

Summary
It is well understood that the STING signalling pathway is critical for generating a robust innate immune response to pathogens. Human and mouse STING signalling pathways are not identical, however. For example, mice lack IFI16, which has been proven important for the human STING pathway. Therefore, we investigated whether humanized mice are an appropriate experimental platform for exploring the human STING signalling cascade in vivo. We found that NOG mice reconstituted with human cord blood haematopoietic stem cells (humanized NOG mice) exhibit human STING signalling responses to an analogue of the cyclic di‐nucleotide cGAMP. There was an increase in the proportions of monocytes in the lungs of mice receiving cGAMP analogue. The most robust levels of STING expression and STING‐induced responses were observed in mice exhibiting the highest levels of human chimerization. Notably, differential levels of STING in lung versus spleen following cGAMP analogue treatment suggest that there are tissue‐specific kinetics of STING activation and/or degradation in effector versus inductive sites. We also examined the mouse innate immune response to cGAMP analogue treatment. We detected that mouse cells in the immunodeficient NOG mice responded to the cGAMP analogue and they do so with distinct kinetics from the human response. In conclusion, humanized NOG mice represent a valuable experimental model for examining in vivo human STING responses.
Keywords: cGAMP, humanized mice, inflammation, innate immunology, monocytes
Abbreviations
- cAIMP
cyclic AMP‐IMP; 3′3′‐cAIMP
- cGAMP
cyclic guanosine monophosphate–adenosine monophosphate
- cGAS
cyclic GMP‐AMP synthase
- IFN
interferon
- ISG
interferon‐stimulated gene
- MCP
monocyte chemoattractant protein
- RBC
red blood cell
- STING
stimulator of interferon genes
Introduction
Elucidating the pathways of innate immune recognition of pathogens is critical for the development of novel therapeutic interventions. One pathway that is gaining interest in recent years is the cGAS‐cGAMP‐STING signalling pathway. STING (stimulator of interferon genes) is an adaptor protein that is of fundamental importance in innate immune responses. In particular, STING, which is expressed in multiple cell lineages (e.g. dendritic cells, macrophages, B‐cells and endothelial cells) is a critical component of innate responses toward detection of cytoplasmic DNA.1, 2 When the DNA sensor cGAS (cyclic GMP‐AMP synthase) senses DNA (foreign or self) in the cytoplasm, it produces the natural cyclic‐di‐nucleotide termed cGAMP (cyclic guanosine monophosphate–adenosine monophosphate).3, 4 cGAMP works as an agonist to STING where it binds into a cavity generated by two homodimeric STING proteins.3 cGAMP‐stimulated STING then traffics to an endoplasmatic‐reticulum‐Golgi interim complex where it activates two transcriptional signalling cascades: the TBK1‐IRF3 and the IKK‐NF‐κB pathways.2, 5, 6, 7 Activation of these pathways typically results in the production of inflammatory cytokines as well as type I interferons (IFNs).3
Much is known about the cGAS‐cGAMP‐STING signalling pathway in vivo in mice as well as in vitro and ex vivo in human cells. However, there is a paucity of information about the in vivo activity of this pathway in humans. It is not appropriate to assume that in vivo findings regarding the cGAS‐cGAMP‐STING pathway that are generated in mice will translate directly to human biology. For example, it is known that IFI16 (IFNγ inducible protein 16) is important for human STING responses, yet we have shown that mice lack IFI16.8 Therefore, we asked whether the human cGAS‐cGAMP‐STING axis can be explored in vivo using humanized mice.
Humanized mice are bioengineered immunodeficient animals that harbour human haematopoietic cells.9 These human cells in the mouse are de novo generated from transplanted human haematopoietic stem cells that reside in the mouse bone marrow.10 Here, we utilized humanized mice that are severely immunodeficient common gamma chain null (NOG) mice transplanted with hCD34+ cord blood‐derived stem cells.11 We hypothesized that these humanized NOG mice would serve as a suitable experimental platform for studying human STING signalling and related innate immune responses in vivo. Tissues of these animals harbour human cells of multiple haematopoietic lineages, including T‐cells, B‐cells, macrophages and dendritic cells.11 Furthermore, these animals have already been well‐characterized with respect to both their human lymphoid cell development and human adaptive immune functions.12 A notable gap in the humanized mice literature, however, is information regarding whether the human cells in these models are able to generate human innate immune responses.13 We sought to address this question in the context of the cGAS‐cGAMP‐STING signalling pathway.
Synthetic cGAMP analogues have potential value as therapeutic STING agonists. cGAMP has guanosine and adenosine as the purine nucleoside pair.14 This is distinct from the synthetic STING agonist used here: cyclic Adenosine monophosphate–Inosine monophosphate (cyclic AMP‐IMP; 3′3′‐cAIMP).15 This 3′3′‐cAIMP (referred to hereafter as cAIMP) is stabilized with a bis‐phosphorothioate linkage of the nucleosides.15 Here, we utilized the synthetic and stabilized cAIMP molecule to investigate human innate immune responses in vivo in humanized NOG mice.
