D1:DMPC ratio of 1:80 with a final cholate concentration of 20 mM. DMPC solubilized in chloroform was transferred to a 10 mL glass test tube and dried under argon gas while rotating the test tube to obtain a thin lipid film. The DMPC film was then lyophilized overnight under 0.05 BAR of vacuum at room temperature (RT). The DMPC powder was redissolved in Ca2+ free Buffer B at pH 7.5. Cholate was added to the DMPC solution and sonicated in an ultrasonic water bath at 60 °C for 30 min or until the solution was clear. MSP1D1 was added to the cholate solubilized DMPC mixture and vortexed gently for 15 s. The MSP1D1-DMPC-cholate mixture was then incubated at RT for 1 h before adding 0.12 g (wet weight) of washed SM-2 Biobeads (Biorad) and incubated overnight at RT with shaking. The sample was centrifuged for 30 s, the supernatant removed and the Biobeads washed with 0.25 mL of Ca2+ free Buffer B at pH 7.5. The supernatants were pooled and concentrated using an Amicon Ultra Centrifuge Filter of 30kDa MWCO (Millipore). MSP1D1 (nanodisc) concentration was determined via UVε280 reading using a Model ND-1000 NanoDrop™ 1000 Spectrophotometer using a coefficient of 21,000 M–1 cm–1 forMSP1D1 (Thermo Fischer, Inc., Waltham, MA). For NMR studies, DMPC nanodiscs were diluted in Ca2+ and DSS free Buffer B, 100 mM NaCl, pH 7.5 to a final concentration of 0.15 mM and a final volume of 0.4 mL. The D2O concentration was readjusted to 6% and DSS was added to a final concentration of 0.05 mM DSS. No CaCl2 was added to the NMR nanodisc samples at this point but the pH was adjusted to 6.5.
2.6. Size exclusion chromatography (SEC)
SEC experiments was performed on a S-200 Superdex column (10 mm × 30 cm, 13 μm) on a Äkta purifier system (GE healthcare). The column was equilibrated with Ca2+ free Buffer B, 100 mM NaCl, pH 7.5 at a flow rate of 0.5 mL/min or until an acceptable baseline was obtained. Dialyzed 15N labeled CaM and DMPC nanodisc samples were individually diluted 1:4 in Ca2+ free Buffer B (100 mM NaCl) pH 7.5 to a final volume of 0.5 mL and injected onto the column to confirm quality, purity and size (for nanodisc, about 10 nm) at a flow rate of 0.5 mL/min.
2.7. Electron microscopy
Electron Microscopy (EM) was performed according to Hagn et al. [25]. DMPC nanodiscs were diluted in Ca2+ free 10 mM Tris pH 8, 100 mM NaCl to a final concentration of 0.025 mM, and adsorbed to glow-discharged, carbon-coated 300 mesh EM grids. Samples were then prepared by conventional negative staining with 1% (w/v) uranylacetate. EM images were collected with an FEI Tecnai 12 transmission electron microscope (FEI Company, Hillsboro, OR, USA) equipped with a LaB6 filament and operated at an acceleration voltage of 120 kV. Images were then recorded with a Gatan 2 × 2 k CCD camera (Gatan, Inc., Pleasanton, CA, USA) at 42000× and a defocus value of ~1.5 μm.
2.8. NMR sample preparation and NMR experiments
The interaction of CEACAM1 peptides (SFWT, SFFA, pSFWT, LFWT and LFRA) with 15N labeled CaM in lipid-free solution were performed by titrating 15N labeled CaM with each of these five peptides to a molar ratio of 1:3 (CaM:Peptide). The stock peptide concentrations were 2–5 mM, and the 15N labeled CaM concentration was 0.2 mM. We performed 2D 1H-15N HSQC [29] using a Bruker 600 MHz spectrometer equipped with a cryogenic probe at pH 6.5 and 25 °C. The spectral width was 14.0 ppm and 31 ppm for proton and nitrogen, respectively. The acquisition time of proton and nitrogen dimensions were 0.122 s, and 0.068 s, respectively, with total experimental time of 40 mins. The lipid free solution NMR buffer (Buffer B) was 15 mM Trisd11 pH 6.5, 6% D2O, 0.05 mM DSS, 2 mM DTT, 5 mM CaCl2, 0.05% NaN3, 100 mM NaCl and protease inhibitor. All the NMR data were processed and analyzed using NMRPipe [30], NMRView [31] or Bruker Topspin.
For the lipid environment (DMPC nanodisc) experiments we used CH3-MUA-CEACAM1 peptides labeled with 15N-Phe that were inserted into the DMPC nanodiscs at a molar ratio of 2.0 to 1.0 (peptide to nanodisc). The anchoring of these peptides into the nanodisc via the CH3-MUA group resulted in a significant line broadening of the 15N-Phe signal. As such, for these lipid environment experiments we used either TROSY [32] or sofast HMQC [33] rather than HSQC experiment. The titration experiments on 15N-Phe CH3-MUA-CEACAM1 peptides incorporated into the DMPC nanodiscs with unlabeled CaM as the titrant were carried out on a Bruker 700 MHz spectrometer equipped with a cryogenic probe at 25 °C. The labeled peptide concentration was 0.12 mM, and the stock CaM concentration was 0.67 mM. The spectral width was 13.3 ppm and 6.0 or 5.0 ppm for proton and nitrogen signals, respectively. The acquisition time of proton and nitrogen dimensions were 0.109 s; and 0.037 s, respectively. The titration was carried to a ratio of CaM to peptide of 2.0:1. The lipid environment NMR buffer (Buffer B) was 15 mM Tris-d11 pH 6.5, 6% D2O, 0.05 mM DSS, 2mM DTT, 2.5 mM CaCl2, 0.05% NaN3, 100 mM NaCl and protease inhibitor.
The ratio of peptide to lipid nanodisc was 2:1 and the ratio of 15N Ca2+/CaM:peptide was 1:1 for the binding study of Ca2+/CaM with unlabeled peptides CH3-MUA-SFWT or CH3-MUA-LFWT in DMPC nanodisc using NMR TROSY experiments. The spectral width was 14.0 ppm and 31 ppm for proton and nitrogen signals, respectively. The acquisition time of proton and nitrogen dimensions were 0.104 s; and 0.036 s, respectively with total acquisition time of 8.5 h.
The dissociation constant KD of the unlabeled peptides titrated to 15N-CaM was derived from a global fitting of chemical shifts of selected residues on CaM versus the CEACAM1 peptide concentrations using Eq. (6) described in a recent review [34]. The fitting was carried out using Prism software. During the fitting, the same KD value was searched for all the residues simultaneously, and one binding site of CEACAM1 peptide on CaM model was assumed.
Measurement of relaxation rate of α and β-spin states of 15N-Phe-SFWT in solution and in the nanodisc was carried out using the 1D version of [15N, 1H]-TRACT experiment [35]. An array of relaxation data with time increment step of 5.4 ms or 10.8 ms was acquired. The peak intensity versus the relaxation time was fitted to derive the Rα and Rβ, which were then used to calculate the total correlation time based on the method described [35].
2.9. SPR studies
Surface plasmon resonance (SPR) analyses were carried out in HBS-P buffer (150 mM NaCl, 0.005% (v/v) Surfactant P20, 10 mM HEPES, pH 7.4) with 2mM CaCl2 using the BIAcore® X100 (BIAcore, Inc.). Briefly, 1 μg CaM was immobilized on a CM5 sensorchip (BIAcore) using the Amine Coupling Kit (BIAcore). The surface of the sensorchip was activated with 30 μL of EDC/NHS (100 mM N-ethyl-N’-(dimethylamino-propyl)-carbodimide hydrochloride, 400 mM N-hydro-xysuccinimide) using a flow rate of 5 μL/min. Subsequently, the sensorchip was deactivated with 30 μL of 1 M ethanolamine hydrochloride, pH 8.5 (flow rate: 5 μL/min), and conditioned with 10 μL of 10 mM Glycine-HCl, pH. 2.0 (flow rate: 20 μL/min). Binding studies and regeneration of the chip surface between injections were carried out at a flow rate of 20 μL/min. SFW and SFFA peptides were diluted in HBS-P buffer immediately prior to injection. Between sample injections the surface was regenerated with 15 μL of 10 mM Glycine-HCl, pH 2.0. Data were analyzed with BIAcore® X100 Evaluation software (BIAcore), and curve fitting was done with the assumption of steady state affinity binding.
