

Summary
Inflammasome activation and subsequent inflammatory cytokine secretion are essential for innate immune defence against multiple stimuli and are regarded as a link to adaptive immune responses. Dysfunction of inflammasome activation has been discovered at the onset or progression of infectious diseases, autoimmune diseases and cancer, all of which are also associated with metabolic factors. Furthermore, many studies concerning the metabolic regulation of inflammasome activation have emerged in recent years, especially regarding the activity of the NLRP3 inflammasome under metabolic reprogramming. In this review, we discuss the molecular mechanisms of the interactions between metabolic pathways and inflammasome activation, which exerts further important effects on various diseases.
Keywords: cancer progression, inflammasome, inflammation, innate immunity, macrophages, metabolic reprogramming, signalling
Introduction
Immunometabolism, which includes interactions between immunity and metabolism, has been widely investigated in recent years.1 Diverse exogenous and endogenous danger signals drive inflammatory responses of immune cells orchestrated with cellular metabolic reprogramming. Inflammasome activation and its downstream cascades, including inflammatory cytokine maturation and secretion, play vital roles in innate immune defence, and its dysregulation is involved in the pathogenesis of many diseases.2 Notably, the significance of metabolic repurposing in regulating inflammasome activation has been reflected in emerging studies,3, 4, 5 which further provide novel therapeutic targets for infectious diseases, autoimmune diseases and cancer.
Regulation of inflammasome activation in inflammation
The inflammasome, a large intracellular multimeric protein complex, consists of cytosolic sensors involving nucleotide‐binding oligomerization domain and leucine‐rich repeat‐containing receptors (NLRs) or absent in melanoma 2‐like receptors (ALRs), adaptor proteins termed apoptotic speck‐containing protein (ASC) or NLR family CARD domain‐containing protein 4 (NLRC4) and the effector protein pro‐caspase‐1 (Fig. 1). Different sensors recognize different pathogen‐associated molecular patterns (PAMPs) or danger‐associated molecular patterns (DAMPs), initiating inflammasome assembly by interacting with ASC. Sensor‐activated ASC aggregates into specks, recruiting pro‐caspase‐1, which leads to proximity‐induced caspase‐1 autoproteolysis,6, 7 subsequently leading to interleukin‐1β (IL‐1β) and IL‐18 maturation as well as gasdermin D (GSDMD) cleavage, ultimately causing inflammatory cytokine secretion and a form of pro‐inflammatory programmed cell death termed pyroptosis.8 Specifically, NLRC4 acts as a bridge between the NLR family, apoptosis inhibitory protein (NAIP) recognition and ASC oligomerization during NLRC4 inflammasome assembly.9
Figure 1.
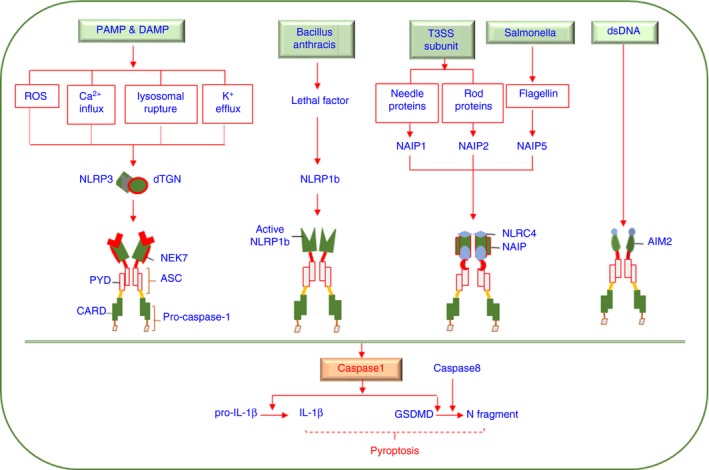
Molecular mechanism of inflammasome activation. Different danger‐associated molecular patterns (DAMPs) and pathogen‐associated molecular patterns (PAMPs) activate distinct sensors, including NLRP3, NLRP1b, NAIPs and AIM2, leading to inflammasome activation and subsequent IL‐1β and GSDMD cleavage by active caspase‐1. The N‐terminal fragment of GSDMD leads to membrane pore formation, which facilitates IL‐1β release and results in pyroptosis under certain conditions. NEK7 and NAIPs are required for activation of the NLRP3 and NLRC4 inflammasomes, respectively. ASC binds with NLRP3 or AIM2 via PYD‐PYD interactions and NLRC4 or NLRP1b via CARD‐CARD interactions.
IL‐1β and IL‐18 secretion potentiate inflammation through promoting both innate and adaptive immune responses,10 such as enhancing neutrophil recruitment and T helper (Th) 17 or Th1 cell differentiation.11, 12, 13 Recently, IL‐18 secretion following inflammasome activation, which is induced by fungi stimulation via the CARD9‐Syk axis, has been shown to protect mice from colon cancer, suggesting its strong capacity for epithelial barrier reparation.13 The GSDMD‐N fragments generated following the cleavage of GSDMD form pores on the cell membrane to facilitate cytokine secretion and induce cell lysis.14 Interestingly, emerging studies have shown that the formation of GSDMD‐dependent pores with detectable cytokine secretion levels does not always result in proptosis.15, 16 Recently, it was reported that active mixed lineage kinase domain‐like pseudokinase (MLKL) signalling, which leads to necroptosis, can promote NLRP3 inflammasome activation with low detected concentrations of intracellular potassium (K+),17 and pyroptosis can be triggered by caspase‐8‐induced cleavage of GSDMD,18, 19 suggesting that the link between inflammasome activation and pyroptosis remains to be clarified.
The well‐studied inflammasomes involving NLRP1, NLRP3, NLRC4 and AIM2 are mainly classified by their capacity to recognize different stimuli (Fig. 1). NLRP1b is cleaved by the protease component of a lethal toxin released by Bacillus anthracis.20 NLRC4 inflammasome activation can be triggered by interactions between rod proteins or needle proteins of the type III secretion system with NAIP2 or NAIP1 independently or with Salmonella flagellin via NAIP5.9, 21 AIM2 directly binds to cytoplasmic double‐stranded DNA and activates ASC by interacting with both pyrin domains (PYD).22, 23 Notably, NLRP3 can be stimulated by numerous PAMPs and DAMPs, such as extracellular ATP, nigericin, uric acid crystals, glucose and cholesterol.24, 25 Because the structures of these ligands are not the same, it is postulated that danger signals have a common downstream to activate NLRP3, and well‐established mechanisms such as reactive oxygen species (ROS) accumulation,26 lysosomal rupture,27 K+ efflux28 and calcium (Ca2+) influx29 are not exclusive to a NLRP3‐activated process. Recently, it has been discovered that disassembly of the trans‐Golgi network (TGN) into dispersed vesicles promotes NLRP3 inflammasome assembly during treatment with various stimuli in which K+ efflux is or is not required, implying that dispersed TGN (dTGN) may be the common upstream signal upon NLRP3 inflammasome activation.30 Notably, NIMA‐related kinase 7 (NEK7), a serine/threonine kinase playing vital roles in mitosis, is required for the NLRP3 inflammasome assembly by binding NLRP3, which occurs after K+ efflux,31 and a recent study has reported a significant increase of NLRP3 and NEK7 in both mRNA and protein levels in patients with systemic lupus erythematosus after drug treatment.32 Furthermore, the polymorphisms in gene levels of NEK7 and toll‐like receptors (TLRs) are associated with change of cholesterol levels,33 suggesting NEK7 is potentially involved in immunometabolism. Collectively, inflammasome activation is crucial for host defence against microbes and implicated in metabolic disorders; thus, tight regulation is required.
The regulation of inflammasome activation at the transcriptional and post‐translational levels has been widely demonstrated by previous studies. Elevated expression levels of inflammasome components are regarded as positive preparation to initiate inflammasome activation. For example, the induction of NLRP3 and pro‐IL‐1β proteins via TLR4‐MyD88‐NF‐κB pathway is crucial for NLRP3 inflammasome activation,34 while type I interferon (IFN), which is also involved in the TLR4‐stimulated downstream pathway, suppresses pro‐IL‐1β production and caspase‐1 activity, resulting in the inhibition of NLRP3 inflammasome activation.35 Interestingly, it has been reported that type I IFN promoted AIM2 inflammasome activation.36 Many protein modification mechanisms, such as phosphorylation and ubiquitylation, are involved in post‐translational control, including the mediation of sensor activation and inflammasome assembly. Phosphorylated ASC caused by the Syk and JNK signals is required for speck formation, which is essential for NLRP3 and AIM2 inflammasome assembly.37 Induction of NLRP3 deubiquitylation is another mechanism involved in TLR4‐dependent NLRP3 abundance.38 Recent studies have revealed that metabolism is implicated in inflammasome activation by providing DAMPs to activate sensors or acting as a mediator regulating both translational and non‐translational levels.
Metabolic reprogramming controls inflammasome activation
Glucose metabolism and inflammasome activation
Glucose metabolism consists of several metabolic pathways, including glycolysis, the tricarboxylic acid (TCA) cycle and the pentose phosphate pathway (PPP), which is an essential source of ATP and reducing equivalent. In addition, glucose metabolism has a complex relationship with fatty acid and amino acid metabolism because they share various biosynthetic intermediates. Because many studies reflect the importance of glucose metabolism reprogramming for immune cells to defend against danger signalling, inflammasome activation may also be a potent target of aberrant glucose metabolism (Fig. 2).
Figure 2.