Methods
Bioengineering of humanized NOG mice
Humanized mouse experiments were conducted according to Danish national, as well as internationally recognized, guidelines. Experiments were approved by The Experimental Animal Inspectorate in Denmark under The Danish Veterinary and Food Administration, Ministry of Food, Agriculture and Fisheries (protocol number: 2013‐15‐2934‐00861). Animal facility conditions were as follows: 12‐hr dark : light cycle; temperature 22 ± 2° and humidity of 55 ± 10%. Mice were maintained, four mice per cage, in a sterile environment. Type III plastic cages (Techniplast, Buguggiate VA, Italy) were prepared with wood chip bedding (Finn Tapvei, Kaavi, Finland) plus enrichment (e.g. hide shelters and gnawing sticks) and then autoclaved. Mice had access to sterile acidified water and sterilized standard chow ad libitum. Animal welfare was monitored daily by experienced animal caretakers. Female NOG (NOD.Cg‐Prkdc scid Il2rg tm1Sug/JicTac) mice (5–8 weeks old) were obtained from Taconic, Denmark. After acclimatization, the mice were γ‐irradiated (75 cGy) and injected with either 3·78 × 105 or 0·75 × 105 cord blood‐derived CD34+ cells/mouse. We leveraged the fact that we had an uncharacteristically large yield of human cord blood‐derived CD34+ cells (EasySep™ Human Cord Blood CD34 Positive Selection Kit II; #17896) from a single female birth and performed these experiments using humanized NOG mice that were all generated from the same human donor. This fact allows for direct comparison between groups with no confounding influence of differing human genetics within each group. Human chimerism was longitudinally monitored by flow cytometry as described below. Mice receiving the high number of stem cells exhibited > 50% human chimerization – defined as the fraction of peripheral white blood cells expressing hCD45. This group is referred to as Hi‐Hu. Mice receiving the low dose of stem cells exhibited 10%–50% human chimerization and are referred to as Low‐Hu. When mice exhibited the indicated chimerization levels (approximately 7 weeks post‐transplant for Hi‐Hu mice and 20 weeks post‐transplant for Low‐Hu mice), they were dosed intravenously with 6·25 mg/kg 3′3′‐cAIMP (3′3′‐cyclic adenosine monophosphate‐inosine monophosphate; InvivoGen, Toulouse, France) and killed 1 or 6 hr after injection.
Flow cytometry
Human chimerism in peripheral blood was tracked longitudinally using flow cytometry analyses as we have previously described.16 Whole peripheral blood from humanized NOG mice was assessed for human chimerism using the BD Biosciences Lyse/Wash protocol (#349202). Whole blood samples were incubated with Human Trustain FcX blocking buffer for 10 min at room temperature (BioLegend, San Diego, CA, USA) and then stained with fluorophore‐conjugated mouse‐anti human antibodies (listed below) for 30 min at room temperature. After antibody labelling, red blood cells (RBCs) were lysed, and the remaining cells were washed in 2% fetal bovine serum‐containing phosphate‐buffered saline (PBS). Lung and spleen mononuclear cells were analysed at harvest. Briefly, lung and spleen mononuclear cells were incubated with Human Trustain FcX blocking buffer for 10 min at room temperature and then stained with fluorophore‐conjugated mouse‐anti‐human antibodies (listed below) for 30 min at room temperature. After antibody labelling, the cells were washed in 0·5% fetal bovine serum‐containing PBS and analysed on the cytometer. The following antibodies were used for labelling: CD8 (SK1) PerCP‐Cy5.5 (Biolegend, #344710); CD4 (SK3) PE‐Cy7 (Biolegend, #344612); CD3 (OKT3), APC‐Cy7 (Biolegend, #317342); CD14 (M5E2) BV421 (Biolegend, #301830); CD33 (RPA‐T4) BV605 (Biolegend, #366612); CD3 (OKT3) FITC (Biolegend, #317306); CD8 (RPA‐T8) PE (BD, #561949); CD16 (3G8) PerCP‐Cy5.5 (Biolegend, #302028); CD19 (sj25c1) PE‐Cy7 (BD, #557835); CD45 (2D1) APC (Biolegend, #368512); and CD4 (RPA‐T4) APC‐Cy7 (BD, #560251). Flow cytometry data were collected on a BD FACSVerse cytometer and analysed in flowjo v.10.
Splenocyte and lung mononuclear cell isolations
Splenocytes and lung cells were isolated as we have previously described.16 Spleen and lung tissues were harvested into ice‐cold suspension media [PBS supplemented with 5 g/l bovine serum albumin (SigmaAldrich, Søborg, Denmark #A8022); 1% final concentration of penicillin/streptomycin solution (Biowest, Nuaillé, France #L0022‐100); and 10 kunitz/unit DNase per ml (SigmaAldrich, #D4263)]. Tissues were transported on ice from the animal facility to the laboratory. For splenocytes, spleens were gently disrupted using a plunger from a 3‐ml syringe on a 70‐μm cell strainer such that individual splenocytes passed through the strainer. Spleen remnants and the strainer were washed with suspension media and resuspended in 10 ml of 1 ×RBC lysis buffer (BioLegend, #420301). The cell suspension was incubated for 10 min at 4°. After RBC lysis, cells were pelleted (5 min; 300 g; 4°), washed in suspension media and resuspended in 1 ml suspension media for viable cell counting using trypan blue exclusion. Humanized NOG mouse lung tissues were processed essentially as we have described before for human sigmoid biopsies.17 Briefly, lung tissues were minced and digested in an enzyme cocktail with constant agitation at 37° for 60 min. After digestion, lung cells were collected at the interface of a 40%–70% Percoll density separation centrifugation. Cells were washed 1× in suspension media and resuspended in 1 ml suspension media for viable cell counting using trypan blue exclusion.