2.10. Molecular model
Molecular modeling (MD) was adapted from our previously published work in which we studied the interaction of G-actin with the short cytoplasmic domain plus the transmembrane domain of CEACAM1 embedded in a liposome [14]. CEACAM1 (TM plus short form cytoplasmic domain) was removed from the POPS lipid bilayer and reinserted into a POPC lipid bilayer, and the short cytoplasmic domain was docked to C-terminal domain of Ca2+/CaM using Glide [36–38]. Choosing the pose that had the aromatic ring of Phe-454 in close proximity to Met-144 and Met-145 was based on the NMR observations using a suitable orientation for the entire C-term domain. The full structure of the peptide was overlaid using anchored Phe-454 as a guide. The structure was then minimized in Maestro [39] to resolve any clashes. We used GROMACS 5.1.0 [40] to further minimize the energy of the model. The CEACAM1 Ca2+/CaM complex was embedded into 256 POPC membrane from Peter Tieleman’s group [41] and packed using inflategro script [42] and equilibrated to 310 Kelvin (K) and 1 atm. Ca2+ bound to CaM was maintained with counter balancing ions (Na+ and Cl–) and water using the SPC/E model [43]. The system was then minimized to 1000 kJ/mol/nm using the steepest descent to achieving optimal packing of lipids and water by holding the protein fixed with a 100 kJ/mol position restraints. Simulations of 500 ps were performed using ensemble with a Nose-Hoover thermostat [44], followed by 2 ns simulations under isothermal-isobaric conditions using ensemble with Parinello-Rahman barostat [45] to equilibrate pressure to approximately 1.05 BAR. This was followed by 50 ns simulations with no restraints. Pymol was used to make the lipid image and Maestro was used to produce the interaction map of CEACAM1-SFWT-Ca2+/ CaM.
3. Results
3.1. Lipid nanodisc anchoring of CEACAM1 peptides
Given the role of Ca2+/CaM in converting CEACAM1 from cis-to trans-dimers on cell surfaces [3,9], and the proximity of the short form cytoplasmic domain to the lipid bilayer [14], we elected to study this association by NMR in the context of lipid nanodiscs. In order to anchor the membrane proximal peptides in the membrane of lipid nanodiscs, peptides were synthesized with an N-terminal CH3-MUA group, similar to the MUA (mercaptoundecanoic acid) we previous used to anchor CEACAM1 peptides to liposomes for NMR studies [14]. The addition of the S-methyl group was to prevent oxidative formation of a disulfide over time, a reaction that abrogated association with both G-actin and Ca2+/CaM (data not shown). As a control, NMR studies were performed on both the lipid free peptides and lipid nanodisc anchored peptides to determine the effect of membrane environment on the association with Ca2+/CaM. Peptides were designed to test the possible role of Phe-454 in Ca2+/CaM binding, a residue previously shown to be critical for G- actin binding [8,14]. A phospho-Thr-457 peptide of the short isoform peptide (pSFWT) was also tested since phosphorylation of this residue was shown to be essential for lumen formation in cell-based studies [8]. A summary of the peptide sequences used in this study is shown in Table 1.
Before the insertion of the peptide into nanodiscs, the quality and the average size of the lipid nanodiscs were determined by SEC and electron microscopy (Supplemental Fig. S1). The CH3-MUA-SFWT peptide was incubated with nanodiscs at a molar ratio of 2:1 (peptide to nanodisc) to insure the majority of nanodiscs contained at least one peptide. The CH3-MUA-SFWT peptide embedding into nanodiscs was verified by several approaches. First, after the insertion of the peptide into the nanodiscs, the retention time on SEC was different between the nanodiscs with and without insertion of the peptide as shown in (Fig. 1A). Secondly, the proton chemical shift of the peptide with 15N-labeled Phe-454 changed significantly between the peptide in free solution and the peptide in the presence of nanodiscs. Furthermore, the line width increased in the presence of nanodiscs due to the much slower tumbling of the peptide after its insertion into nanodiscs. These results are manifested in (Fig. 1B), the overlay of proton dimensions from 1H-15N 2D correlation experiments. To further demonstrate the insertion of peptide into nanodisc, the apparent rotational correlation time of 15N-labeled CH3-MUA-SFWT peptide was estimated using 1H-15N-TRACT NMR experiments [35] to be 1.1 and 11.2 ns for peptide in free solution and in the presence of nanodiscs, respectively (see Fig. 1C and D). The significantly increased rotational time of the peptide in the presence of nanodiscs supports the conclusion that peptide inserted into the nanodisc, and thus tumbled much like a higher molecular weight molecule. It is worth mentioning that the rotational correlation time measured from the peptide may not necessary correspond to the actual molecular weight of nanodiscs considering the peptide is still mobile at the surface of the membrane.
Fig. 1.
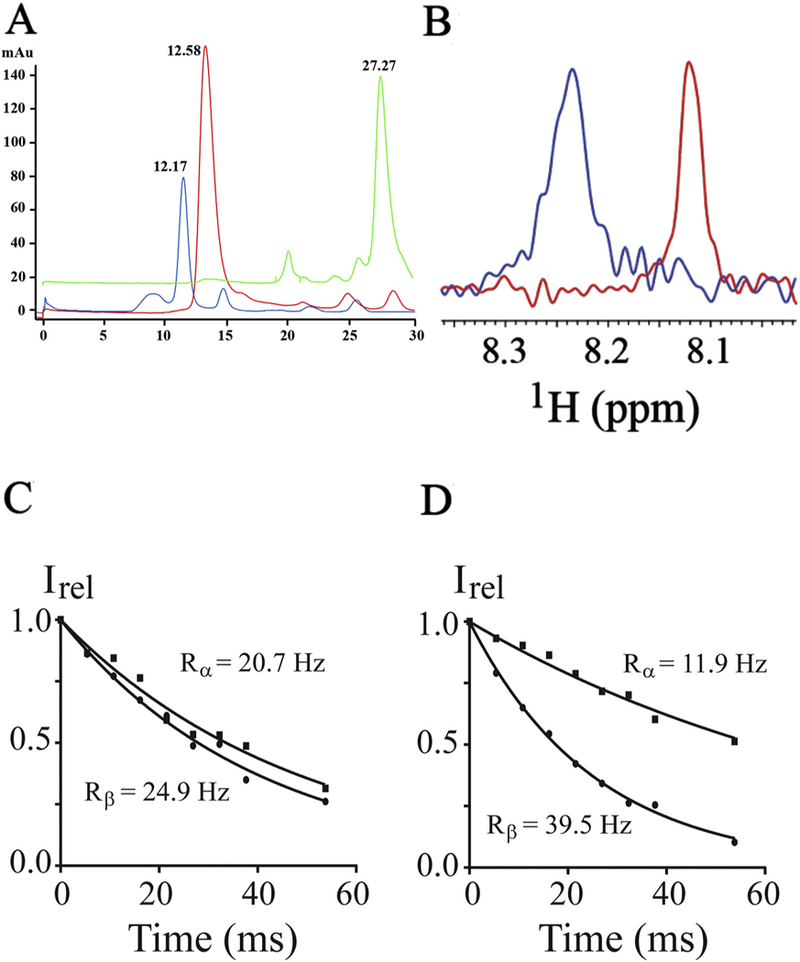
Insertion of CH3MUA-SFWT peptide into nanodiscs. A: SEC chromatograms of nanodiscs before (in red) and after (in blue) addition of CH3MUA-SFWT peptide. (Free CH3MUA-SFWT peptide is shown in green). B: The 1H slice from 2D of 1H-15N HSQC acquired on 15N-Phe CH3MUA-SFWT peptide before (in red) and after (in blue) incubation with nanodiscs. C: The relaxation rate of α- (squares) and β-spin (circles) states of 15N from the [15N, 1H]-TRACT experiment on 15N-Phe CH3MUA-SFWT peptide in solution. D: The relaxation rate of α-(squares) and β-spin (circles) states of 15N from the [15N, 1H]-TRACT experiment on 15N-Phe CH3MUA-SFWT peptide embedded in nanodiscs.