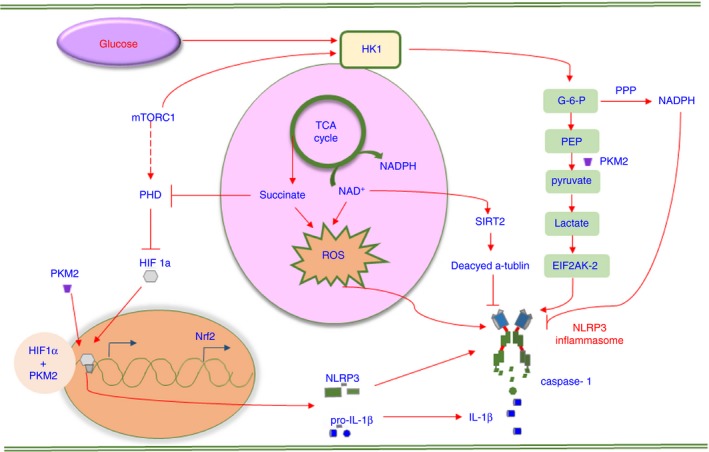
The signalling pathways involved in glucose metabolism‐regulated NLRP3 inflammasome activation. Glucose metabolism mainly involves glycolysis, the pentose phosphate pathway (PPP) and the tricarboxylic acid (TCA) cycle. The metabolites participating in glycolysis are HK1 and PKM2. HK1 promotes NLRP3 inflammasome activation by enhancing glycolysis levels, requiring mTORC1 activation. Active PKM2 activates NLRP3 via lactate‐induced EIF2AK2 phosphorylation, and dimeric PKM2 combined with HIF1‐α promotes pro‐IL‐1β transcription. NADPH mostly derived from the PPP downregulates the expression of the NLRP3 inflammasome components, and a decreased amount of NAD + facilitates NLRP3 and ASC assembly by suppressing α‐tubulin deacetylase SIRT2 activity. The metabolites involved in the TCA cycle are citrate and succinate, both of which active the NLRP3 inflammasome. Succinate promotes HIF1‐α stabilization by reducing prolyl hydroxylase (PHD) release and induces reactive oxygen species (ROS) generation. Active Nrf2 translocates into the nucleus and enhances the expression of proteins to suppress ROS.
Glycolysis and inflammasome activation
The preference for glycolysis under aerobic conditions is termed the Warburg effect, which is often regarded as the hallmark of tumour cells.39 Nonetheless, numerous studies have shown that stimuli‐activated immune cells rapidly generate ATP, mainly owing to increased cytosolic glycolysis instead of oxidative phosphorylation.40, 41
Hexokinase (HK), the primary enzyme involved in glycolysis, is a key factor regulating the level of glycolysis. It was demonstrated that the mammalian target of rapamycin complex 1 (mTORC1), as a supervisor and regulator of energy metabolism42 and immune cell activation,43, 44 participated in HK1‐mediated glycolysis in macrophages. The inhibition of mTORC1 decreased the expression of HK1 protein, thus suppressing the NLRP3 inflammasome response, suggesting that a mTORC1/HK1‐dependent glycolysis axis is involved in inducing NLRP3 inflammasome activation but not AIM2 inflammasome activation. Consistent with this, glucose deprivation and 2‐deoxyglucose (2‐DG) treatment can suppress caspase‐1 activation and IL‐1β secretion independently through a similar effect on the inhibition of glycolysis.45 However, in another study, 2‐DG was shown to promote IL‐1β maturation in a dose‐dependent manner, and excess glucose‐6‐phosphate (G6P), which inhibits HK activation via a negative feedback loop, can also induce NLRP3 inflammasome activation. In addition, HK combines with N‐acetylglucosamine (NAG), which is one of the fundamental units of peptidoglycan (PGN), subsequently inducing HK dissociation from the mitochondrial membrane towards the cytosol and sufficiently activating the NLRP3 inflammasome. The underlying mechanism is that direct binding of NAG to the active site of HK causes enzymatic inhibition and a change in intracellular localization, both of which lead to NLRP3 inflammasome activation independent of K+ efflux and proptosis.46 The paradox between these different studies suggests a complex relationship between glycolysis and the NLRP3 inflammasome.
Pyruvate kinase M2 (PKM2), which contributes to converting phosphoenolpyruvate to pyruvate and ATP generation, is associated with rate limitation in glycolysis. Lipopolysaccharides (LPS)‐primed macrophages show a potentiated abundance of PKM2. The PKM2 enzymatically inactive dimer directly binds to hypoxia‐inducible factor 1α (HIF1‐α), promoting the stabilization of HIF1‐α, which facilitates the transcription of genes including pro‐IL‐1β. In contrast, the activation of PKM2 leads to the formation of a tetramer that cannot enter the nucleus, which eventually results in HIF1‐α destabilization and biosynthesis limitation.47 A later study reported that PKM2‐dependent glycolysis leads to lactate production, which strongly induces phosphorylation of the eukaryotic translation initiation factor 2 alpha kinase 2 (EIF2AK2), and activated EIF2AK2 can promote the activation of the NLRP3 or AIM2 inflammasome in LPS‐activated bone marrow‐derived macrophages (BMDMs) treated with ATP or poly (dA : dT), respectively, which means this is specific to the regulation of NLRP3 and AIM2 inflammasome activation.48
Because glycolysis is an essential energy‐generating pathway with a complex association with other forms of metabolism, a subtle change can cause a perturbation in glycolytic flux. In addition to HK and PKM2 mentioned above, the inhibition of glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) and α‐enolase by the small molecule GB111‐NH2 has also been found to lead to NLRP3 inflammasome activation and pyroptosis, in which an increased ratio of NAD+/NADH is responsible for NLRP3 inflammasome activation.49
The pentose phosphate pathway and inflammasome activation
The PPP, which utilizes G6P to produce NADPH and ribose, is important to maintain cellular redox balance and convert intermediates generated from glycolysis into the precursors of amino acids or nucleotides. NADPH performs a complex role in redox maintenance under different conditions and can act as a co‐enzyme to produce glutathione (GSH), which is an antioxidant to diminish ROS50 or, in contrast, can be utilized by NADPH oxidases (NOXs) to promote superoxide and ROS generation.51 A recent study revealed that treating microglial cells with NADPH and the NOX inhibitor apocynin alone or together downregulated the expression levels of NLRP3, ASC, caspase‐1, IL‐18 and IL‐1β significantly in a murine stroke model.52 Additionally, PKM2 has been found to be extensive in activated immune cells and tumour cells.47, 53 In cancer cells, PKM2 catalysis features a slower rate of glycolysis with more substrates diverted into pathways that can generate NADPH compared with catalysis of another isoform, PKM1.42 Although previous studies have demonstrated the importance of NADPH in oxidative maintenance, the role of the PPP in regulating inflammasome activation remains poorly understood.
The tricarboxylic acid cycle and inflammasome activation
The TCA cycle, an important pathway occurring in mitochondria, connects with oxidative phosphorylation by providing NADH and FADH2, and connects with glycolysis and fatty acid metabolism by consuming acetyl coenzyme A (acetyl‐CoA). It was reported that microbial TCA cycle mutants can have an effect on NLRP3 inflammasome activation, which suggests that bacterial metabolites also act as stimuli of inflammasomes. In detail, Salmonella typhimurium (Stm), which dominantly triggers NLRC4 activation by flagellin, can also stimulate the NLRP3 inflammasome owing to excessive citrate metabolism through promoting the formation of mitochondrial ROS, whereas aconitase has an opposite effect.54
Succinate, an intermediate accumulated in the TCA cycle in LPS‐stimulated microphages, has been shown to lead to HIF1‐α stabilization through inhibiting prolyl hydroxylase (PHD) activity, resulting in increased IL‐1β generation.55 Consistently, succinate accumulation under hypoxic conditions in synovial tissue led to NLRP3 inflammasome activation with the stabilization of HIF1‐α.56 Further studies may reveal the relationship between succinate and inflammasome activation in immune cells. Metabolite derivatives of substrates in the TCA cycle, such as dimethyl fumarate (DMF), are capable of inducing NF‐E2‐related factor 2 (Nrf2)‐associated gene expression, which further leads to mitochondrial ROS reduction, explaining the reduced activation of the NLRP3 inflammasome in a colitis mouse model.5 Its redox analogue, ethyl pyruvate (EP), inhibits mitochondrial DNA translocation towards the cytoplasm, resulting in the suppression of NLRP3 inflammasome activation.57, 58
NLRP3 inflammasome activation is always linked to damaged mitochondrial integrity.59, 60 One mechanism of mitochondrial dysfunction that triggers inflammasome activation is related to a low concentration of NAD+, which acts as a co‐enzyme of sirtuin 2 (SIRT2). A decrease in the amount of cellular NAD+ suppresses the deacetylation of α‐tubulin by downregulating SIRT2 activation, which in turn results in a higher abundance of activated acetylated α‐tubulin, facilitating the assembly of NLRP3 and ASC.61 Additionally, a decreased NAD+/NADH ratio has been found in LPS‐primed BMDMs, which limited the desuccinylase and demalonylase activity of SIRT5.55 Succinylation is also an important mechanism to regulate enzyme activity beyond acetylation;62 this suggests the potential involvement of SIRT5 in mediating inflammasome activation.
Fatty acid metabolism and inflammasome activation
Fatty acid synthesis (FAS) and fatty acid oxidation (FAO) seem to have distinct functions in regulating the inflammatory status of immune cells. In contrast to FAS, which is always linked to pro‐inflammatory cells, FAO leads to lipid derivative accumulation and ATP supplementation, which contributes to cell proliferation and growth. Thus, fatty acid metabolism plays an important role in the polarization and activation of innate immune cells (Fig. 3).
Figure 3.
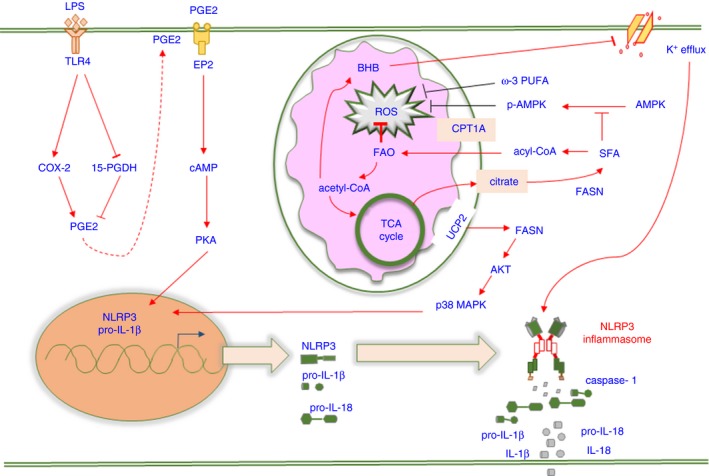
The signalling pathways involved in fatty acid metabolism‐regulated NLRP3 inflammasome activation. Fatty acid metabolism involves fatty acid synthesis (FAS), fatty acid oxidation (FAO) and cholesterol metabolism. Saturated fatty acids (SFAs) and ω‐3 polyunsaturated fatty acids (PUFAs) exert opposite effects on NLRP3 inflammasome activation via the regulation of reactive oxygen species (ROS) production, and SFA enhances ROS generation by downregulating AMPK activity. Fatty acid synthase (FASN) induces NLRP3 and pro‐IL‐1β expression via the AKT/p38 MAPK axis. β‐Hydroxybutyrate (BHB) decreases NLRP3 inflammasome activation by inhibiting K+ efflux. An increased abundance of PGE 2 leads to pro‐IL‐1β expression via the EP2‐cAMP‐PKA axis.