Olink proteomics
Serum samples were analysed using the ‘Immuno‐Oncology’ panel (n = 92 analytes) by Olink® by Olink Proteomics (BioXpedia, Aarhus, Denmark). Within this human analyte panel, there are some Olink detection antibodies that can recognize either human or mouse proteins given homology between species. Therefore, our experimental setup also included non‐humanized NOG mice that received the cAIMP just as we performed with the humanized NOG mice. Serum from the non‐humanized NOG mice was also analysed by Olink. Analytes that were positive in the non‐humanized mice were excluded from the presented data (Fig. 1a,b). Olink data values were calculated by subtracting the average NPX (log2 of the absolute measurement) of the measurement of unstimulated control mice from the respective group (Hi‐Hu or Low‐Hu) from the NPX of each of the three animals in the stimulated groups [ln2(fold change) = NPX(stimulation) − NPX(control)].18 Respective baseline controls from the Low‐Hu and Hi‐Hu group were utilized to properly account for any potential differences that could have existed between the groups at dosing. This analysis approach allows us to distinguish cGAMP analogue‐specific effects within each group. Measurements of proteins below the detection limit were set to 0. An upregulation or downregulation of log fold change equal to 1 or −1 indicates a twofold difference in absolute values.
Figure 1.
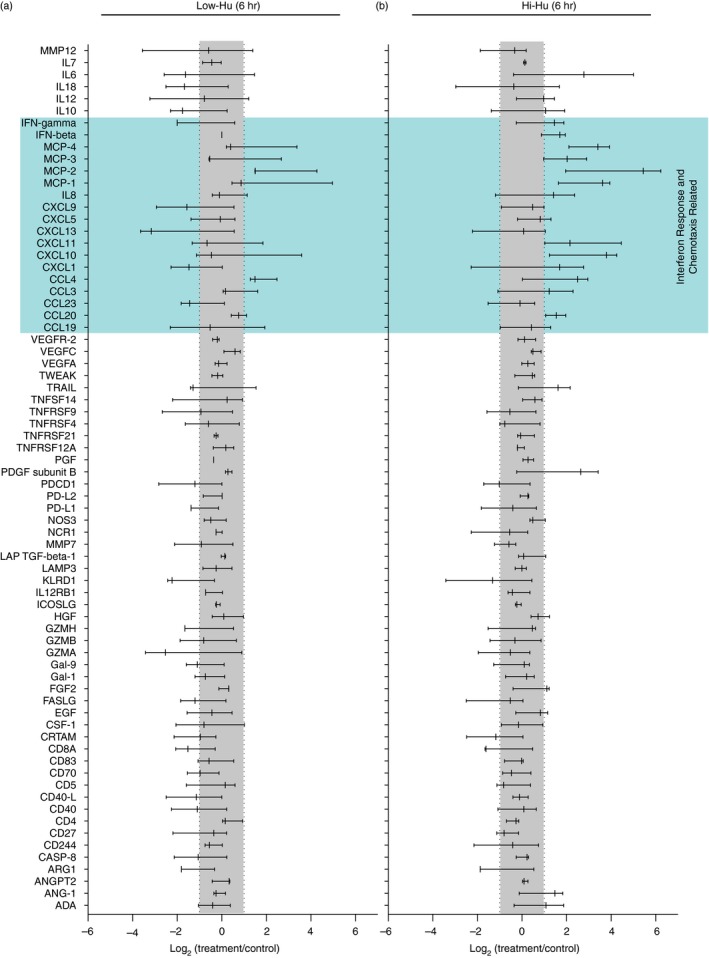
STING stimulation with the cGAMP analogue 3′3′‐cAIMP induced the production of human chemotaxis‐related proteins in humanized mice. (a, b) Forest plot presentations of log2 fold changes in 72 analytes detected with Olink technology. Serum samples were collected for this analysis 6 hr following cAIMP dosing of Low‐Hu (a; n = 3) and Hi‐Hu (b; n = 3) mice. The forest plots incorporate non‐treated control (n = 3, Low‐Hu; n = 2, Hi‐Hu) values into the data presentation as the basis for the grey ‘no change’ bar that bisects the graphs from top to bottom reflects. The log2 fold change presented is interpreted as upregulated if the 90% confidence interval presented for each analyte is outside the bounds of the −1 to 1 interval highlighted by the grey bar. Confidence intervals in the forest plot span from 5% to 95%. Serum from cAIMP‐treated (6 hr) non‐humanized NOG mice was also analysed, and analytes that were positive in the non‐humanized mice were excluded such that the reported analyte concentrations reflect the levels of human molecules.
Type I IFN bioassay
Human type I IFN was measured using the HEK‐Blue IFN‐α/β (InvivoGen) bioassay according to manufacturer protocols.8 Briefly, HEK‐Blue cells were seeded in 96‐well plates in 150 μl medium (without blasticidin or zeocin). After an overnight incubation, 50 μl of humanized NOG mouse serum was added to wells in duplicate. Bioactive human IFN‐α/β in the serum activates the ISG54 promotor in these cells inducing secreted embryonic alkaline phosphatase production, which is quantitated against an IFN‐α (A2) (PBL Assay Science, Piscataway, NJ, USA) standard curve using optical density (620 nm; ELx808; BioTEK, Winooski, VT, USA).