3.2. Association of lipid nanodisc anchored 15N-Labeled CH3-MUA-SFWT CEACAM1 peptide with Ca2+/CaM
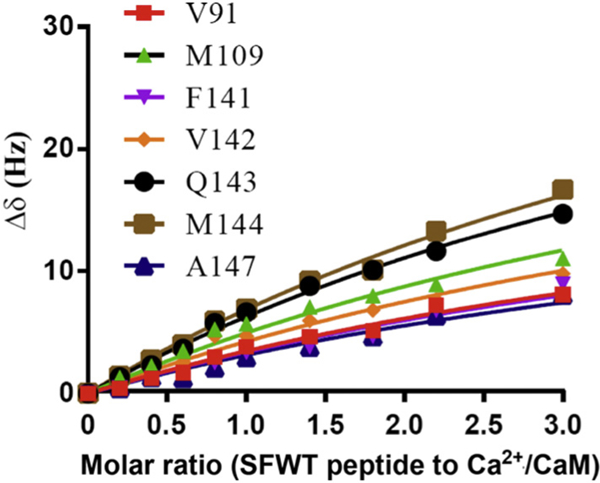
Since Ca2+/CaM has an overall negative charge, it was important to determine if Ca2+/CaM interacts with the positively charged choline head group in DMPC nanodiscs. To address this issue, we compared the 2D 1H-15N HSQC spectra of Ca2+/CaM in the presence and absence of nanodisc (Fig. 2). The chemical shift assignment of Ca2+/CaM [46,47] was mapped from the original results obtained at 37 °C through the temperature titration to 25 °C. There are no significant chemical shift perturbations (CSPs) between 15N-labeled Ca2+/CaM in the presence and absence nanodiscs except for the residue Q3. Since Q3 is in the N-terminal non-structural region, this isolated chemical shift perturbation is most likely due to random and transient contact with lipid head groups. In addition to the lack of CSP, the line width of Ca2+/CaM cross peaks were not increased in the presence of nanodiscs. These observations suggest that there was little or no association between Ca2+/CaM and lipid nanodisc under the conditions used in this study. We then performed titration experiments in which the unlabeled Ca2+/CaM was added to 15N-Phe labeled CH3-MUA-SFWT CEACAM1 peptide incorporated into the DMPC nanodiscs (Fig. 3A). The KD of Ca2+/CaM complex with CH3-MUA-SFWT peptide inserted in nanodisc was determined to be 0.22 ± 0.06 mM from the curve fitting of the combined 1H and 15N CSPs of 15N-Phe vs the concentration of Ca2+/CaM (Fig. 4).
Fig. 2.
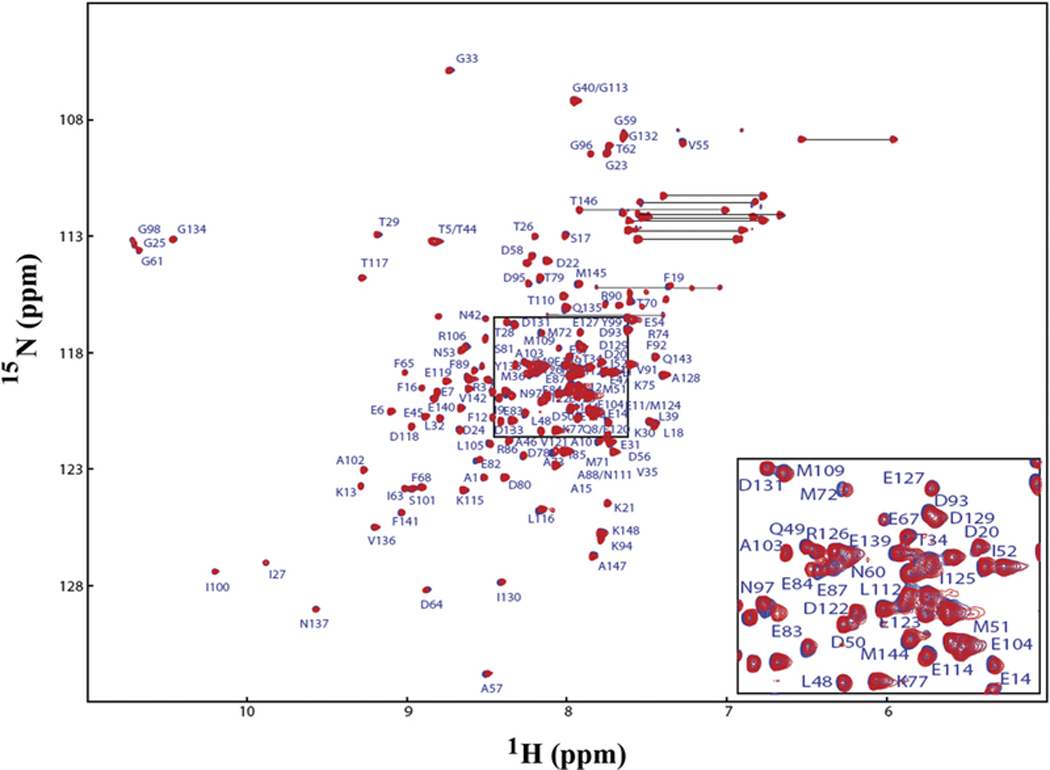
Superimposed 2D 1H-15N HSQC spectra of Ca2+/CaM in the presence and absence of nanodiscs. No nanodiscs (red), plus nanodiscs (blue). The side chain —CONH2 cross peaks are not labeled.
Fig. 3.
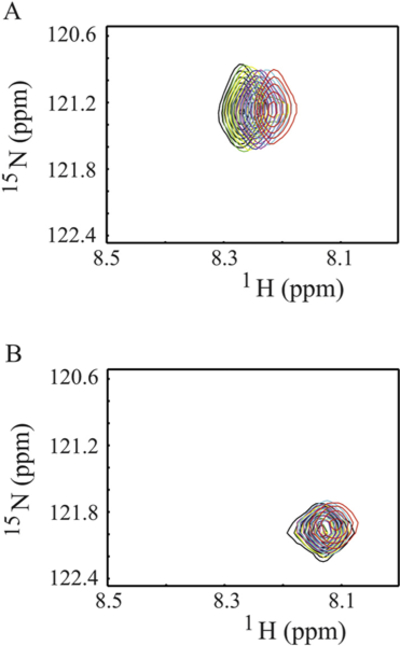
Titration of Ca2+/CaM to 15N-Phe labeled CH3MUA-SFWT and CH3MUA-LFWT peptides embedded in nanodiscs. A: Overlay of 2D 1H-15N correlation spectra of CH3MUA-SFWT peptide. B: Overlay of 2D 1H-15N correlation spectra of CH3MUA-LFWT peptide. The ratio of peptide to nanodisc was 2:1. The color of cross peaks corresponds to the molar ratio of Ca2+/CaM to peptide as follows: Black (0:1); green (0.1:1); yellow (0.3:1); blue (0.6:1); purple (1:1); cyan (1.5:1) and red (2:1).
Fig. 4.