Fatty acid synthesis and inflammasome activation
Fatty acid synthesis has been revealed to positively regulate innate and adaptive immune responses.63, 64, 65 De novo FAS is tightly regulated by fatty acid synthase (FASN), which facilitates fatty acid elongation. The increased expression of FASN has been observed in cancer cells accompanied by aberrant glycolytic metabolism.66, 67 Consistent with this, the uncoupling protein‐2 (UCP2)‐dependent induction of FASN is also remarkably found in LPS‐stimulated BMDMs, which promotes NLRP3 and pro‐IL‐1β transcription by activating the AKT/p38 MAPK signalling axis.68 As the increased induction of FASN contributes to the generation of long‐chain saturated fatty acids (SFAs),66 SFAs also act as critical mediators for inflammasome activation.
Saturated fatty acids, such as palmitate (PA), are regarded as stimuli that promote inflammatory responses in high abundance.69, 70 The dual treatment of macrophages with PA and LPS led to NLRP3 inflammasome activation and IL‐1β and IL‐18 secretion. The precise mechanism by which PA activates inflammasomes involves ROS generation and K+ efflux, in which the enhancement of ROS relies on suppressed adenosine monophosphate‐activated protein kinase (AMPK) activity, while P2X7 receptor (P2X7R), which is essential for inflammasome activation after ATP treatment, is dispensable for inducing K+ efflux in this context.71 In contrast, the activation of AMPK facilitates ATP‐induced NLRP3 inflammasome activation and pyroptosis. Because AMPK acts as a negative mediator of lipid and protein synthesis, which are required for cell survival,72, 73 this suggests that mediating the activity of AMPK signalling may be crucial for inflammasome‐induced pyroptosis.
In contrast to SFAs, polyunsaturated fatty acids (PUFAs) play complex roles in mediating inflammation. Docosahexaenoic acid (DHA, ω‐3 UFAs) is a potent inhibitor of nigericin‐induced NLRP3 inflammasome activation and anthrax lethal toxin‐induced NLRP1b inflammasome activation. The deficiency of G‐protein‐coupled receptor 120 (GPR120) and GPR40 predominantly suppresses the influence of DHA in NLRP3 inflammasome activation, and β‐arrestin 2, a protein downstream of GPR120,74 can directly interact with NLRP3 or NLRP1b but not NLRC4 or AIM2, which may explain the exclusive inhibition of NLRP3 and NLRP1b inflammasome activation.75 A recent study reported that ω‐3 UFAs in dietary PUFAs alleviated NLRP3 inflammasome activation.76 In addition, derivatives of PUFAs generated via the lipoxygenase (LOX) and cyclooxygenase (COX) pathways may also partially influence inflammatory responses.77, 78, 79 It has been shown that 15‐LOX metabolites of α‐linolenic acid (ALA, ω‐3 UFAs) inhibited NLRP3 inflammasome activation in a peroxisome proliferator‐activated receptor‐γ (PPAR‐γ)‐dependent manner, which in turn induced apoptosis through suppressing autophagy.80 However, the metabolites of ω‐6 UFAs, such as arachidonic acid (AA), have been confirmed to promote inflammatory processes.79
Fatty acid oxidation and inflammasome activation
Because FAO has been shown to promote M2 macrophage polarization,81 FAO is always linked to anti‐inflammatory responses. In line with this hypothesis, the enhanced expression of carnitine palmitoyltransferase 1A (CPT1A), which transports fatty acids into mitochondria, remarkably promotes FAO in PA‐induced macrophages, resulting in decreased triglyceride levels and reduced ROS levels.82 Interestingly, a later study revealed that FAO potentiates NLRP3 inflammasome activation in a NOX4‐dependent pathway with no effect on NLRC4, NLRP1 and AIM2 inflammasome activation. The enhanced expression of NOX4 along with mitochondrial ROS contributes to CPT1A protein expression in LPS‐primed macrophages triggered by ATP or nigericin, leading to increased FAO, which ultimately promotes NLRP3‐induced ASC oligomerization.83 This further suggests the potential that FAS is critical for initiating NLRP3 inflammasome activation and that FAO acts as a positive regulator during downstream cascade amplification.68, 83
The oxidation of fatty acids leads to the generation of NADH, FADH2 and acetyl‐CoA, which are further utilized to efficiently produce ATP. Increased concentrations of acetyl‐CoA are critical for regulating the TCA cycle and ketone generation under certain conditions.83, 84 β‐Hydroxybutyrate (BHB), a main member of ketone bodies, is a major alternative metabolic fuel to provide ATP during prolonged starvation, and a strong capacity for anti‐inflammation has been found in neurological diseases and peripheral tissue inflammation.85, 86, 87 Accordingly, BHB has been shown to suppress K+ efflux, thus dampening NLRP3 inflammasome activation in macrophages.88 A later study also has revealed BHB blocks the priming and assembly steps of the NLRP3 inflammasome in neutrophils.89
Other inflammatory lipid mediators and inflammasome activation
Cholesterol metabolism exerts sufficient influence on cellular homeostasis, including the regulation of inflammasome activation.90, 91, 92 25‐Hydroxycholesterol (25‐HC), a soluble oxysterol derived from cholesterol by cholesterol 25‐hydroxylase (CH25H),93 has been shown to be present at increased levels because of the enhanced expression of CH25H under the phenotype of X‐linked adrenoleukodystrophy, which contributes to microglial recruitment and NLRP3 inflammasome assembly.94 Furthermore, the activation of TLR3 and TLR4 signalling promotes CH25H expression via the type I IFN‐IFNR‐STAT1 axis in dendritic cells (DCs) and macrophages, respectively.95 Interestingly, SCAP‐SREBP2 translocation, which shows a positive function in both NLRP3 inflammasome activity and cholesterol biosynthesis, can be inhibited by 25‐HC or cholesterol overexposure, suggesting 25‐HC and cholesterol may also act as inhibitors of NLRP3 inflammasome activation in LPS‐treated BMDMs96 in contrast to numerous studies showing the strong ability of cholesterol to promote inflammasome activation.91, 97 Collectively, the different observed effects of these two substances may be associated with different stages of the inflammatory response and different types of inflammasomes as the upregulation of 25‐HC is known to inhibit AIM2 inflammasome activation through suppressing cholesterol biosynthesis, but its effect on NLRP3 inflammasome activation is ambiguous so far.97 Bile acid, an important inflammatory mediator derived from oxysterols, negatively regulates nigericin‐induced NLRP3 inflammasome activation through the transmembrane G‐protein‐coupled receptor‐5 (TGR5)‐cAMP‐PKA axis in non‐differentiated BMDMs.98 Intriguingly, a recent study reported that cholestasis aggravates sepsis, and bile acids not only potentiate NLRP3 and IL‐1β expression but also stimulate NLRP3 inflammasome activation by driving a prolonged Ca2+ influx.3 It is noteworthy that this positive influence of bile acids is detected in a wide spectrum of macrophages but not in monocytes.
Eicosanoids, including prostaglandins (PGs) and leukotrienes, are derived from AA via the COX and 5‐LOX pathways, respectively.99 PGE2, an abundant eicosanoid present in LPS‐induced BMDMs owing to COX‐2 activation and 15‐hydroxydehydrogenase inhibition, is required for IL‐1β transcription through interaction with the EP2 receptor, which leads to increased cAMP signalling.100 It is noteworthy that in macrophages, exogenous PGE2 added after LPS negatively regulates NLRP3 inflammasome activation,101 whereas PGE2 added before LPS promotes this process.100 Additionally, a rapid increase in eicosanoids, termed an ‘eicosanoid storm’, is detected during NAIP5/NLRC4 inflammasome activation,102 which facilitates the recruitment of neutrophils to clear pathogens after pore‐induced intracellular traps are formed following pyroptosis.103
Activated inflammasomes control cell metabolism
The mechanisms we mentioned above are mainly about the cross‐talk between aberrant metabolism and sensor activation or inflammasome assembly, and all emphasize that metabolic reprogramming is essential for mediating inflammatory responses. Furthermore, inflammasome activation leads to the cleavage of caspase‐1, which subsequently converts IL‐1β and IL‐18 into their mature forms, which can also have a dominant influence on cellular metabolism in turn.
Caspase‐1, which directly cleaves at least 41 substrates, has been shown to target several glycolytic enzymes, including GAPDH and α‐enolase, during Salmonella‐induced NLRC4 inflammasome activation, which subsequently leads to pyroptosis. Because the cleavage of GAPDH by caspase‐1 is less efficient than that of pro‐IL‐1β in vitro, it implies stricter conditions for processing GAPDH in which abundant active caspase‐1 is required. The degradation of glycolytic enzymes leads to a lower rate of glycolysis, which is in contrast with the common perception that activated macrophages exert a pro‐inflammatory function with increased glycolysis;104, 105 thus, the reduction in glycolysis may explain the shift towards pyroptosis.106 NLRP3 inflammasome activation is correlated with GAPDH proteolysis in mouse skeletal muscle during ageing.107 Because fatty acid metabolism plays diverse roles in mediating inflammasome activation, tight constraints are essential for maintaining the balance between different lipid mediators. For example, an eicosanoid storm that facilitates neutrophil recruitment is induced via caspase‐1‐dependent Ca2+ influx that subsequently activates Ca2+‐dependent cytosolic phospholipase A2 (cPLA2) during NAIP5/NLRC4 inflammasome activation.102
Recently, a positive feedback loop between IL‐1β and insulin during postprandial stimulation involving glucose and microbial products has been shown. Food intake leads to IL‐1β secretion by peritoneal macrophages, and the binding of IL‐1β and IL‐1 receptor (IL‐1R) expressed on the cell surface of β‐cells triggers insulin secretion, causing enhanced secretion of IL‐1β in macrophages via NLRP3 inflammasome activation. Collectively, this loop is important for regulating the glucose uptake rate and immune response.108
Metabolic regulation of inflammasome activation in inflammatory diseases
Inflammasome activation and metabolism regulation are required for many inflammatory diseases (Table 1).