Western blot analyses
STING immunoblotting was performed as previously described.8 Briefly, splenocytes or lung cells were lysed in RIPA buffer (Thermo Scientific, Roskilde, Denmark) containing proteases (Roche, Hvidovre, Denmark), 0·2% sodium dodecyl sulphate (SDS), 1 × XT Sample Buffer (Bio‐Rad, Copenhagen, Denmark) and 1 × XT Reducing Agent (Bio‐Rad) and cryopreserved. Upon thawing, lysates were sonicated and separated using a 10% SDS–polyacrylamide gel electrophoresis (Criterion TGX gels; Bio‐Rad). Following transfer and washing, membranes were blocked with 5% skim‐milk (Sigma Aldrich 70166‐500G). After blocking, membranes were probed with anti‐STING (Cell Signaling Technologies, Leiden, The Netherlands, #13647) or anti‐vinculin (Sigma Aldrich v9131) antibodies diluted 1 : 1000 in 1% skim‐milk solutions. Secondary antibodies (dilution 1:15 000): peroxidase‐conjugated Affinipure F(abŕ)2 donkey anti‐rabbit IgG (H + L) or peroxidase‐conjugated F(abŕ)2 donkey anti‐mouse IgG (H + L) (Jackson Immuno Research, Cambridgeshire, UK). Signal was detected using Clarity Western ECL Blotting Substrate according to manufacturer protocols.
Statistics
For the Olink data presented in Fig. 1, the log2 fold change presented is upregulated if the 90% confidence interval presented for each analyte is outside the bounds of the −1 to 1 interval highlighted by the grey bar. All treatment groups were compared with control values using Mann–Whitney U‐tests or, if also compared with each other, using one‐way ANOVA Kruskal–Wallis with Dunn's multiple comparisons. The utilized statistical test is specified in each figure legend. Statistical comparisons that met the alpha of 0·05 are noted with P‐values above the data within the figure.
Results
Human serum proteins related to chemotaxis are elevated 6 hr after cAIMP treatment
We utilized Olink technology to quantify human protein levels (n = 72) in humanized NOG mouse serum 6 hr after cAIMP administration (Fig. 1a,b). Mice were stratified based on human chimerism levels: Low‐Hu had 10%–50% blood human chimerization and Hi‐Hu had > 50% blood human chimerization. The blue field in Fig. 1 encompasses the bulk of cAIMP‐modulated proteins, and draws attention to several particularly relevant analytes in the context of IFN responses and chemotaxis. The greatest overall response was observed in the Hi‐Hu group, which exhibited upregulation of many analytes including MCP‐1, MCP‐2, MCP‐4 and CXCL10 (Fig. 1b). While the cAIMP effect was less pronounced in Low‐Hu mice, these animals also showed increased levels of some chemotaxis‐related molecules (e.g. MCP‐2 and CCL4; Fig. 1a). These results show that the human immune system in humanized NOG mice can respond to cAIMP stimulation.
Human and mouse tissue cells exhibit an IFN response following cAIMP treatment
In addition to the peripheral blood analysis presented in Fig. 1, we examined humanized NOG mouse tissues for evidence of a response to cAIMP treatment. We examined spleen and lung as important sites for distinct immune functions. The frequency of human CD45+ cells in the different tissues is presented in Fig. S1. The human CD45+ frequencies mirror the frequencies found in peripheral blood. The spleen, as a secondary lymphoid tissue, is an immune inductive site,19 and the lungs represent an immune effector site that exhibits IFN responses to invading pathogens.20, 21 Transcriptional analysis was focused on IFNβ (a type I IFN) and CXCL10 [an IFN‐stimulated gene (ISG)]. Consistent with the serum Olink data trend towards higher IFNß in Hi‐Hu mice at 6 hr post‐cAIMP dosing (Fig. 1b), we found that splenocytes from Hi‐Hu mice expressed human IFNβ mRNA at 6 hr post‐cAIMP dosing (Fig. 2a). We also observed that splenocytes and to a lesser extent lung cells from Hi‐Hu mice produced human CXCL10 (also known as IFNγ‐induced protein 10) mRNA at 6 hr post‐cAIMP dosing (Fig. 2b), which mirrors the observed CXCL10 protein levels in serum (Fig. 1b). With these data, we confirmed the systemic nature of the human cAIMP response in humanized NOG mice.
Figure 2.
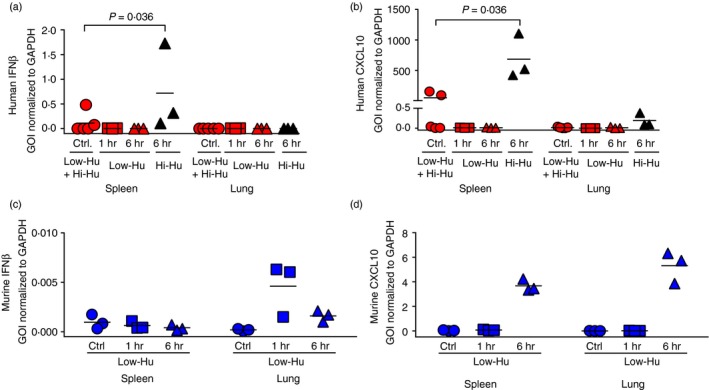
Both mouse and human interferon (IFN) responses were observed in cAIMP‐treated humanized mice. (a–d) Expression of the human IFNβ (a) and human CXCL10 (b) as well as murine IFNβ (c) and murine CXCL10 (d) genes as measured by qPCR analysis. Data are normalized to GAPDH mRNA levels. Mann–Whitney U‐test comparisons that met the alpha of 0·05 are noted with P‐values above the data within the figure. Each symbol represents one humanized NOG mouse.