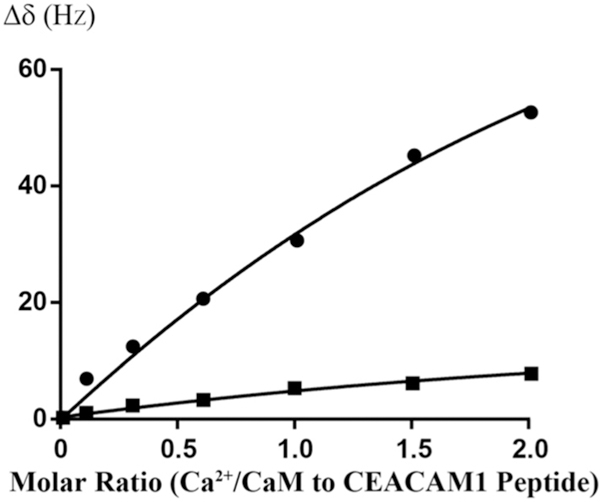
Curve fitting of Ca2+/CaM titrating to 15N-Phe labeled CH3MUA-SFWT and CH3MUA-LFWT peptides embedded in nanodiscs. The KD for CH3MUA-SFWT binding to Ca2+/CaM was determined to be 0.22 ± 0.06 mM from the curve fitting CSP of 15N-Phe versus the concentration of Ca2+/CaM (circles). No KD was determined for the binding of CH3MUA-LFWT to Ca2+/CaM (squares) since it was far from saturation under the same condition.
3.3. Association of lipid nanodisc anchored CH3-MUA-SFWT CEACAM1 peptide with 15N-labeled Ca2+/CaM
Having established that the CH3-MUA conjugated peptide inserted into lipid nanodiscs and interacted with unlabeled Ca2+/CaM, we were interested in determining which residues of Ca2+/CaM interacted with unlabeled peptides. Therefore, we acquired 2D 1H-15N TROSY spectra on uniformly 15N-labeled CaM in the presence of unlabeled CH3-MUA-SFWT CEACAM1 peptide (Table 1) anchored into lipid nanodiscs. Spectral overlay (Fig. 5) of 15N-labeled Ca2+/CaM in the presence or absence of nanodisc anchored CH3-MUA-SFWT show many significant chemical shift perturbations (CSPs) indicating binding of Ca2+/CaM to the peptide. Since Ca2+/CaM alone does not interact with the nanodisc (Fig. 2), the excessive CSPs for Ca2+/CaM were likely caused by close contacts of Ca2+/CaM to the membrane surface due to the binding of Ca2+/CaM to the peptide inserted into the nanodiscs.
Fig. 5.
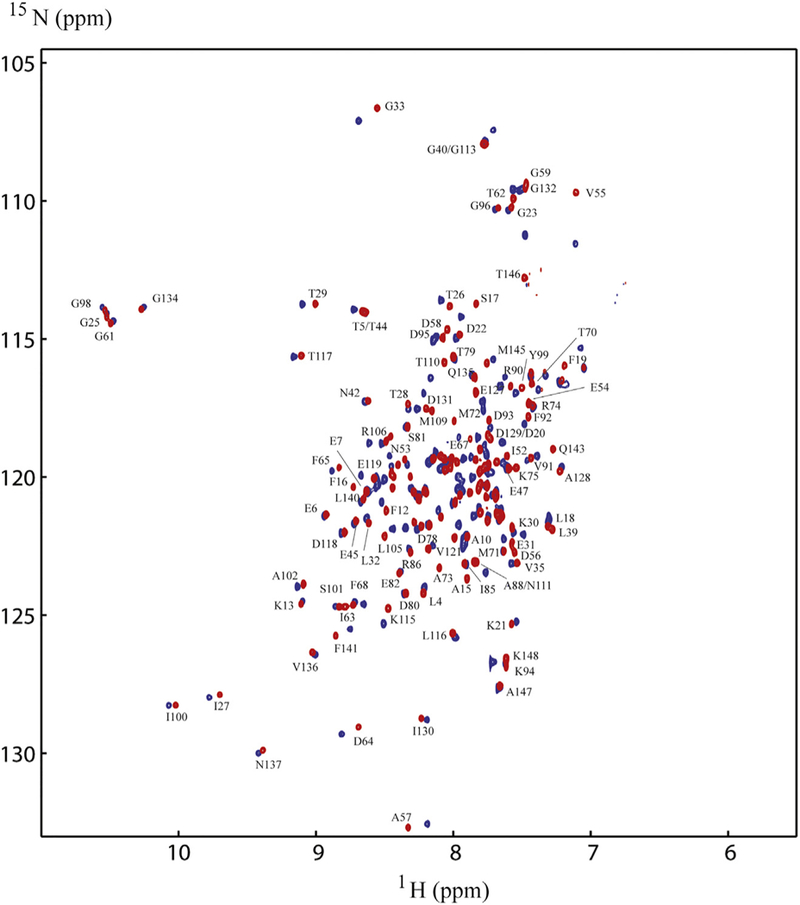
Comparison ofNMR spectra of15N-labeled Ca2+/CaM with and without CH3MUA-SFWT peptide inserted into nanodiscs. The spectrum in red is in the absence of the CH3MUA-SFWT peptide, and the spectrum in blue is in the presence of the CH3MUA-SFWT. Only those well resolved residues for the red peaks are labeled in the figure for simplicity.
3.4. Association of CEACAM1 short form peptides with 15N-labeled CaM in lipid nanodisc free solution
Since a goal of this study was to determine if the binding of Ca2+/ CaM to CEACAM1 cytoplasmic domain peptides was affected by their proximity to a lipid bilayer environment, it was essential to measure their binding to Ca2+/CaM in the absence of nanodisc. The interaction of CEACAM1 short form wild type peptide (SFWT) with 15N-labeled Ca2+/CaM in nanodisc free conditions was investigated with a 1H-15N HSQC experiment. A representative region of spectrum overlay of 15N-labeled Ca2+/CaM with and without SFWT is shown in Fig. 6A. The KD of the complex was determined to be 1.44 ± 0.3 mM (Fig. 7) from the titration of the peptide to Ca2+/CaM. This KD value is significantly higher than that obtained when the peptide was inserted in the nanodiscs. A plot of combined 1H and 15N CSPs of the amide groups of Ca2+/ CaM in the presence of SFWT peptide (Supplemental Fig. S2A) identified 14 residues that were affected significantly by peptide binding. Two of the fourteen residues are in the hinge and N-lobe region, while the remaining twelve residues are in the C lobe region. This suggests that in free solution, the SWFT peptide mainly binds the C-lobe of CaM.
Fig. 6.
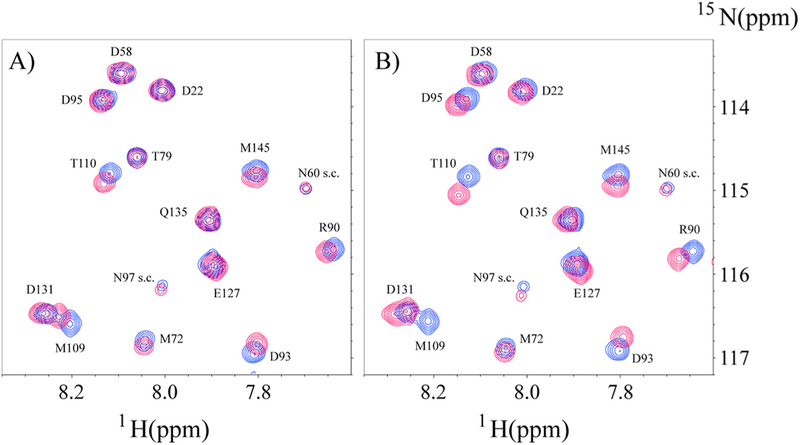
Overlay of selective spectra region of N-labeled Ca2+/CaM in the presence or absence of unlabeled SFWT (A) and LFWT (B) in solution. In both A) and B) the cross peaks in blue color are from free 15N-CaM, and the cross peaks in red color are from the mixture of Ca2+/CaM with SFWT in (A), and with LFWT in (B). The molar ratio between Ca2+/ CaM and peptide is 1:3. The s.c. label stands for side chain.
Fig. 7.