Table 1.
The role of metabolic regulation on inflammasome activation in diseases
| Disease classify | Phenotype | Effect factor | Functions | Type of inflammasome | Disease |
|---|---|---|---|---|---|
| Infectious diseases | Chronic infection | Aconitase | Converts citrate to isocitrate | NLRP3 inflammasome | Mice infected by acnB‐deficient Stm have higher level of extracellular IL‐1851 |
| Sepsis | Bile acids | Promote calcium influx | NLRP3 inflammasome | LPS‐stimulated mice pretreated with bile acid sequestrant are detected decreased serum level of IL‐1β and reduced mortality3 | |
| Sepsis | UCP2 | UCP2 promotes NLRP3 expression via FASN‐Akt‐p38 MAPK axis | NLRP3 inflammasome | UCP2 deficiency mice has decreased serum levels of pro‐inflammatory cytokines and lower mortality during sepsis65 | |
| Sterile inflammation | Gout | BHB | Inhibition of NLRP3 inflammasome priming and assembly | NLRP3 inflammasome | KD induces remission of knee swelling and severe inflammation at joints86 |
| Ischaemic stroke | BHB | Induction of suppressed capacity of mitochondrial fission and deceased ROS production | NLRP3 inflammasome | Feeding KD is essential for reducing infarct area and neuron injury in mice subjected to ischaemic stroke122 | |
| FCAS | BHB | Blockade of inflammasome assembly | NLRP3 inflammasome | BHB contributes to decreased IL‐1β secretion in a dose‐dependent manner86 | |
| CIA | Dimethyl malonate | SDH inhibitor but reduce succinate accumulation in this case | NLRP3 inflammasome | Dimethyl malonate treatment is associated with decreased level of TGF‐β1‐induced IL‐1β production53 | |
| Cancer | Cervical cancer | SIRT1 | Protein deacetylation to promote metabolic reprogramming and AIM2 inflammasome dysfunction | AIM2 inflammasome | si‐SIRT1 injection reduced tumour growth and tumour weight in cervical cancer mice model4 |
| Breast cancer | Obesity | Obesity‐induced NLRC4 inflammasome activation in macrophages | NLRC4 inflammasome | IL‐1β/IL‐1R signalling promotes VEGFA gene expression, facilitating angiogenesis in obese mice with breast cancer164 | |
| Colorectal cancer | Cholesterol | Cholesterol induces NLRP3 inflammasome activation via cathepsin B‐AMPK‐ROS axis | NLRP3 inflammasome | Mice treated with high cholesterol and AOM have higher tumour load. In contrast, NLRP3 deficiency can ameliorate the effect27 |
BHB, β‐hydroxybutyrate; KD, ketogenic diet; LPS, lipopolysaccharide; ROS, reactive oxygen species; SDH, succinate dehydrogenase.
Infectious diseases
Because inflammation is vital for the host defence against microbial infection, the mechanism by which inflammatory caspases and inflammasomes protect the host from severe infectious diseases has been widely discussed before.109 In addition to the efficient recognition of microbial components by pattern recognition receptors (PRRs), metabolic disruption by pathogens can also regulate inflammasome activation. Stm, which evades NLRC4 inflammasome surveillance by downregulating flagellin expression, can lead to delayed NLRP3 inflammasome activation.110 Mice infected by AcnB‐deficient Stm have a lower abundance of bacteria in their spleens and higher levels of IL‐18 secretions compared with mice infected with aconitase‐sufficient Stm, and the underlying mechanism for this is that the absence of AcnB‐encoded aconitase can promote NLRP3 inflammasome activation by enhancing mitochondrial ROS.54
Sepsis is a common result of microbial infection when it is resistant to host defence, which is mostly caused by facultative anaerobes, leading to septic shock and even death.111 Bacterial toxin‐like LPS produced by gram‐negative bacteria is significant in the pathogenesis of sepsis.112, 113 Bile acid accumulation is regarded as a common consequence after the onset of sepsis due to the altered expression of multidrug resistance‐associated protein (MRP)3, which suggests the poor prognosis for sepsis.114 Recently, bile acids have been shown to play an active role in NLRP3 inflammasome activity by promoting the transcription of IL‐1β and calcium mobilization in macrophages. For LPS‐induced sepsis, exogenous bile acid addition results in a notable shorter survival time for LPS‐treated mice, while pretreatment with cholestyramine resin, a sequestrant of bile acid, significantly decreased the serum level of IL‐1β and reduced mortality.3 An earlier study reported that EP treatment blocked NF‐κB signalling in macrophages and decreased high‐mobility group box 1 (HMGB1) in circulation in septic mice.115 EP suppresses NLRP3 inflammasome activation in LPS‐induced macrophages following treatment with various stimuli by sustaining the integrity of the mitochondria but not preventing mitochondrial ROS, leading to the reduced secretion of HMGB1.57 Further study of the direct link between EP‐induced sepsis remission and NLRP3 inflammasome activation is required.
Sterile inflammation
Sterile inflammation includes a wide spectrum of inflammatory disorders in which pathogenesis is independent of pathogen invasion, such as gout, atherosclerosis and neuroinflammation. Because multiple endogenous signals can trigger NLRP3 inflammasome activation, the NLRP3 inflammasome has been implicated in the development of various inflammatory diseases.91, 116 For example, hypoxia involved in the etiopathogenesis of atherosclerosis is known to be a potent inducer of HIF1‐α, which has been confirmed to promote the expression of IL‐1β.55, 117 Cholesterol‐treated hypoxic human macrophages secrete excessive active IL‐1β. Meanwhile, LPS‐induced hypoxic macrophages have increased NLRP3 expression,118 suggesting that the NLRP3 inflammasome has the potential to participate in chronic inflammation, but further investigation is required.
Gout is a chronic form of inflammatory arthritis with the hallmark of monosodium urate (MSU) crystal deposition in the joints,119 and clinical manifestations are predominantly aggravated during ageing.120 Mechanistically, MSU triggers NLRP3 inflammasome activation due to MSU‐induced intracellular damage, such as mitochondrial dysfunction,24 which results in constant IL‐1β secretion121 and ultimately leads to intense pain. Early epidemiological studies have reported that various dietary factors, such as alcohol and fructose, are relevant to the progression of gout,122 and therapeutic strategies for metabolic mediation, such as caloric restriction, have been confirmed to effectively reduce serum urate levels in gout.123 Ketogenic diets (KDs), which elevate endogenous BHB levels, sufficiently induce the remission of knee swelling and severe inflammation of joints in an MSU‐injected rat model, and aged mice suffering MSU‐induced peritonitis display downregulation of Nlrp3 and Il1b gene expression when treated with KD, suggesting that metabolic regulation is crucial for alleviating inflammation via altering innate immune responses.89
Ischaemic stroke, which features aberrant blood flow and decreased abundance of serum glucose, can lead to brain ischaemia‐reperfusion injury accompanied by chronic inflammation.124, 125 It has been shown that NLRP3 deficiency is sufficient to ameliorate neuronal injury after ischaemia with restored blood–brain barrier damage.126, 127 Furthermore, mediating NLRP3 inflammasome activation by feeding a KD is essential for reducing infarct area and neuronal injury in mice subjected to ischaemic stroke. Mechanistically, oxygen‐glucose deprivation/reoxygenation (OGD/R)‐treated SH‐SY 5Y human neuroblastoma cells exhibit suppressed capacity for mitochondrial fission and deceased ROS production with the addition of BHB.125 The importance of inflammasome activation in neuropathology involving neuroinflammation and neurodegenerative disease has been extensively discussed recently.128, 129, 130, 131 Chronic alcohol intake has been regarded as a vital risk factor for the liver and brain damage.132, 133 Alcohol‐treated mice have increased microglial activation, enhanced expression of NLRP3 and elevated levels of IL‐1β in the cerebellum compared with those of control mice, and Nlrp3 or ASC gene deficiency is beneficial to protect against alcohol‐induced brain injury. It is known that TLR4 signalling only partially regulates cytokine production, and increased active HMGB1 is assumed to play a crucial role in triggering NLRP3 inflammasome activation.134 For microglial cells, alcohol augments the activation of P2X7R, which is an ATP‐gated cation channel often involved in ATP‐triggered NLRP3 inflammasome activation.135, 136 Neuroinflammation in mice fed a mix of ethanol and a high‐fat diet (HFD) is inhibited by treatment with P2X7R antagonist.137 Further studies about diet and its correlation with sterile inflammation may focus on its mechanism of mediating the activation of the inflammasome to formulate specific diet regulations.