Next, we examined whether there was also a mouse STING response to cAIMP treatment given that the cAIMP molecule utilized here targets both human and murine STING.15, 22 We postulated that there would be a mouse response given that the common gamma chain mutation found in NOG mice precludes T‐cells, B‐cell and NK cell development, but not myeloid cell development.11 However, the common gamma chain is broadly involved in many critical immune signalling pathways,23 so its absence could impact STING signalling. We measured mouse IFNβ and mouse CXCL10 mRNA levels in humanized NOG mice tissues. The levels of mouse IFNβ mRNA were increased in lung, but not spleen, at 1 hr post‐cAIMP dosing and had returned to baseline by 6 hr post‐administration (Fig. 1c). The transcription of mouse CXCL10 was elevated at the 6‐hr time point in both spleen and lung (Fig. 1d). Thus, STING agonist administration stimulated both mouse and human STING responses in humanized NOG mice.
The proportion of human monocytes in humanized NOG mouse lungs increases following cAIMP treatment
One hallmark of efficient immune function is the translocation of effector cells to sites of immune effector activity (e.g. lungs). Furthermore, we observed in Fig. 1 that multiple monocyte chemoattractant protein (MCP) chemokine levels are increased with cAIMP treatment. These proteins have roles in the recruitment of monocytes and macrophages, as well as other leucocytes, to sites of inflammation.24 Therefore, we quantitated changes in the fraction of myeloid‐derived lung cells in Low‐Hu (Hi‐Hu mouse lung cells were prioritized to other assays) mice according to surface expression patterns of CD33, CD14 and CD16 (Fig. 3a–e).25 Frequencies of lung cell subtypes from individual Low‐Hu mice are presented in Table S1. Our approach did not allow us to specifically characterize CD33+ alveolar macrophages as was recently reported based upon CyTOF data.26 We noted a separation of cells into either CD33neg, CD33dim or CD33bright populations (Fig. 3b), and a specific increase in the classical monocytes (the CD33bright myeloid cells; Fig. 3e) following cAIMP administration (P = 0·02; Fig. 3f). This finding is consistent with prior data showing a majority of classical monocytes at effector sites early in the inflammatory response.27 Moreover, neutrophils are actively recruited to sites of inflammation as a response to IFN‐stimulation in the early phases of inflammation,28 and are known to develop in humanized NOD/SCID gamma chain null mice.13, 29 In line with this, we found the lung CD33dimCD16+ CD14neg population (consistent with a neutrophil granulocyte population25) to be significantly upregulated (P = 0·04) 1 hr post‐cAIMP administration (Fig. 3g). These human myeloid cell population dynamics in the lungs are consistent with the increased human chemotactic protein levels observed in the serum Olink analyses (Fig. 1).
Figure 3.
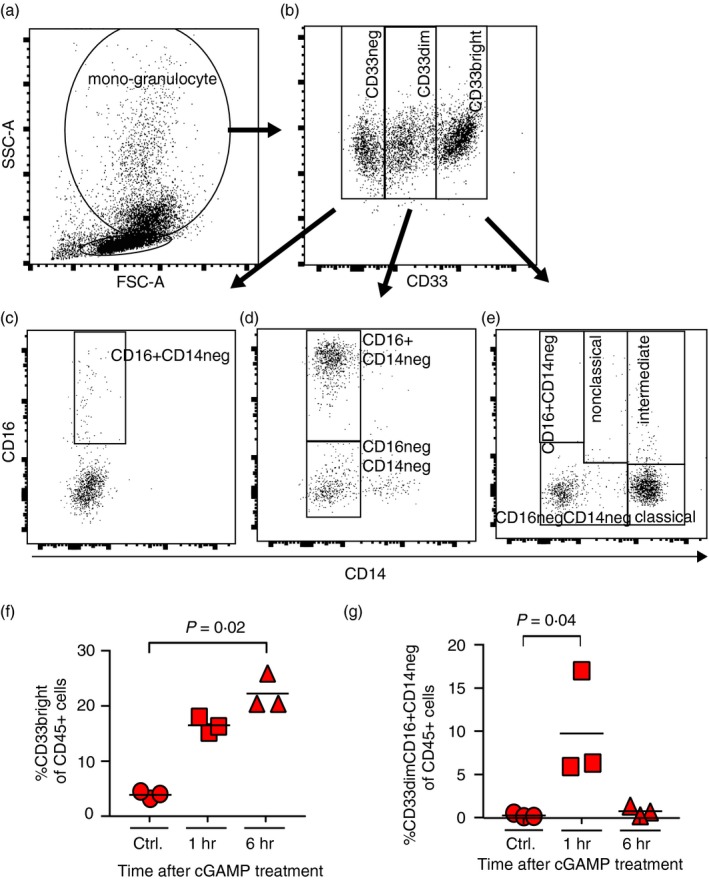
Classical monocyte proportions increased in the lungs of cAIMP‐treated humanized mice. (a–e) These panels present the mouse lung cell flow cytometry gating strategy. Gates were set based on singlets (SSC‐A/SSC‐H), CD45+ and subsequently on FSC/SSC to divide cells into a lymphocyte and a mono‐granulocyte compartment (a). Within this gate, cells were divided into CD33neg, CD33dim, CD33bright populations (b). These populations were further characterized based on CD14 and CD16 expression (c–e). (f,g) Proportions of CD33bright cells in Low‐Hu mice (b) are shown for each mouse in (f). The proportions of CD33dimCD16+ CD14neg cells in these same animals (d) are shown in (g). Values are presented as percentages of total CD45+ cells. Each symbol represents one mouse. One‐way ANOVA Kruskal–Wallis with Dunn's multiple comparisons results that met the alpha of 0·05 are noted with P‐values above the data within the figure.