Curve fitting of SFWT titration to 15N-labeled Ca2+/CaM in the absence of nanodisc. The KD (1.44 ± 0.3 mM) of complex between SFWT and 15N-labeled Ca2+/CaM was derived from the global fitting of CSP from selected residues of Ca2+/CaM versus the concentration increment of SFWT peptide.
It was also important to determine the binding of the null mutant Phe454Ala (SFFA) and phosphorylated pThr457 (pSFWT) peptides to Ca2+/CaM in the absence of nanodisc. The titration of pSFWT to 15N-labeled Ca2+/CaM under the same conditions as the SFWT peptide did not reach saturation, thus yielding no meaningful KD (Supplemental Fig. S3A). The weaker binding of phosphorylated peptide, pSFWT to 15N-labeled Ca2+/CaM could be due to the negative charge of the phosphate group on Thr-457 and the acidic nature of Ca2+/CaM. However, in the case of the SFFA mutant, saturation was reached at 1:1 molar ratio, yielding a KD of 0.27 μM (Supplemental Fig. S3B).
To find out why the mutant peptide binds Ca2+/CaM tighter, the CSPs of Ca2+/CaM caused by the addition of SFFA were analyzed (Supplemental Fig. S2B). Unlike the interaction with SFWT, residues in both N- and C-lobs of Ca2+/CaM are involved in the interaction with SFFA. The KD value was determined based on the CSPs of residues in the N-lobe of CaM (Supplemental Fig. S3C), and the value was the same as that derived based on the CSPs of residues in the C-lobe of CaM. This suggested that both N-and C-lobe are involved in the SFFA peptide binding collectively, and this explains why the binding is much tighter than the SFWT. The residues with significant CSP values in complex with either SFWT or SFFA were mapped on a Ca2+/CaM structure and presented in Supplemental Fig. S4. Since the tighter binding of SFFA was unexpected, the KDs were determined by a second approach, using SPR in which Ca2+/CaM was immobilized on the biosensor chip, and either SFWT or SFFA was flowed over immobilized CaM in the presence of 2 mM Ca2+ (Fig. S5). Once again, the binding of the mutant SFFA was substantially tighter than the wild type SFWT peptide, in general agreement with the NMR results. It should be noted that the KDs were determined at equilibrium due to the rapid off rates observed for both peptides. Thus, while the tighter binding of the mutant peptide is surprising, the results are consistent with the idea that Phe-454 is a critical residue in that stronger binding to Ca2+/CaM (in the case of the mutant) may prevent its dissociation, a requirement for the subsequent binding of actin. Since Phe-454 is a key residue in the short form peptide binding to G-actin [14], its affinity to Ca2+/CaM cannot be greater than for G-actin.
3.5. Association of CEACAM1 long form peptides with 15N-labeled CaM in lipid nanodisc and free solution
Obrink and coworkers [3], found that the membrane proximal region of the long form isoform of CEACAM1 also bound Ca2+/CaM. Since its amino acid sequence diverges from that of the short form (Table 1) immediately after Gly-458 due to alternative mRNA splicing of exon 7, it was of interest to compare the LFWT peptide to the SFWT peptide.
Similar to SFWT, the 15N-Phe labeled CH3-MUA-LFWT was used to investigate the interaction between CH3-MUA-LFWT inserted into lipid nanodiscs and Ca2+/CaM. The 1H-15N cross peak of 15N-Phe labeled CH3-MUA-LFWT was monitored over the titration of Ca2+/CaM, and the CSPs are presented in Fig. 3B. Under the same conditions, the KD could not be derived from the titration of Ca2+/CaM to the CH3-MUA-LFWT which was far from saturation (Fig. 4). Thus, the binding of CH3- MUA-LFWT inserted into lipid nanodiscs to Ca2+/CaM is weaker than that of CH3-MUA-SFWT. The interaction of CEACAM1 LFWT with 15N-labeled Ca2+/CaM in nanodisc free conditions was investigated using an 1H-15N HSQC experiment. A representative spectral region in the absence and presence of LFWT is shown in Fig. 6B. The residues with significant chemical shift perturbations in Ca2+/CaM are presented in Supplemental Fig. S2C. The residues with significant CSP in Ca2+/CaM are located in the C lobe except for one residue in the hinge region. Thus, similar to SFWT, the LFWT also binds to the C lobe of Ca2+/CaM. The analysis of LFWT titration to Ca2+/CaM provided a KD of 0.76 ±0.1 mM (Supplemental Fig. S6), indicating that the LFWT peptide binds Ca2+/CaM with two fold higher affinity than the SFWT peptide in nanodisc free conditions. This result is consistent with the observation made by Obrink and coworkers from the study of the rat homologs of CEACAM1 [3].
According to the studies by Obrink and coworkers, Arg-459 was identified as a critical residue. Therefore, we compared the binding of the long form wild type peptide (LFWT) and the Arg-459-Ala mutated peptide (LFRA) to Ca2+/CaM, in addition to comparison of the LFWT and SFWT. Under similar conditions, the titration of Arg-459 mutated long form peptide (LFRA) to Ca2+/CaM gave a KD of 0.75 mM (Supplemental Fig. S3D). Thus, the Arg-459-Ala mutation had a minimum effect of the long form peptide binding to Ca2+/CaM, in contrast to the observation made by Obrink and coworkers [3] for the rat homolog.
3.6. A model of the association of the SFWT peptide with Ca2+/CaM in a lipid bilayer
Since the binding of the SFWT peptide to Ca2+/CaM in the absence of nanodisc was mainly localized to the C-lobe of Ca2+/CaM, while in the nanodisc anchored study, both domains were equally affected (due to lipid membrane interactions), we decided to model the interaction using the nanodisc free NMR data. In that analysis, significant CSPs were observed for Met-109, Val-142 and Met-144 in the C-lobe. Based on the canonical association of a Met cluster in Ca2+/CaM with aromatic residues in their binding partners [48,49], we first modeled the complete transmembrane plus cytoplasmic domain of CEACAM1-SF in a lipid bilayer (Fig. 8A) binding to Ca2+/CaM with Phe-454 in CEACAM1-SF in close proximity to Met-144 of Ca2+/CaM (Fig. 8B). In this model Ca2+/CaM is pressed up against the lipid bilayer, making multiple contacts that may account for the many CSPs observed for Ca2+/CaM when interacting with CEACAM1-SF anchored to a nanodisc. As shown earlier, no such CSPs are found in the absence of peptide anchored to the nanodisc. In order to exclude the possibility that extra CSPs could come from the interaction between the CH3-MUA moiety and Ca2+/CaM in a similar way that was found for the interaction between CaM and a myristoylated viral peptide [50], the chemical shift of 15N-Phe-CH3MUA-SFWT was monitored in the presence and absence of Ca2+/CaM in lipid nanodisc free solution. Two cross peaks appeared when Ca2+/CaM was added to 15N-Phe-CH3MUA-SFWT (Fig. 9A), suggesting that both CH3-MUA moiety and SFWT peptide interact with Ca2+/CaM. When inserted into nanodisc, the CH3-MUA moiety was not available for Ca2+/CaM binding as shown in Fig. 9B, in which the cross peak of 15N-Phe-CH3MUA-SFWT did not split into two peaks after the addition of Ca2+/CaM. Furthermore, the model is consistent with a role for Thr457, which is in close proximity to Phe454. Upon phosphorylation, the model predicts a steric clash, pushing the residue away from the membrane and disrupting the interaction between Phe454 and Ca2+/CaM.
Fig. 8.