Autoinflammatory and autoimmune disorders
Chronic or excessive self‐directed immune system activation can lead to autoinflammation or autoimmune disorders, among which the largest difference is whether adaptive immune responses, such as high concentrations of autoantibodies, play essential roles in pathogenesis.138 Inflammasome activation and IL‐1β secretion have been detected in various autoinflammatory diseases,139, 140, 141 and multiple reagents targeting IL‐1 have a predominantly positive function in the remission of disease symptoms.142, 143 The potential of inflammasome activation to participate in the pathogenesis of autoimmune diseases mainly focuses on the production of inflammatory cytokines in myeloid cells, which further influence T‐ and B‐cell amplification and activation.144, 145, 146 For example, IL‐1β is crucial for the development of experimental autoimmune encephalomyelitis (EAE) by promoting Th17 cell expansion and granulocyte‐macrophage colony‐stimulating factor (GM‐CSF) production.147, 148, 149 Accordingly, NLRP3 deficiency also leads to the delayed progression of EAE.150
Cryopyrin‐associated periodic syndromes (CAPS), a monogenic autoinflammatory disease, feature various mutations of the Nlrp3 gene that lead to overactivation of the NLRP3 inflammasome and subsequent IL‐1β secretion.151 In a mouse model of familial cold autoinflammatory syndrome (FCAS), a type of CAPS, mice displayed decreased IL‐1β secretion when treated with BHB in a dose‐dependent manner, suggesting that BHB is capable of suppressing NLRP3 inflammasome activation,89 which supports a previous finding that BHB blocked the NF‐κB pathway by attenuating the degradation of IκB‐α.152
Rheumatoid arthritis (RA) is a progressive autoimmune disorder characterized by joint pain, fibrosis in synovial tissue and skeletal destruction accompanied by the induction of numerous inflammatory cytokines that contribute to disease symptom aggravation. Succinate, as an intermediate of the TCA cycle, is known to induce IL‐1β production via HIF‐1α activation in macrophages.55 Consistent with this, dimethyl malonate, an inhibitor of succinate dehydrogenase (SDH), suppresses succinate accumulation, suggesting that SDH promotes the reversal of succinate production in synovial tissue under hypoxic conditions, leading to a reduction in IL‐1β production in a mouse collagen‐induced arthritis (CIA) model, and it is known that succinate accumulation enhances HIF‐1α production to activate the NLRP3 inflammasome in fibroblasts.56 In a later study, it was revealed that the succinate‐HIF‐1α axis is also involved in synovial angiogenesis by contributing to the expression of vascular endothelial growth factor (VEGF), which is regarded as a crucial molecule of angiogenesis.153, 154 Although blood vessel formation is capable of changing the anoxic environment, it is also linked with increased immune cell migration that maintains or exacerbates the symptoms of RA.155 Therefore, succinate‐induced HIF‐1α activation plays a potent role in the progression of RA by participating in inflammatory responses and neoangiogenesis, which further provides novel insight into metabolic disruption during RA pathogenesis.
Type 1 diabetes (T1D) is a prevalent autoimmune disease featuring the dysfunction of pancreatic β cells that secrete insulin essential for glucose deprivation.156 In a streptozotocin (STZ)‐induced T1D mouse model, NLRP3 inflammasome activation has been detected during the onset of T1D, and IL‐1 secretion promotes antigen‐specific T‐cell amplification in pancreatic lymph nodes.157 Interestingly, STZ‐treated IL‐1R‐associated kinase‐M (IRAK‐M)‐deficient mice developed T1D much sooner than wild‐type STZ‐treated mice, and the mRNA levels of NLRP3 and NLRP1 are decreased in the mononuclear blood cells (PBMCs) from patients with T1D,158 suggesting that IL‐1 signalling is dispensable for the early onset of T1D. This highlights the importance of NLRP3 inflammasome activation in autoimmune diseases with metabolic disorders, and further studies are needed to figure out the role of NLRP3 inflammasome activation in the onset and persistence of T1D.
Cancer
The effect of inflammasome activation in different cancers has been determined in previous studies, including NLRC4 inflammasome activation in colorectal cancer (CRC),159 NLRP1 inflammasome activation in metastatic melanoma,160 and NLRP3 inflammasome activation in metastatic CRC161 as well as in breast cancer.162 It is intriguing that inflammasome activation can either play a positive or negative role in the tumourigenesis of different cancers.
Cervical cancer is a high‐incidence malignancy in women with high mortality, with most clinical cases caused by human papillomavirus (HPV) infection.163 SIRT1, an NAD+‐dependent histone or non‐histone deacetylase, is important for regulating cellular metabolism and immune defence via protein deacetylation along with other SIRT family members.164 It has been reported that in patient groups classified by relatively high or low expression of SIRT1 during the progression of cervical cancer, higher mortality is seen in the high‐SIRT1 expression group. In this context, SIRT1 leads to the destabilization of RelB mRNA, which can suppress AIM2 expression, and further contributes to the inhibition of AIM2 inflammasome‐mediated pyroptosis, promoting cancer cell survival in the SiHa human cervical cancer cell line. Notably, SIRT1 knockdown increased glycolysis levels and decreased the levels of ATP in SiHa cells, suggesting that metabolic reprogramming may also be involved in SIRT1‐induced AIM2 inflammasome dysfunction.4
Breast cancer is the most common cancer among women worldwide, and its incidence is remarkably correlated with obesity.165 Data have shown that obesity‐induced chronic inflammation and insulin resistance are responsible for the increased risk of breast cancer development.166 Consistent with this, enhanced NLRC4 and IL‐1β expression are specifically detected in obese mice that suffer from breast cancer cell transplantation into the mammary fat pad, and deficiency of NLRC4 or both caspase‐1 and caspase‐11 significantly reduced tumour growth. It is noteworthy that IL‐1β secretion from tumour‐infiltrating myeloid cells indirectly promotes tumour development by enhancing VEGFA mRNA levels in adipocytes, which further leads to increased vascularity.167 This study provides a novel direction to study the influence of inflammasome activation in cancer beyond the direct effects of cell death and the IL‐1/IL‐1R signalling‐induced enhanced immune response.
Colorectal cancer is linked to multiple gene mutations and environmental risk factors.168 In particular, a HFD is strongly associated with severe intestinal inflammation and tumourigenesis.169, 170 A HFD‐induced increase of faecal deoxycholic acid (DCA), a kind of second bile acid, shows the capacity to exacerbate DSS‐induced murine colitis by inducing NLRP3 inflammasome activation in macrophages, requiring cathepsin B release into the cytoplasm and bile acid recognition by sphingosine‐1‐phosphate receptor 2 (S1PR2) but not TGR5.171 For CRC, DCA treatment enhanced the expression of NLRP3 and pro‐inflammatory cytokines accompanied by structural and functional damage of the intestinal mucosal barrier in ApcMin/+ mice, which induced spontaneous intestinal tumourigenesis,172 and this further highlights that excessive fat intake impacts intestinal metabolic homeostasis, leading to dysfunction of the intestinal immune system and elevated incidence of CRC. Consistent with this, high cholesterol‐induced NLRP3 inflammasome activation directly facilitates tumourigenesis in an AOM‐treated CRC mouse model. Mechanistically, cholesterol triggers NLRP3 inflammasome activation in LPS‐treated THP‐1 cell‐derived macrophages (THP‐Ms), requiring cathepsin B release and AMPK inhibition that induces the activation of mitochondrial ROS, and secreted IL‐1β is responsible for CRC cell proliferation in vitro.27
Conclusions and future perspectives
In this review, we summarized many previous studies about the molecular mechanisms of inflammasome activation, cross‐talk in metabolic reprogramming, including aberrant switches in glucose and lipid metabolism, as well as the abundance of their metabolic derivatives with the inflammasome response and the metabolic regulation of inflammasome activation in disease onset or development. Because the NLRP3 inflammasome can be activated by diverse stimuli, including many metabolites, the metabolic regulation of its activity is well studied in macrophages. Nevertheless, other inflammasomes also have the potential to exert vital influence because the identified stimuli recognized by inflammasome sensors are currently limited, and microbiota changes may also be involved in promoting metabolic disorders. For example, NLRC4 inflammasome activation is known to be essential in diabetic nephropathy and obesity‐associated breast cancer.167, 173 Regarding disease regulation, substantial evidence has shown that the initiation or dysregulation of inflammasome activation is responsible for the magnitude of disease severity, and while most studies have focused on the molecular mechanism of triggering or inhibiting inflammasome activation, few have uncovered the interaction between metabolic regulation and inflammasome activation in a wide spectrum of diseases. For example, DMF has been shown to reduce IL‐1β expression efficiently in LPS‐treated microglia, supporting its approval as a clinical drug for multiple sclerosis,58, 174 and its capacity for reducing mRNA levels of NLRP3 in THP‐Ms, which also leads to a decrease in IL‐1β, was reported,5 suggesting that DMF likely suppresses pro‐inflammatory cytokine production by blocking inflammasome activation, which further drives us to study cross‐talk between metabolism and inflammasome activation and may provide insight into the aetiology and pathology of those diseases. Collectively, the activity of the inflammasome and metabolic balance are both known to be important in inflammatory disorders; thus, in‐depth investigations about the metabolic regulation of inflammasome activation in molecular mechanisms and disease development are urgently required for developing better therapeutic methods targeting inflammasome‐involved inflammation.
Disclosures
The authors declare no competing financial interests.
Acknowledgements
The authors’ research is supported by grants from the National Natural Science Foundation for Key Programs of China (31730024, G.L.) and the National Natural Science Foundation for General Programs of China (31671524, G.L.).
Contributor Information
Yujing Bi, Email: byj7801@sina.com.
Guangwei Liu, Email: liugw@bnu.edu.cn.