cAIMP‐induced serum type I human IFNs in humanized NOG mice are bioactive
We extended the protein findings in Fig. 1 with an assay for quantitating functional human type I IFN. We observed that bioactive human IFN levels were not significantly increased in the Low‐Hu group, but were higher in the Hi‐Hu mice (Fig. 4a). A potential mechanistic explanation for these findings was revealed via Western blot analyses of the spleen and lung. The key comparisons in Fig. 4(b,c) are the relative differences in the levels of STING between the treated versus untreated mice within a given group. We observed lower total STING levels in Low‐Hu versus Hi‐Hu mice (Fig. 4b,c). This observation indicates a relative lack of binding targets for cAIMP in the Low‐Hu animals. The sole outlier to these observations was control mouse ‘a’, which was experiencing systemic immune activation in the absence of cAIMP stimulation due to an inflammatory condition unrelated to the study. Importantly, we did not detect STING (human or mouse) in control NOG mice that were not humanized as presented in Fig. S2. Together, these data lead us to conclude that high levels of human chimerism elevate human STING levels, which facilitate an increased human innate immune response to cAIMP administration.
Figure 4.
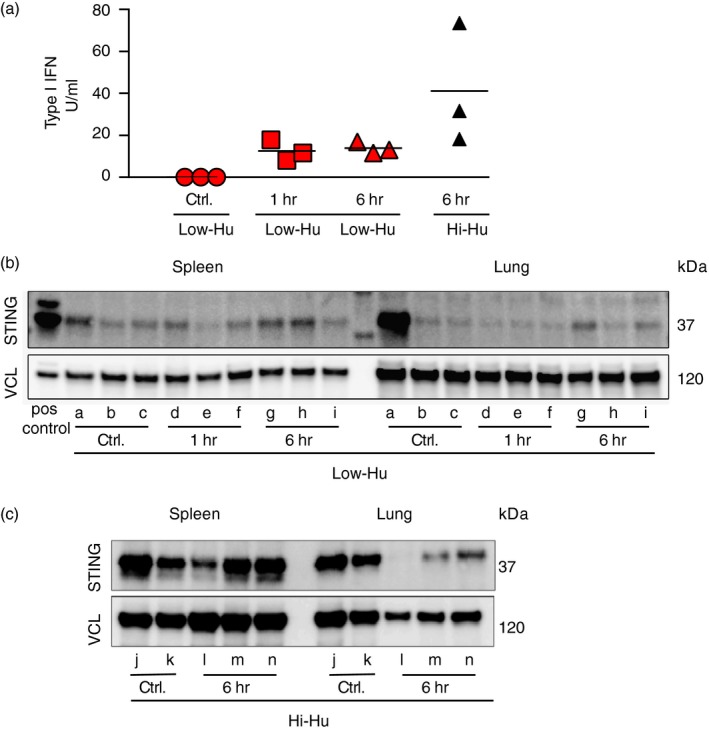
Functional human type I interferon (IFN) production improved with STING expression. (a) Bioassay results for functional type I IFN levels in serum are shown. Mann–Whitney U‐test comparisons that met the alpha of 0·05 are noted with P‐values above the data within the figure. Each symbol represents one humanized NOG mouse. (b, c) Western blot analyses are shown for STING protein as well as for vinculin (VCL) expression in splenocytes and lung cells from humanized NOG mice. Each lower‐case letter label identifies a unique mouse. Group inclusion and treatment protocols for each animal are also noted.
Discussion
Here, we present data that answer a key question: Is it possible to study the human cGAS‐cGAMP‐STING signalling pathway in vivo using humanized mice? Specifically, we show that the cGAMP analogue cAIMP elicits a STING‐dependent human IFN response in humanized NOG mice. We also find that mouse cells are able to respond to this same stimulation in the absence of mouse IL2 receptor common gamma chain signalling. Beyond this pair of important observations, our data raise several interesting hypotheses and suggest multiple lines of future investigations.
There are three insights to be gleaned from the fact that we observed both mouse and human innate responses to cAIMP treatment. First is the novel observation that the mouse common gamma chain signalling is not necessary for mouse cells to respond to this cGAMP analogue in vivo. Second, mouse and human cytokines are present at the same time in cAIMP stimulated humanized mice. This observation is noteworthy because it highlights the potential for cross‐talk between mouse and human cells/proteins in humanized mice. This is a potential complication that is typically unexplored in humanized mouse research despite bioinformatic predictions of the probabilities of such cross‐talk.30 In our dataset, the relatively low mouse cytokine responses, versus human, are likely due to the fact that the mouse strain already has defects in immune pathways and subsequently received irradiation that could further impair immune function. While our data stop short of proving inter‐species cellular communication in vivo, we confirm that this is a potential confounder that should be considered in preclinical humanized mouse experiments. Lastly, the IFNß response in human and mouse cells was quite distinct when examined kinetically. The mouse response was present at 1 hr post cAIMP dosing and gone by 6 hr. In contrast, the human response was only observed at 6 hr post‐dosing. This differential in vivo response time is novel and should be investigated to determine a mechanism that can explain this observation.