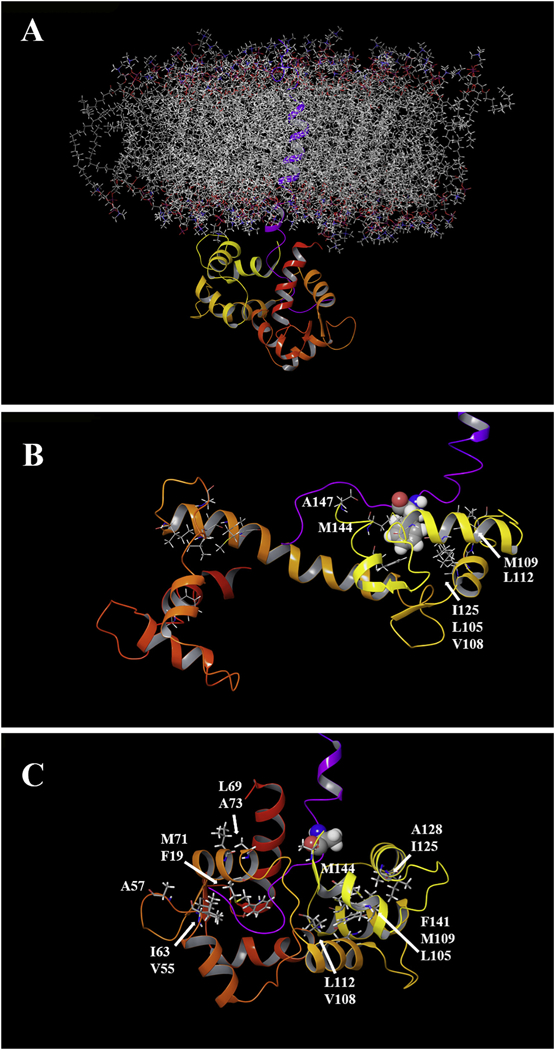
Computer modeling of Ca2+/CaM and CH3MUA-SFWT embedded in a lipid bilayer. A: The transmembrane and short form cytoplasmic domains of CEACAM1 (purple trace) were embedded in a POPC lipid bilayer and the cytoplasmic domain docked to the C-terminal domain of Ca2+/CaM (see Methods). B: The key residue Phe-454 (space filling) of CEACAM1 is shown in close proximity with hydrophobic clusters of Ca2+/CaM in the C-lobe C: A model of the F454A mutant in close proximity to hydrophic clusters in both lobes of Ca2+/CaM.
Fig. 9.
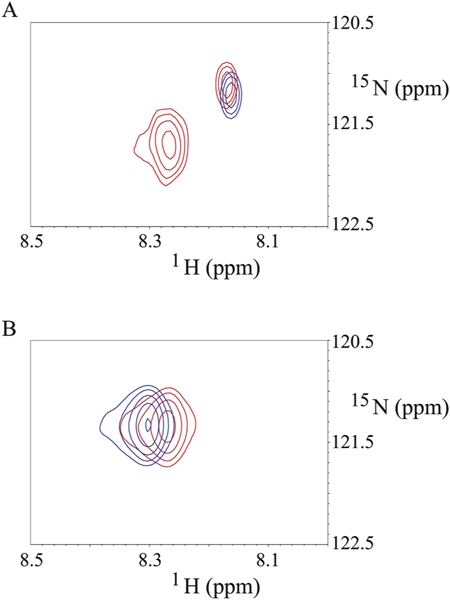
Binding of Ca2+/CaM to N-Phe-CH3MUA-SFWT peptides in the absence and presence of nanodisc. A: the blue and red cross peaks are from 15N-Phe-CH3MUA-SFWT peptide not inserted in the nanodiscs in the absence and presence of Ca2+/CaM, respectively. B: the blue and red cross peaks are from 15N-Phe-CH3MUA-SFWT peptide inserted in the nanodiscs in the absence and presence of Ca2+/CaM, respectively. The molar ratio of 15N-Phe-CH3MUA-SFWT peptide to Ca2+/CaM is 1:1 in both A and B spectra.
Since the mutation F454A resulted in stronger binding to Ca2+/CaM than SFWT (Fig. 7 and Fig. S3B & 3C) and large CSPs in both the N- and C-lobes of Ca2+/CaM (Fig. S2B), a second model was generated to reflect this mode of binding (Fig. 8C). To accommodate the multi-lobe interactions, the collapsed N-/C-lobe structure of Ca2+/CaM was used [51]. Thus, based on the CSP data, the F454A mutant binds more strongly to Ca2+/CaM due to more interactions across Ca2+/CaM than the SFWT. Importantly, this mutant would be predicted to interfere with the actin binding function of SFWT. Since Ca2+/CaM binding is considered to be the pre-step to actin binding, the implications of the F454A mutant to Ca2+/CaM binding may be there is a need for weaker binding of SFWT to Ca2+/CaM than to actin.
4. Discussion
Our functional studies on the role of CEACAM1-SF in actin binding define a critical role for two residues in its 12 amino acid cytoplasmic domain, namely Phe-454 and Thr-457 [8]. Since CEACAM1-SF resides on cells in an inactive cis-dimer conformation [9], the first step in its activation is conversion of cis- to trans-dimers. The current model for this conversion requires the breaking of the cis-dimers at the cytoplasmic domain by Ca2+/CaM [5,9]. The breaking of cis-dimers would then allow the formation of trans-dimers, enabling CEACAM1-SF to function as a cell-cell adhesion molecule. The next step in cell signaling involves the binding of G-actin to membrane proximal Phe-454 [8]. A clue to the mechanism of this step is the observation that nearby Thr-457 must be phosphorylated by CaMK2D for lumen formation [8]. We have now shown that the 12 amino acid cytoplasmic domain can bind Ca2+/CaM either inserted into a lipid nanodisc (Fig. 3A) or in free solution (Fig. 6A). In the lipid nanodisc, we used 15N-labeled Phe-454 as an NMR reporter and showed this residue is involved in the interaction with Ca2+/CaM. This result was anticipated because other peptide-Ca2+/CaM interactions require an aromatic residue of the peptide to interact with a cluster of methionines in Ca2+/CaM [48,49]. However, unlike our studies on G-actin binding to the cytoplasmic domain of CEACAM1-SF in which the mutation Phe454Ala abrogated binding, in the case of Ca2+/CaM, the Phe454Ala mutation enhanced binding significantly (Supplemental Fig. S3B and S3C). This result suggests that the role of F454 in actin binding is critical, while in Ca2+/ CaM, dispensable. In this respect, the Phe454Ala mutation demonstrates that other residues in the cytoplasmic domain contribute to the binding to Ca2+/CaM as demonstrated by multiple CSPs in both lobes of Ca2+/CaM. Furthermore, it appears that the phosphorylation of nearby Thr-457 plays a functional role in that it abolishes Ca2+/CaM binding, allowing actin binding to proceed.
Since the cytoplasmic domain of CEACAM-SF is only 12 amino acids in length, its proximity to the membrane requires a close approach of its binding partners to the lipid bilayer. Thus the cytoplasmic domain serves as docking site for both Ca2+/CaM and G-actin. In order to convey specificity to the docking site, one would surmise that neither protein would have an intrinsic association to the membrane and that some state change in the cytoplasmic domain of CEACAM1-SF would expose its critical Phe-454. In this respect, we have shown the role of Ca2+ signaling, in which Phe-454 can flip its aromatic phenyl ring out of the membrane in the presence of Ca2+ [14]. In support of these concepts, we now show that Ca2+/CaM has no association with the lipid nanodisc in the absence of inserted peptide (Fig. 2), but multiple interactions in its presence (Fig. 5). Furthermore, Ca2+ ion can serve two purposes in this mechanism, activating CaM by Ca2+ and exposing Phe-454 to the outer leaflet of the lipid bilayer. As previously discussed, the postulated mechanism for the aromatic ring flip relies on a cation-pi interaction [14]. It is likely that the Ca2+ first associates with the phospholipid head group to shed water of solvation and to place it in close proximity to the membrane inserted aromatic ring of Phe-454. While specific evidence for this is lacking, it is well known that Ca2+ released from the ER rapidly associates with the plasma membrane [52].