References
- 1. O'Neill LA, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol 2016; 16:553–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med 2015; 21:677–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hao H, Cao L, Jiang C, Che Y, Zhang S, Takahashi S et al Farnesoid X receptor regulation of the NLRP3 inflammasome underlies cholestasis‐associated sepsis. Cell Metab 2017; 25:856–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. So D, Shin HW. Cervical cancer is addicted to SIRT1 disarming the AIM2 antiviral defense. Oncogene 2018; 37:5191–204. [DOI] [PubMed] [Google Scholar]
- 5. Liu X, Zhou W, Zhang X, Lu P, Du Q, Tao L et al Dimethyl fumarate ameliorates dextran sulfate sodium‐induced murine experimental colitis by activating Nrf2 and suppressing NLRP3 inflammasome activation. Biochem Pharmacol 2016; 112:37–49. [DOI] [PubMed] [Google Scholar]
- 6. Broz P, von Moltke J, Jones JW, Vance RE, Monack DM. Differential requirement for Caspase‐1 autoproteolysis in pathogen‐induced cell death and cytokine processing. Cell Host Microbe 2010; 8:471–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Huang MT, Taxman DJ, Holley‐Guthrie EA, Moore CB, Willingham SB, Madden V et al Critical role of apoptotic speck protein containing a caspase recruitment domain (ASC) and NLRP3 in causing necrosis and ASC speck formation induced by Porphyromonas gingivalis in human cells. J Immunol 2009; 182:2395–404. [DOI] [PubMed] [Google Scholar]
- 8. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H et al Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015; 526:660–5. [DOI] [PubMed] [Google Scholar]
- 9. Zhao Y, Shao F. The NAIP‐NLRC4 inflammasome in innate immune detection of bacterial flagellin and type III secretion apparatus. Immunol Rev 2015; 265:85–102. [DOI] [PubMed] [Google Scholar]
- 10. Keyel PA. How is inflammation initiated? Individual influences of IL‐1, IL‐18 and HMGB1. Cytokine 2014; 69:136–45. [DOI] [PubMed] [Google Scholar]
- 11. Mills KH, Dungan LS, Jones SA, Harris J. The role of inflammasome‐derived IL‐1 in driving IL‐17 responses. J Leuko Biol 2013; 93:489–97. [DOI] [PubMed] [Google Scholar]
- 12. Sedimbi SK, Hagglof T, Karlsson MC. IL‐18 in inflammatory and autoimmune disease. Cell Mol Life Sci 2013; 70:4795–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Malik A, Sharma D, Malireddi RKS, Guy CS, Chang TC, Olsen SR et al SYK‐CARD9 signaling axis promotes gut fungi‐mediated inflammasome activation to restrict colitis and colon cancer. Immunity 2018; 49:515–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. He WT, Wan H, Hu L, Chen P, Wang X, Huang Z et al Gasdermin D is an executor of pyroptosis and required for interleukin‐1beta secretion. Cell Res 2015; 25:1285–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Evavold CL, Ruan J, Tan Y, Xia S, Wu H, Kagan JC. The pore‐forming protein gasdermin D regulates interleukin‐1 secretion from living macrophages. Immunity 2018; 48:35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Heilig R, Dick MS, Sborgi L, Meunier E, Hiller S, Broz P. The Gasdermin‐D pore acts as a conduit for IL‐1beta secretion in mice. Eur J Immunol 2018; 48:584–92. [DOI] [PubMed] [Google Scholar]
- 17. Conos SA, Chen KW. Active MLKL triggers the NLRP3 inflammasome in a cell‐intrinsic manner. Proc Natl Acad Sci USA 2017; 114:E961–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Orning P, Weng D, Starheim K, Ratner D. Pathogen blockade of TAK1 triggers caspase‐8‐dependent cleavage of gasdermin D and cell death. Science 2018; 362:1064–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sarhan J, Liu BC, Muendlein HI, Li P, Nilson R, Tang AY et al Caspase‐8 induces cleavage of gasdermin D to elicit pyroptosis during Yersinia infection. Proc Natl Acad Sci USA 2018; 115:E10 888–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hellmich KA, Levinsohn JL, Fattah R, Newman ZL, Maier N, Sastalla I et al Anthrax lethal factor cleaves mouse nlrp1b in both toxin‐sensitive and toxin‐resistant macrophages. PLoS ONE 2012; 7:e49741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kofoed EM, Vance RE. Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature 2011; 477:592–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jin T, Perry A, Smith P, Jiang J, Xiao TS. Structure of the absent in melanoma 2 (AIM2) pyrin domain provides insights into the mechanisms of AIM2 autoinhibition and inflammasome assembly. J Biol Chem 2013; 288:13 225–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hornung V, Ablasser A, Charrel‐Dennis M, Bauernfeind F, Horvath G, Caffrey DR et al AIM2 recognizes cytosolic dsDNA and forms a caspase‐1‐activating inflammasome with ASC. Nature 2009; 458:514–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nomura J, So A, Tamura M, Busso N. Intracellular ATP decrease mediates NLRP3 inflammasome activation upon nigericin and crystal stimulation. J Immunol 2015; 195:5718–24. [DOI] [PubMed] [Google Scholar]
- 25. Camell C, Goldberg E, Dixit VD. Regulation of Nlrp3 inflammasome by dietary metabolites. Semi Immunol 2015; 27:334–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Heid ME, Keyel PA, Kamga C, Shiva S, Watkins SC, Salter RD. Mitochondrial reactive oxygen species induces NLRP3‐dependent lysosomal damage and inflammasome activation. J Immunol 2013; 191:5230–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Du Q, Wang Q, Fan H, Wang J, Li X, Wang H et al Dietary cholesterol promotes AOM‐induced colorectal cancer through activating the NLRP3 inflammasome. Biochem Pharmacol 2016; 105:42–54. [DOI] [PubMed] [Google Scholar]
- 28. Petrilli V, Papin S, Dostert C, Mayor A, Martinon F, Tschopp J. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ 2007; 14:1583–9. [DOI] [PubMed] [Google Scholar]
- 29. Lee GS, Subramanian N, Kim AI, Aksentijevich I, Goldbach‐Mansky R, Sacks DB et al The calcium‐sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature 2012; 492:123–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chen J, Chen ZJ. PtdIns4P on dispersed trans‐Golgi network mediates NLRP3 inflammasome activation. Nature 2018; 564:71–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. He Y, Zeng MY, Yang D, Motro B, Nunez G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 2016; 530:354–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ma ZZ, Sun HS, Lv JC, Guo L, Yang QR. Expression and clinical significance of the NEK7‐NLRP3 inflammasome signaling pathway in patients with systemic lupus erythematosus. J Inflamm 2018; 15:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gomes Torres A, Leite N, Tuerk LV, de Souza RLR, Titski ACK, Milano‐Gai GE et al Association between Toll‐like receptors (TLR) and NOD‐like receptor (NLR) polymorphisms and lipid and glucose metabolism. Gene 2019; 685:211–21. [DOI] [PubMed] [Google Scholar]
- 34. Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D et al Cutting edge: NF‐kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol 2009; 183:787–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Guarda G, Braun M, Staehli F, Tardivel A, Mattmann C, Förster I et al Type I interferon inhibits interleukin‐1 production and inflammasome activation. Immunity 2011; 34:213–23. [DOI] [PubMed] [Google Scholar]
- 36. Man SM, Karki R, Malireddi RK, Neale G, Vogel P, Yamamoto M et al The transcription factor IRF1 and guanylate‐binding proteins target activation of the AIM2 inflammasome by Francisella infection. Nat Immunol 2015; 16:467–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hara H, Tsuchiya K, Kawamura I, Fang R, Hernandez‐Cuellar E, Shen Y et al Phosphorylation of the adaptor ASC acts as a molecular switch that controls the formation of speck‐like aggregates and inflammasome activity. Nat Immunol 2013; 14:1247–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Juliana C, Fernandes‐Alnemri T, Kang S. Non‐transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J Biol Chem 2012; 287:36 617–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Warburg O. On the origin of cancer cells. Science 1956; 123:309–14. [DOI] [PubMed] [Google Scholar]
- 40. Kelly B, O'Neill LA. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res 2015; 25:771–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rodriguez‐Prados JC, Traves PG, Cuenca J, Rico D, Aragonés J, Martín‐Sanz P et al Substrate fate in activated macrophages: a comparison between innate, classic, and alternative activation. J Immunol 2010; 185:605–14. [DOI] [PubMed] [Google Scholar]
- 42. Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer 2011; 11:85–95. [DOI] [PubMed] [Google Scholar]
- 43. Marcais A, Cherfils‐Vicini J, Viant C, Degouve S, Viel S, Fenis A et al The metabolic checkpoint kinase mTOR is essential for IL‐15 signaling during the development and activation of NK cells. Nat Immunol 2014; 15:749–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Liu Y, Zhang DT, Liu XG. mTOR signaling in T cell immunity and autoimmunity. Int Rev Immunol 2015; 34:50–66. [DOI] [PubMed] [Google Scholar]
- 45. Moon JS, Hisata S, Park MA, DeNicola GM, Ryter SW, Nakahira K et al mTORC1‐Induced HK1‐dependent glycolysis regulates NLRP3 inflammasome activation. Cell Rep 2015; 12:102–15. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 46. Wolf AJ, Reyes CN, Liang W, Becker C, Shimada K, Wheeler ML et al Hexokinase is an innate immune receptor for the detection of bacterial peptidoglycan. Cell 2016; 166:624–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Palsson‐McDermott EM, Curtis AM, Goel G, Lauterbach MAR, Sheedy FJ, Gleeson LE et al Pyruvate kinase M2 regulates Hif‐1alpha activity and IL‐1beta induction and is a critical determinant of the Warburg effect in LPS‐activated macrophages. Cell Metabol 2015; 21:65–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Xie M, Yu Y, Kang R, Zhu S, Yang L, Zeng L et al PKM2‐dependent glycolysis promotes NLRP3 and AIM2 inflammasome activation. Nat Commun 2016; 7:13 280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sanman LE, Qian Y, Eisele NA, Ng TM, van der Linden WA, Monack DM et al Disruption of glycolytic flux is a signal for inflammasome signaling and pyroptotic cell death. Elife 2016; 5:e13663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hughes MM, McGettrick AF, O'Neill LAJ. Glutathione and glutathione transferase omega 1 as key posttranslational regulators in macrophages. Microbiol Spectrum 2017; 5:1. [DOI] [PubMed] [Google Scholar]