The fact that there is more STING detected in Hi‐Hu versus Low‐Hu mice is attributed to the increased number of human cells present. Based on this observation plus other data indicating that Hi‐Hu mice responded more vigorously to cAIMP stimulation, we conclude that higher human levels (and the accompanying higher STING levels) are necessary to generate robust innate immune responses to a cGAMP analogue in vivo. Further, within the STING Western blot and the IFNβ mRNA data, there is an interesting and hypothesis‐generating tissue‐specific finding. It is known that intracellular trafficking and proteasomal degradation regulate activated STING levels.31 We noted that mice ‘l’, ‘m’ and ‘n’ exhibited differential levels of STING in splenocytes versus lung cells in the Western blot, and versus control mice ‘j’ and ‘k’ in lung cells. At the same 6‐hr time point, we observed that splenocytes exhibited higher IFNß versus lung cells. When examined together, these observations suggest that STING activation was detected at the 6‐hr time point in splenocytes (inductive site), while STING degradation was detected at the 6‐hr time point in lung cells (effector site). Future experiments are necessary to begin dissecting whether there are differential kinetics of STING activation and degradation following cGAMP stimulation at immune effector versus inductive sites.
Limitations of this work are noted. This study was possible because the human haematopoietic stem cell yield that we obtained from a single cord blood sample was sufficient to generate both Hi‐Hu and Low‐Hu groups with identical human genetics (a strength of the study); nevertheless, a limitation of this work is that we assessed humanized mice bioengineered from a single human donor, which limits the generalizability of these results. Second, we had an insufficient number of Hi‐Hu animals to also perform the 1‐hr cAIMP stimulation in this group. In particular, this precludes assessing differences between 1‐ versus 6‐hr cAIMP treatment in Hi‐Hu mice and 1‐hr versus 1‐hr between groups. We did detect a significant upregulation of IFNß mRNA in Hi‐Hu after 6 hr of treatment in the spleen, but the Olink assay did not show a clear upregulation of serum protein levels of IFNβ after only 6 hr of treatment. Thus, the time points we analysed permitted us to observe the mRNA production phase without detecting peak systemic levels of this cytokine in the blood.
In conclusion, we show that a single systemic administration of the cAIMP, a cGAMP analogue, mobilizes and activates a human innate response via STING signalling in a chimerization‐level‐dependent manner in humanized NOG mice. Our results outline an important relationship between chimerization status and innate immune responses in humanized mice. Further, because we show that both mouse and human cells respond to the cAIMP treatment, our results highlight the potential of cross‐talk between mouse and human signalling‐ and chemotaxis‐related molecules, especially in the myeloid compartment of humanized mice. This is an important consideration when using humanized mice in preclinical research, particularly when innate immunity is the focus of those efforts.
Author contributions
PWD, MRJ, AHFA, RO, MT and LØ conceived and designed the study. AHFA, RO, KLJ, JFH, CK, KM, FD‐H, MRJ and PWD bioengineered humanized mice and performed experiments. MKT formulated cAIMP dosing. AHFA wrote the first manuscript draft. All authors participated in data analyses, data interpretations as well as manuscript preparation. All authors approved the final manuscript version.
Disclosure
The authors declare no conflicts of interest.
Supporting information
Figure S1. Proportions of human CD45+ cells in total lung and spleen tissues from Low‐Hu and Hi‐Hu mice are shown as a mean of each group for each organ (± SD).
Figure S2. Western blot analyses are shown for STING protein as well as for vinculin (VCL) expression in splenocytes and lung cells from non‐humanized NOG mice.
Table S1. Frequencies of lung cell subtypes from individual Low‐Hu mice
Acknowledgements
The authors thank the AU Biomedicine Animal Facility staff, particularly Ms Jani Kær, for animal care and colony maintenance efforts. The authors thank InvivoGen for the design, synthesis and production of endotoxin‐free 3′3′‐cAIMP material. This study was funded by Aarhus University Hospital and an Aarhus University Faculty of Health graduate student scholarship (AHFA), a grant from Aarhus University Research Foundation (AUFF‐E‐2016‐FLS‐8‐9) (PWD), and The Lundbeck Foundation (MRJ).
A.H.F.A., R.O. and K.L.J. contributed equally to this study.
Contributor Information
Martin R. Jakobsen, Email: mrj@biomed.au.dk
Paul W. Denton, Email: pade@clin.au.dk.