A further consideration is that the KD values measured for the cytoplasmic domain of CEACAM1-SF to Ca2+/CaM are mM to sub-mM, indicating that they are inherently weak. We can rationalize these results as follows: the measurements are for bimolecular interactions, whether in the presence or absence of nanodisc, while in the cell, CEACAM1-SF molecules are found clustered to the inner surface of a two-dimensional membrane due to their extra-cellular domain interactions. Thus, the relative concentrations of the cytoplasmic domains would be expected to be much higher than in free solution. Estimates of the actual concentrations are easily in the mM range given the estimated “volumes” at the membrane [14].
Since Obrink and coworkers [3] were the first to describe the binding of Ca2+/CaM to membrane proximal peptides of CEACAM1, we wanted to determine if their findings for the SF and LF peptides could be reproduced under more biological relevant conditions. This was an issue since their study relied on immobilizing the peptides on a cellulose solid support in the opposite orientation than is expected on a lipid bilayer. Indeed, we were able to confirm their results for SF and LF membrane proximal peptides in solution, and for the SF membrane proximal peptide anchored to lipid nanodisc, but not for the LF membrane proximal peptide anchored to lipid nanodisc. This discrepancy between the two studies may require further studies on full-length LF peptides, since their study and ours utilized truncated LF membrane proximal peptides that can be viewed as artificial. However, as seen from Table 1, the membrane proximal sequences of the SF and LF differ immediately after the critical sequence 453-HFGKTG-458 in which F- 454 was shown by us to play a role in binding G-actin but not Ca2+/ CaM. Furthermore, phosphorylation of T-457 was shown by us to a play a role in actin dependent lumen formation. Thus, it is of interest to consider the consequences of the amino acid sequence divergence in LF close to these residues in the context of this study where pT-457 was shown to affect binding to Ca2+/CaM. First, the point of divergence is a consequence of an mRNA splicing event in which one may speculate that the site of divergence has intended biological consequences. Since according to Obrink, both the SF and LF proximal membrane peptides bound calmodulin, it can be concluded that both sequences contained a minimal calmodulin binding site. Again, according to Obrink, the introduction of Arg immediately after G-458 in the LF peptide increased calmodulin binding. It was for this reason that we mutated this residue to Ala, a change that had little effect on Ca2+/CaM binding in our study. Since Obrink’s study presented the peptide to CaM in the biologically incorrect orientation C → N rather than N → C as in our study, the absence or presence of the basic residue at this position may have played a different role. These considerations become more complicated in the context of the lipid nanodisc where calmodulin residues were shown to interact with the lipid nanodisc in, and only in, the context of peptide anchored to the lipid nanodisc. In fact, it would be a surprise if calmodulin could bind lipid nanodisc anchored peptide and not interact with the lipid nanodisc, given the short length of the peptides. It was for this reason that we built a model that included a lipid bilayer to help visualize the consequences of calmodulin binding a peptide in this context. The model demonstrates that indeed Ca2+/CaM can approach the membrane close enough to interact with all residues of the CEACAM1-SF cytosolic domain.
5. Conclusion
This study demonstrates the utility of nanodisc anchored peptides and their mutants to study the binding of Ca2+/CaM to cytoplasmic domain targets that are anchored to the lipid bilayer. While it does not take into account the multivalency of the targets, it does allow measurements of their bimolecular interactions, including calculations of their KDs using NMR approaches. The results are consistent with cell- based studies that showed the importance of key residues previously identified on CEACAM-SF.
Supplementary Material
Acknowledgements
This research was supported by NIH grant CA84202. We are grateful to the support from NMR, EM and synthetic and biopolymer chemistry core. We thank Dr. Donghan Lee for the script calculating the tau values.
Footnotes
Conflict of interest
The authors declare that they have no conflicts of interest with the contents of this article.
Transparency document
The Transparency document associated with this article can be found, in online version.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bbamem.2019.01.004.
References
- [1].Zebhauser R, Kammerer R, Eisenried A, McLellan A, Moore T, Zimmermann W, Identification of a novel group of evolutionarily conserved members within the rapidly diverging murine Cea family, Genomics 86 (2005) 566–580. [DOI] [PubMed] [Google Scholar]
- [2].Gray-Owen SD, Blumberg RS, CEACAM1: contact-dependent control of immunity, Nat. Rev. Immunol. 6 (2006) 433–446. [DOI] [PubMed] [Google Scholar]
- [3].Edlund M, Blikstad I, Obrink B, Calmodulin binds to specific sequences in the cytoplasmic domain of C-CAM and down-regulates C-CAM self-association, J. Biol. Chem. 271 (1996) 1393–1399. [DOI] [PubMed] [Google Scholar]
- [4].Schumann D, Chen CJ, Kaplan B, Shively JE, Carcinoembryonic antigen cell adhesion molecule 1 directly associates with cytoskeleton proteins actin and tropomyosin, J. Biol. Chem. 276 (2001) 47421–47433. [DOI] [PubMed] [Google Scholar]
- [5].Hunter I, Sawa H, Edlund M, Obrink B, Evidence for regulated dimerization of cell-cell adhesion molecule (C-CAM) in epithelial cells, Biochem. J. 320 (Pt 3) (1996) 847–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Huber M, Izzi L, Grondin P, Houde C, Kunath T, Veillette A, Beauchemin N, The carboxyl-terminal region of biliary glycoprotein controls its tyrosine phosphorylation and association with protein-tyrosine phosphatases SHP-1 and SHP-2 in epithelial cells, J. Biol. Chem. 274 (1999) 335–344. [DOI] [PubMed] [Google Scholar]
- [7].Nguyen T, Chen CJ, Shively JE, Phosphorylation of CEACAM1 molecule by calmodulin kinase IID in a three-dimensional model of mammary gland lumen formation, J. Biol. Chem. 289 (2014) 2934–2945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Chen CJ, Kirshner J, Sherman MA, Hu W, Nguyen T, Shively JE, Mutation analysis of the short cytoplasmic domain of the cell-cell adhesion molecule CEACAM1 identifies residues that orchestrate actin binding and lumen formation, J. Biol. Chem. 282 (2007) 5749–5760. [DOI] [PubMed] [Google Scholar]
- [9].Patel PC, Lee HS, Ming AY, Rath A, Deber CM, Yip CM, Rocheleau JV, Gray-Owen SD, Inside-out signaling promotes dynamic changes in the carcinoembryonic antigen-related cellular adhesion molecule 1 (CEACAM1) oligomeric state to control its cell adhesion properties, J. Biol. Chem. 288 (2013) 29654–29669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Obrink B, CEA adhesion molecules: multifunctional proteins with signal-regulatory properties, Curr. Opin. Cell Biol. 9 (1997) 616–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Chin D, Means AR, Calmodulin: a prototypical calcium sensor, Trends Cell Biol. 10 (2000) 322–328. [DOI] [PubMed] [Google Scholar]
- [12].Blikstad I, Wikstrom T, Aurivillius M, Obrink B, C-CAM (Cell-CAM 105) is a calmodulin binding protein, FEBS Lett. 302 (1992) 26–30. [DOI] [PubMed] [Google Scholar]
- [13].Edlund M, Obrink B, Evidence for calmodulin binding to the cytoplasmic domains of two C-CAM isoforms, FEBS Lett. 327 (1993) 90–94. [DOI] [PubMed] [Google Scholar]