- 51. Chen H, Song YS, Chan PH. Inhibition of NADPH oxidase is neuroprotective after ischemia‐reperfusion. J Cereb Blood Flow Metab 2009; 29:1262–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Qin YY, Li M, Feng X, Wang J, Cao L, Shen XK et al Combined NADPH and the NOX inhibitor apocynin provides greater anti‐inflammatory and neuroprotective effects in a mouse model of stroke. Free Radic Biol Med 2017; 104:333–45. [DOI] [PubMed] [Google Scholar]
- 53. Dayton TL, Gocheva V, Miller KM, Bhutkar A, Lewis CA, Bronson RT et al Isoform‐specific deletion of PKM2 constrains tumor initiation in a mouse model of soft tissue sarcoma. Cancer Metabol 2018; 6:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wynosky‐Dolfi MA, Snyder AG, Philip NH, Doonan PJ, Poffenberger MC, Avizonis D et al Oxidative metabolism enables Salmonella evasion of the NLRP3 inflammasome. J Exp Med 2014; 211:653–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Tannahill GM, Curtis AM, Adamik J, Palsson‐McDermott EM, McGettrick AF, Goel G et al Succinate is an inflammatory signal that induces IL‐1beta through HIF‐1alpha. Nature 2013; 496:238–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Li Y, Zheng JY, Liu JQ, Yang J, Liu Y, Wang C et al Succinate/NLRP3 inflammasome induces synovial fibroblast activation: therapeutical effects of clematichinenoside AR on arthritis. Front Immunol 2016; 7:532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Li S, Liang F, Kwan K, Tang Y, Wang X, Tang Y et al Identification of ethyl pyruvate as a NLRP3 inflammasome inhibitor that preserves mitochondrial integrity. Mol Med 2018; 24:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Miljkovic D, Blazevski J, Petkovic F, Djedović N, Momčilović M, Stanisavljević S et al A comparative analysis of multiple sclerosis‐relevant anti‐inflammatory properties of ethyl pyruvate and dimethyl fumarate. J Immunol 2015; 194:2493–503. [DOI] [PubMed] [Google Scholar]
- 59. Tschopp J, Schroder K. NLRP3 inflammasome activation: the convergence of multiple signalling pathways on ROS production? Nat Rev Immunol 2010; 10:210–5. [DOI] [PubMed] [Google Scholar]
- 60. Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC et al Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 2011; 12:222–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Misawa T, Takahama M, Kozaki T, Lee H, Zou J, Saitoh T et al Microtubule‐driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat Immunol 2013; 14:454–60. [DOI] [PubMed] [Google Scholar]
- 62. Hirschey MD, Zhao Y. Metabolic regulation by lysine malonylation, succinylation, and glutarylation. Mol Cell Proteom 2015; 14:2308–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Everts B, Amiel E, Huang SC, Smith AM, Chang CH. TLR‐driven early glycolytic reprogramming via the kinases TBK1‐IKKvarepsilon supports the anabolic demands of dendritic cell activation. Nat Immunol 2014; 15:323–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Dufort FJ, Gumina MR, Ta NL, Tao Y, Heyse SA, Scott DA et al Glucose‐dependent de novo lipogenesis in B lymphocytes: a requirement for atp‐citrate lyase in lipopolysaccharide‐induced differentiation. J Biol Chem 2014; 289:7011–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL et al The NLRP3 inflammasome instigates obesity‐induced inflammation and insulin resistance. Nat Med 2011; 17:179–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer 2007; 7:763–77. [DOI] [PubMed] [Google Scholar]
- 67. Wang M, Han J, Xing H, Zhang H, Li Z, Liang L et al Dysregulated fatty acid metabolism in hepatocellular carcinoma. Hepatic Oncol 2016; 3:241–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Moon JS, Lee S, Park MA, Siempos II, Haslip M, Lee LE et al UCP2‐induced fatty acid synthase promotes NLRP3 inflammasome activation during sepsis. J Clin Invest 2015; 125:665–80. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 69. Boden G. Interaction between free fatty acids and glucose metabolism. Curr Opin Clin Nutr Metab Care 2002; 5:545–9. [DOI] [PubMed] [Google Scholar]
- 70. Snodgrass RG, Huang S, Choi IW, Rutledge JC, Hwang DH. Inflammasome‐mediated secretion of IL‐1beta in human monocytes through TLR2 activation; modulation by dietary fatty acids. J Immunol 2013; 191:4337–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Wen H, Gris D, Lei Y, Jha S, Zhang L, Huang MT et al Fatty acid‐induced NLRP3‐ASC inflammasome activation interferes with insulin signaling. Nat Immunol 2011; 12:408–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hardie DG. The AMP‐activated protein kinase pathway–new players upstream and downstream. J Cell Sci 2004; 117:5479–87. [DOI] [PubMed] [Google Scholar]
- 73. Hardie DG, Pan DA. Regulation of fatty acid synthesis and oxidation by the AMP‐activated protein kinase. Biochem Soc Trans 2002; 30:1064–70. [DOI] [PubMed] [Google Scholar]
- 74. Miller WE, Lefkowitz RJ. Expanding roles for beta‐arrestins as scaffolds and adapters in GPCR signaling and trafficking. Curr Opin Cell Biol 2001; 13:139–45. [DOI] [PubMed] [Google Scholar]
- 75. Yan Y, Jiang W, Spinetti T, Tardivel A, Castillo R, Bourquin C et al Omega‐3 fatty acids prevent inflammation and metabolic disorder through inhibition of NLRP3 inflammasome activation. Immunity 2013; 38:1154–63. [DOI] [PubMed] [Google Scholar]
- 76. Shen L, Yang Y, Ou T, Key CC, Tong SH, Sequeira RC et al Dietary PUFAs attenuate NLRP3 inflammasome activation via enhancing macrophage autophagy. J Lipid Res 2017; 58:1808–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Shibata T, Kondo M, Osawa T, Shibata N, Kobayashi M, Uchida K. 15‐Deoxy‐delta 12,14‐prostaglandin J2. A prostaglandin D2 metabolite generated during inflammatory processes. J Biol Chem 2002; 277:10 459–66. [DOI] [PubMed] [Google Scholar]
- 78. Arthur S, Singh S. Cyclooxygenase pathway mediates the inhibition of Na‐glutamine co‐transporter B0AT1 in rabbit villus cells during chronic intestinal inflammation. PLoS ONE 2018; 13:e0203552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Korotkova M, Lundberg IE. The skeletal muscle arachidonic acid cascade in health and inflammatory disease. Nat Rev Rheumatol 2014; 10:295–303. [DOI] [PubMed] [Google Scholar]
- 80. Kumar N, Gupta G, Anilkumar K, Fatima N, Karnati R, Reddy GV et al 15‐Lipoxygenase metabolites of alpha‐linolenic acid, [13‐(S)‐HPOTrE and 13‐(S)‐HOTrE], mediate anti‐inflammatory effects by inactivating NLRP3 inflammasome. Sci Rep 2016; 6:31 649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Huang SC, Everts B, Ivanova Y, O'Sullivan D, Nascimento M, Smith AM et al Cell‐intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat Immunol 2014; 15:846–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Malandrino MI, Fucho R, Weber M, Calderon‐Dominguez M, Mir JF, Valcarcel L et al Enhanced fatty acid oxidation in adipocytes and macrophages reduces lipid‐induced triglyceride accumulation and inflammation. Am J Physiol Endocrinol Metab 2015; 308:E756–69. [DOI] [PubMed] [Google Scholar]
- 83. Moon JS, Nakahira K, Chung KP, DeNicola GM, Koo MJ, Pabón MA et al NOX4‐dependent fatty acid oxidation promotes NLRP3 inflammasome activation in macrophages. Nat Med 2016; 22:1002–12. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 84. Cahill GF Jr. Fuel metabolism in starvation. Annu Rev Nutr 2006; 26:1–22. [DOI] [PubMed] [Google Scholar]
- 85. Hazem SH, Hamed MF, Saad MA, Gameil NM. Comparison of lactate and beta‐hydroxybutyrate in the treatment of concanavalin‐A induced hepatitis. Int Immunopharmacol 2018; 61:376–84. [DOI] [PubMed] [Google Scholar]
- 86. Chen Y, Ouyang X, Hoque R, Garcia‐Martinez I, Yousaf MN, Tonack S et al Beta‐hydroxybutyrate protects from alcohol‐induced liver injury via a Hcar2‐cAMP dependent pathway. J Hepatol 2018; 69:687–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Rahman M, Muhammad S, Khan MA, Chen H, Ridder DA, Müller‐Fielitz H et al The beta‐hydroxybutyrate receptor HCA2 activates a neuroprotective subset of macrophages. Nat Commun 2014; 5:3944. [DOI] [PubMed] [Google Scholar]
- 88. Youm YH, Nguyen KY, Grant RW, Goldberg EL, Bodogai M, Kim D et al The ketone metabolite beta‐hydroxybutyrate blocks NLRP3 inflammasome‐mediated inflammatory disease. Nat Med 2015; 21:263–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Goldberg EL, Asher JL, Molony RD, Shaw AC, Zeiss CJ, Wang C et al Beta‐hydroxybutyrate deactivates neutrophil NLRP3 inflammasome to relieve gout flares. Cell Rep 2017; 18:2077–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Min HK, Kapoor A, Fuchs M, Mirshahi F, Zhou H, Maher J et al Increased hepatic synthesis and dysregulation of cholesterol metabolism is associated with the severity of nonalcoholic fatty liver disease. Cell Metab 2012; 15:665–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG et al NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010; 464:1357–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Lehmann JM, Kliewer SA, Moore LB, Smith‐Oliver TA, Oliver BB, Su JL et al Activation of the nuclear receptor LXR by oxysterols defines a new hormone response pathway. J Biol Chem 1997; 272:3137–40. [DOI] [PubMed] [Google Scholar]
- 93. Russell DW. Oxysterol biosynthetic enzymes. Biochim Biophys Acta 2000; 1529:126–35. [DOI] [PubMed] [Google Scholar]
- 94. Jang J, Park S, Jin Hur H, Cho HJ, Hwang I, Pyo Kang Y et al 25‐Hydroxycholesterol contributes to cerebral inflammation of X‐linked adrenoleukodystrophy through activation of the NLRP3 inflammasome. Nat Commun 2016; 7:13 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Park K, Scott AL. Cholesterol 25‐hydroxylase production by dendritic cells and macrophages is regulated by type I interferons. J Leuk Biol 2010; 88:1081–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Guo C, Chi Z, Jiang D, Xu T, Yu W, Wang Z et al Cholesterol homeostatic regulator SCAP‐SREBP2 integrates NLRP3 inflammasome activation and cholesterol biosynthetic signaling in macrophages. Immunity 2018; 49:842–56. [DOI] [PubMed] [Google Scholar]