References
- 1. Burdette DL, Monroe KM, Sotelo‐Troha K, Iwig JS, Eckert B, Hyodo M et al STING is a direct innate immune sensor of cyclic di‐GMP. Nature 2011; 478:515–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 2008; 455:674–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chen Q, Sun L, Chen ZJ. Regulation and function of the cGAS‐STING pathway of cytosolic DNA sensing. Nat Immunol 2016; 17:1142–9. [DOI] [PubMed] [Google Scholar]
- 4. Li XD, Wu J, Gao D, Wang H, Sun L, Chen ZJ. Pivotal roles of cGAS‐cGAMP signaling in antiviral defense and immune adjuvant effects. Science 2013; 341:1390–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT et al IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol 2003; 4:491–6. [DOI] [PubMed] [Google Scholar]
- 6. Sharma S, tenOever BR, Grandvaux N, Zhou GP, Lin R, Hiscott J. Triggering the interferon antiviral response through an IKK‐related pathway. Science 2003; 300:1148–51. [DOI] [PubMed] [Google Scholar]
- 7. Tanaka Y, Chen ZJ. STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci Signal 2012; 5:ra20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jonsson KL, Laustsen A, Krapp C, Skipper KA, Thavachelvam K, Hotter D et al IFI16 is required for DNA sensing in human macrophages by promoting production and function of cGAMP. Nat Commun 2017; 8:14391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Manz MG, Di Santo JP. Renaissance for mouse models of human hematopoiesis and immunobiology. Nat Immunol 2009; 10:1039–42. [DOI] [PubMed] [Google Scholar]
- 10. Goyama S, Wunderlich M, Mulloy JC. Xenograft models for normal and malignant stem cells. Blood 2015; 125:2630–40. [DOI] [PubMed] [Google Scholar]
- 11. Ito M, Hiramatsu H, Kobayashi K, Suzue K, Kawahata M, Hioki K et al NOD/SCID/gamma(c)(null) mouse: an excellent recipient mouse model for engraftment of human cells. Blood 2002; 100:3175–82. [DOI] [PubMed] [Google Scholar]
- 12. Ito R, Takahashi T, Katano I, Ito M. Current advances in humanized mouse models. Cell Mol Immunol 2012; 9:208–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tanaka S, Saito Y, Kunisawa J, Kurashima Y, Wake T, Suzuki N et al Development of mature and functional human myeloid subsets in hematopoietic stem cell‐engrafted NOD/SCID/IL2rgammaKO mice. J Immunol 2012; 188:6145–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gao D, Wu J, Wu YT, Du F, Aroh C, Yan N et al Cyclic GMP‐AMP synthase is an innate immune sensor of HIV and other retroviruses. Science 2013; 341:903–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lioux T, Mauny MA, Lamoureux A, Bascoul N, Hays M, Vernejoul F et al Design, synthesis, and biological evaluation of novel cyclic adenosine‐inosine monophosphate (cAIMP) analogs that activate stimulator of interferon genes (STING). J Med Chem 2016; 59:10253–67. [DOI] [PubMed] [Google Scholar]
- 16. Denton PW, Nochi T, Lim A, Krisko JF, Martinez‐Torres F, Choudhary SK et al IL‐2 receptor gamma‐chain molecule is critical for intestinal T‐cell reconstitution in humanized mice. Mucosal Immunol 2012; 5:555–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Krarup AR, Abdel‐Mohsen M, Schleimann MH, Vibholm L, Engen PA, Dige A et al The TLR9 agonist MGN1703 triggers a potent type I interferon response in the sigmoid colon. Mucosal Immunol 2017; 11:449–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Quackenbush J. Microarray data normalization and transformation. Nat Genet 2002; 32(Suppl):496–501. [DOI] [PubMed] [Google Scholar]
- 19. Bronte V, Pittet MJ. The spleen in local and systemic regulation of immunity. Immunity 2013; 39:806–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kopf M, Schneider C, Nobs SP. The development and function of lung‐resident macrophages and dendritic cells. Nat Immunol 2015; 16:36–44. [DOI] [PubMed] [Google Scholar]
- 21. Kumagai Y, Takeuchi O, Kato H, Kumar H, Matsui K, Morii E et al Alveolar macrophages are the primary interferon‐alpha producer in pulmonary infection with RNA viruses. Immunity 2007; 27:240–52. [DOI] [PubMed] [Google Scholar]
- 22. Skouboe MK, Knudsen A, Reinert LS, Boularan C, Lioux T, Perouzel E et al STING agonists enable antiviral cross‐talk between human cells and confer protection against genital herpes in mice. PLoS Pathog 2018; 14:e1006976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ma A, Koka R, Burkett P. Diverse functions of IL‐2, IL‐15, and IL‐7 in lymphoid homeostasis. Annu Rev Immunol 2006; 24:657–79. [DOI] [PubMed] [Google Scholar]
- 24. McQuibban GA, Gong JH, Wong JP, Wallace JL, Clark‐Lewis I, Overall CM. Matrix metalloproteinase processing of monocyte chemoattractant proteins generates CC chemokine receptor antagonists with anti‐inflammatory properties in vivo . Blood 2002; 100:1160–7. [PubMed] [Google Scholar]
- 25. Gustafson MP, Lin Y, Maas ML, Van Keulen VP, Johnston PB, Peikert T et al A method for identification and analysis of non‐overlapping myeloid immunophenotypes in humans. PLoS One 2015; 10:e0121546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Morrell ED, Wiedeman A, Long SA, Gharib SA, West TE, Skerrett SJ et al Cytometry TOF identifies alveolar macrophage subtypes in acute respiratory distress syndrome. JCI Insight 2018; 3:99281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ingersoll MA, Platt AM, Potteaux S, Randolph GJ. Monocyte trafficking in acute and chronic inflammation. Trends Immunol 2011; 32:470–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mantovani A, Cassatella MA, Costantini C, Jaillon S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol 2011; 11:519–31. [DOI] [PubMed] [Google Scholar]
- 29. Coughlan AM, Freeley SJ, Robson MG. Humanised mice have functional human neutrophils. J Immunol Methods 2012; 385:96–104. [DOI] [PubMed] [Google Scholar]
- 30. Rongvaux A, Takizawa H, Strowig T, Willinger T, Eynon EE, Flavell RA et al Human hemato‐lymphoid system mice: current use and future potential for medicine. Annu Rev Immunol 2013; 31:635–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gonugunta VK, Sakai T, Pokatayev V, Yang K, Wu J, Dobbs N et al Trafficking‐mediated STING degradation requires sorting to acidified endolysosomes and can be targeted to enhance anti‐tumor response. Cell Rep 2017; 21:3234–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Proportions of human CD45+ cells in total lung and spleen tissues from Low‐Hu and Hi‐Hu mice are shown as a mean of each group for each organ (± SD).
Figure S2. Western blot analyses are shown for STING protein as well as for vinculin (VCL) expression in splenocytes and lung cells from non‐humanized NOG mice.
Table S1. Frequencies of lung cell subtypes from individual Low‐Hu mice