- [14].Lu R, Niesen MJ, Hu W, Vaidehi N, Shively JE, Interaction of actin with carcinoembryonic antigen-related cell adhesion molecule 1 (CEACAM1) receptor in liposomes is Ca2+- and phospholipid-dependent, J. Biol. Chem. 286 (2011) 27528–27536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Groves JT, Dustin ML, Supported planar bilayers in studies on immune cell adhesion and communication, J. Immunol. Methods 278 (2003) 19–32. [DOI] [PubMed] [Google Scholar]
- [16].Baksh MM, Jaros M, Groves JT, Detection of molecular interactions at membrane surfaces through colloid phase transitions, Nature 427 (2004) 139–141. [DOI] [PubMed] [Google Scholar]
- [17].Nam JM, Nair PM, Neve RM, Gray JW, Groves JT, A fluid membrane-based soluble ligand-display system for live-cell assays, Chembiochem 7 (2006) 436–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kallick DA, Tessmer MR, Watts CR, Li CY, The use of dodecylphosphocholine micelles in solution NMR, J. Magn. Reson. B 109 (1995) 60–65. [DOI] [PubMed] [Google Scholar]
- [19].Roumestand C, Louis V, Aumelas A, Grassy G, Calas B, Chavanieu A, Oligomerization of protegrin-1 in the presence of DPC micelles. A proton high-resolution NMR study, FEBS Lett. 421 (1998) 263–267. [DOI] [PubMed] [Google Scholar]
- [20].Bayburt TH, Sligar SG, Single-molecule height measurements on microsomal cytochrome P450 in nanometer-scale phospholipid bilayer disks, Proc. Natl. Acad. Sci. U. S. A. 99 (2002) 6725–6730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Tinoco LW, Da Silva A Jr., A. Leite, A.P. Valente, F.C. Almeida, NMR structure of PW2 bound to SDS micelles. A tryptophan-rich anticoccidial peptide selected from phage display libraries, J. Biol. Chem. 277 (2002) 36351–36356. [DOI] [PubMed] [Google Scholar]
- [22].Denisov IG, Grinkova YV, Lazarides AA, Sligar SG, Directed self-assembly of monodisperse phospholipid bilayer Nanodiscs with controlled size, J. Am. Chem. Soc. 126 (2004) 3477–3487. [DOI] [PubMed] [Google Scholar]
- [23].Sharma MK, Gilchrist ML, Templated assembly of biomembranes on silica microspheres using bacteriorhodopsin conjugates as structural anchors, Langmuir 23 (2007) 7101–7112. [DOI] [PubMed] [Google Scholar]
- [24].Baginski L, Gobbo OL, Tewes F, Salomon JJ, Healy AM, Bakowsky U, Ehrhardt C, In vitro and in vivo characterisation of PEG-lipid-based micellar complexes of salmon calcitonin for pulmonary delivery, Pharm. Res. 29 (2012) 1425–1434. [DOI] [PubMed] [Google Scholar]
- [25].Hagn F, Etzkorn M, Raschle T, Wagner G, Optimized phospholipid bilayer nanodiscs facilitate high-resolution structure determination of membrane proteins, J. Am. Chem. Soc. 135 (2013) 1919–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Puthenveetil R, Vinogradova O, Optimization of the design and preparation of nanoscale phospholipid bilayers for its application to solution NMR, Proteins 81 (2013) 1222–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Denisov IG, Sligar SG, Nanodiscs in membrane biochemistry and biophysics, Chem. Rev. 117 (2017) 4669–4713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Ritchie TK, Grinkova YV, Bayburt TH, Denisov IG, Zolnerciks JK, Atkins WM, Sligar SG, Chapter 11 - reconstitution of membrane proteins in phospholipid bilayer nanodiscs, Methods Enzymol. 464 (2009) 211–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Bodenhausen G, Ruben DJ, Natural abundance nitrogen-15 NMR by enhanced heteronuclear spectroscopy, Chem. Phys. Lett. 69 (1980) 185–189. [Google Scholar]
- [30].Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A, NMRPipe: a multidimensional spectral processing system based on UNIX pipes, J. Biomol. NMR 6 (1995) 277–293. [DOI] [PubMed] [Google Scholar]
- [31].Johnson BA, Blevins RA, NMR view: a computer program for the visualization and analysis of NMR data, J. Biomol. NMR 4 (1994) 603–614. [DOI] [PubMed] [Google Scholar]
- [32].Pervushin K, Riek R, Wider G, Wuthrich K, Transverse relaxation–optimized spectroscopy (TROSY) for NMR studies of aromatic spin systems in 13C-labeled proteins, J. Am. Chem. Soc. 120 (1998) 6394–6400. [Google Scholar]
- [33].Schanda P, Brutscher B, Very fast two-dimensional NMR spectroscopy for realtime investigation of dynamic events in proteins on the time scale of seconds, J. Am. Chem. Soc. 127 (2005) 8014–8015. [DOI] [PubMed] [Google Scholar]
- [34].Williamson MP, Using chemical shift perturbation to characterise ligand binding, Prog. Nucl. Magn. Reson. Spectrosc. 73 (2013) 1–16. [DOI] [PubMed] [Google Scholar]
- [35].Lee D, Hilty C, Wider G, Wuthrich K, Effective rotational correlation times of proteins from NMR relaxation interference, J. Magn. Reson. 178 (2006) 72–76. [DOI] [PubMed] [Google Scholar]
- [36].Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS, Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy, J. Med. Chem. 47 (2004) 1739–1749. [DOI] [PubMed] [Google Scholar]
- [37].Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, Banks JL, Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening, J. Med. Chem. 47 (2004) 1750–1759. [DOI] [PubMed] [Google Scholar]
- [38].Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT, Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes, J. Med. Chem. 49 (2006) 6177–6196. [DOI] [PubMed] [Google Scholar]
- [39].Laimer J, Hofer H, Fritz M, Wegenkittl S, Lackner P, MAESTRO – multi agent stability prediction upon point mutations, BMC Bioinf. 16 (2015) 116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Abraham MJ, Murtola T, Schulz R, Pall S, Smith JC, Hess B, Lindahl E, GROMACS: High Performance Molecular Simulations Through Multi-Level Parallelism From Laptops to Supercomputers. SoftwareX1–2, (2015), pp. 19–25. [Google Scholar]
- [41].Sapay N, Bennett WFD, Tieleman DP, Thermodynamics of flip-flop and desorption for a systematic series of phophatidylcholine lipids, Soft Matter 5 (2009) 3295–3302. [Google Scholar]
- [42].Kandt C, Ash WL, Tieleman DP, Setting up and running molecular dynamics simulations of membrane proteins, Methods 41 (2007) 475–488. [DOI] [PubMed] [Google Scholar]
- [43].Berendsen HJC, Grigera JR, Straatsma TP, The missing term in effective pair potentials, J. Chem. Phys. 91 (1987) 6269–6271. [Google Scholar]
- [44].Evans DJ, Holian BL, The nose-hoover thermostat, J. Chem. Phys. 83 (1985) 4069. [Google Scholar]
- [45].Parrinello M, Rahman A, Polymorphic transitions in single crystals: a new molecular dynamics method, J. Appl. Phys. 52 (1981) 7182. [Google Scholar]
- [46].Ikura M, Kay LE, Krinks M, Bax A, Triple-resonance multidimensional NMR study of calmodulin complexed with the binding domain of skeletal muscle myosin light-chain kinase: indication of a conformational change in the central helix, Biochemistry 30 (1991) 5498–5504. [DOI] [PubMed] [Google Scholar]
- [47].Kainosho M, Torizawa T, Iwashita Y, Terauchi T, Mei Ono A, Guntert P, Optimal isotope labelling for NMR protein structure determinations, Nature 440 (2006) 52–57. [DOI] [PubMed] [Google Scholar]
- [48].Vogel HJ, Zhang M, Protein engineering and NMR studies of calmodulin, Mol. Cell. Biochem. 149–150 (1995) 3–15. [DOI] [PubMed] [Google Scholar]
- [49].Siivari K, Zhang M, Palmer AG 3rd, Vogel HJ, NMR studies of the methionine methyl groups in calmodulin, FEBS Lett. 366 (1995) 104–108. [DOI] [PubMed] [Google Scholar]
- [50].Matsubara M, Nakatsu T, Kato H, Taniguchi H, Crystal structure of a myristoylated CAP-23/NAP-22 N-terminal domain complexed with Ca2+/calmodulin, EMBO J. 23 (2004) 712–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Ikura M, Clore GM, Gronenborn AM, Zhu G, Klee CB, Bax A, Solution structure of a calmodulin-target peptide complex by multidimensional NMR, Science 256 (1992) 632–638. [DOI] [PubMed] [Google Scholar]
- [52].Clapham DE, Calcium signaling, Cell 131 (2007) 1047–1058. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.