- 97. Dang EV, McDonald JG, Russell DW, Cyster JG. Oxysterol restraint of cholesterol synthesis prevents AIM2 inflammasome activation. Cell 2017; 171:1057–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Guo C, Xie S, Chi Z, Zhang J, Liu Y, Zhang L et al Bile acids control inflammation and metabolic disorder through inhibition of NLRP3 inflammasome. Immunity 2016; 45:802–16. [DOI] [PubMed] [Google Scholar]
- 99. Funk CD. Prostaglandins and leukotrienes: advances in eicosanoid biology. Science 2001; 294:1871–5. [DOI] [PubMed] [Google Scholar]
- 100. Zaslona Z, Palsson‐McDermott EM, Menon D, Haneklaus M. The induction of Pro‐IL‐1beta by lipopolysaccharide requires endogenous prostaglandin E2 production. J Immunol 2017; 198:3558–64. [DOI] [PubMed] [Google Scholar]
- 101. Sokolowska M, Chen LY, Liu Y, Martinez‐Anton A, Qi HY, Logun C et al Prostaglandin E2 inhibits NLRP3 inflammasome activation through EP4 receptor and intracellular cyclic AMP in human macrophages. J Immunol 2015; 194:5472–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. von Moltke J, Trinidad NJ, Moayeri M, Kintzer AF, Wang SB, van Rooijen N et al Rapid induction of inflammatory lipid mediators by the inflammasome in vivo . Nature 2012; 490:107–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Jorgensen I, Lopez JP, Laufer SA, Miao EA. IL‐1beta, IL‐18, and eicosanoids promote neutrophil recruitment to pore‐induced intracellular traps following pyroptosis. Euro J Immunol 2016; 46:2761–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Cramer T, Yamanishi Y, Clausen BE, Förster I, Pawlinski R, Mackman N et al HIF‐1alpha is essential for myeloid cell‐mediated inflammation. Cell 2003; 112:645–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. El Kasmi KC, Stenmark KR. Contribution of metabolic reprogramming to macrophage plasticity and function. Semin Immunol 2015; 27:267–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Shao W, Yeretssian G, Doiron K, Hussain SN, Saleh M. The caspase‐1 digestome identifies the glycolysis pathway as a target during infection and septic shock. J Biol Chem 2007; 282:36 321–9. [DOI] [PubMed] [Google Scholar]
- 107. McBride MJ, Foley KP, D'Souza DM, Li YE, Lau TC, Hawke TJ et al The NLRP3 inflammasome contributes to sarcopenia and lower muscle glycolytic potential in old mice. Am J Physiol Endocrinol Metab 2017; 313:E222–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Dror E, Dalmas E, Meier DT, Wueest S, Thévenet J, Thienel C et al Postprandial macrophage‐derived IL‐1beta stimulates insulin, and both synergistically promote glucose disposal and inflammation. Nat Immunol 2017; 18:283–92. [DOI] [PubMed] [Google Scholar]
- 109. Man SM, Karki R, Kanneganti TD. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol Rev 2017; 277:61–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Broz P, Newton K, Lamkanfi M, Mariathasan S, Dixit VM, Monack DM. Redundant roles for inflammasome receptors NLRP3 and NLRC4 in host defense against Salmonella . J Exp Med 2010; 207:1745–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Minasyan H. Sepsis and septic shock: pathogenesis and treatment perspectives. J Critic Care 2017; 40:229–42. [DOI] [PubMed] [Google Scholar]
- 112. Balk RA. Severe sepsis and septic shock. Definitions, epidemiology, and clinical manifestations. Critic Care Clin 2000; 16:179–92. [DOI] [PubMed] [Google Scholar]
- 113. Horn KD. Evolving strategies in the treatment of sepsis and systemic inflammatory response syndrome (SIRS). QJM 1998; 91:265–77. [DOI] [PubMed] [Google Scholar]
- 114. Vanwijngaerden YM, Wauters J, Langouche L, Vander Perre S, Liddle C, Coulter S et al Critical illness evokes elevated circulating bile acids related to altered hepatic transporter and nuclear receptor expression. Hepatology 2011; 54:1741–52. [DOI] [PubMed] [Google Scholar]
- 115. Ulloa L, Ochani M, Yang H, Tanovic M, Halperin D, Yang R, et al Ethyl pyruvate prevents lethality in mice with established lethal sepsis and systemic inflammation. Proc Natl Acad Sci USA 2002; 99:12 351–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Bauer C, Duewell P, Mayer C, Lehr HA, Fitzgerald KA, Dauer M et al Colitis induced in mice with dextran sulfate sodium (DSS) is mediated by the NLRP3 inflammasome. Gut 2010; 59:1192–9. [DOI] [PubMed] [Google Scholar]
- 117. Frede S, Stockmann C, Freitag P, Fandrey J. Bacterial lipopolysaccharide induces HIF‐1 activation in human monocytes via p44/42 MAPK and NF‐kappaB. Biochem J 2006; 396:517–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Folco EJ, Sukhova GK, Quillard T, Libby P. Moderate hypoxia potentiates interleukin‐1beta production in activated human macrophages. Circ Res 2014; 115:875–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Pillinger MH, Goldfarb DS, Keenan RT. Gout and its comorbidities. Bull NYU Hosp Jt Dis 2010; 68:199–203. [PubMed] [Google Scholar]
- 120. Lee JH, Yang JA. Elderly patients exhibit stronger inflammatory responses during gout attacks. J Korean Med Sci 2017; 32:1967–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout‐associated uric acid crystals activate the NALP3 inflammasome. Nature 2006; 440:237–41. [DOI] [PubMed] [Google Scholar]
- 122. Roddy E, Choi HK. Epidemiology of gout. Rheum Dis Clin North Am 2014; 40:155–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Dessein PH, Shipton EA, Stanwix AE, Joffe BI, Ramokgadi J. Beneficial effects of weight loss associated with moderate calorie/carbohydrate restriction, and increased proportional intake of protein and unsaturated fat on serum urate and lipoprotein levels in gout: a pilot study. Ann Rheum Dis 2000; 59:539–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Shichita T, Sakaguchi R, Suzuki M, Yoshimura A. Post‐ischemic inflammation in the brain. Front Iimmunol 2012; 3:132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Guo M, Wang X, Zhao Y, Yang Q, Ding H, Dong Q et al Ketogenic diet improves brain ischemic tolerance and inhibits NLRP3 inflammasome activation by preventing Drp1‐mediated mitochondrial fission and endoplasmic reticulum stress. Front Mol Neurosci 2018; 11:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Yang F, Wang Z, Wei X, Han H, Meng X, Zhang Y et al NLRP3 deficiency ameliorates neurovascular damage in experimental ischemic stroke. J Cereb Blood Flow Metab 2014; 34:660–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Gao L, Dong Q, Song Z, Shen F, Shi J, Li Y. NLRP3 inflammasome: a promising target in ischemic stroke. Inflamm Res 2017; 66:17–24. [DOI] [PubMed] [Google Scholar]
- 128. Nakanishi A, Kaneko N, Takeda H, Sawasaki T, Morikawa S, Zhou W et al Amyloid beta directly interacts with NLRP3 to initiate inflammasome activation: identification of an intrinsic NLRP3 ligand in a cell‐free system. Inflamm Regen 2018; 38:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Zhou Y, Lu M, Du RH, Qiao C, Jiang CY, Zhang KZ et al MicroRNA‐7 targets Nod‐like receptor protein 3 inflammasome to modulate neuroinflammation in the pathogenesis of Parkinson's disease. Mol Neurodegen 2016; 11:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Zhang CJ, Jiang M, Zhou H, Liu W, Wang C, Kang Z et al TLR‐stimulated IRAKM activates caspase‐8 inflammasome in microglia and promotes neuroinflammation. J Clin Invest 2018; 128:5399–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Freeman LC, Ting JP. The pathogenic role of the inflammasome in neurodegenerative diseases. J Neurochem 2016; 136(Suppl 1):29–38. [DOI] [PubMed] [Google Scholar]
- 132. Szabo G, Mandrekar P, Petrasek J, Catalano D. The unfolding web of innate immune dysregulation in alcoholic liver injury. Alcohol Clin Exp Res 2011; 35:782–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Qin L, He J, Hanes RN, Pluzarev O, Hong JS, Crews FT. Increased systemic and brain cytokine production and neuroinflammation by endotoxin following ethanol treatment. J Neuroinflamm 2008; 5:10–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Lippai D, Bala S, Petrasek J, Csak T, Levin I, Kurt‐Jones EA et al Alcohol‐induced IL‐1beta in the brain is mediated by NLRP3/ASC inflammasome activation that amplifies neuroinflammation. J Leuk Biol 2013; 94:171–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Asatryan L, Ostrovskaya O, Lieu D, Davies DL. Ethanol differentially modulates P2X4 and P2X7 receptor activity and function in BV2 microglial cells. Neuropharmacology 2018; 128:11–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Di Virgilio F. Liaisons dangereuses: P2X(7) and the inflammasome. Trends Pharmacol Sci 2007; 28:465–72. [DOI] [PubMed] [Google Scholar]
- 137. Freire D, Reyes RE, Baghram A, Davies DL, Asatryan L. P2X7 receptor antagonist A804598 inhibits inflammation in brain and liver in C57BL/6J mice exposed to chronic ethanol and high fat diet. J Neuroimmune Pharmacol 2018; 12:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Shaw PJ, McDermott MF, Kanneganti TD. Inflammasomes and autoimmunity. Trends Mol Med 2011; 17:57–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Chae JJ, Cho YH, Lee GS, Cheng J, Liu PP, Feigenbaum L et al Gain‐of‐function Pyrin mutations induce NLRP3 protein‐independent interleukin‐1beta activation and severe autoinflammation in mice. Immunity 2011; 34:755–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Repa A, Bertsias GK, Petraki E, Choulak C, Vassou D, Kambas K et al Dysregulated production of interleukin‐1beta upon activation of the NLRP3 inflammasome in patients with familial Mediterranean fever. Hum Immunol 2015; 76:488–95. [DOI] [PubMed] [Google Scholar]
- 141. Manthiram K, Zhou Q, Aksentijevich I, Kastner DL. The monogenic autoinflammatory diseases define new pathways in human innate immunity and inflammation. Nat Immunol 2017; 18:832–42. [DOI] [PubMed] [Google Scholar]
- 142. Lachmann HJ, Kone‐Paut I, Kuemmerle‐Deschner JB, Leslie KS, Hachulla E, Quartier P et al Use of canakinumab in the cryopyrin‐associated periodic syndrome. N Engl J Med 2009; 360:2416–25. [DOI] [PubMed] [Google Scholar]
- 143. Canna SW, de Jesus AA, Gouni S, Brooks SR, Marrero B, Liu Y et al An activating NLRC4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome. Nat Genet 2014; 46:1140–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Agostini L, Martinon F, Burns K, McDermott MF, Hawkins PN, Tschopp J. NALP3 forms an IL‐1beta‐processing inflammasome with increased activity in Muckle‐Wells autoinflammatory disorder. Immunity 2004; 20:319–25. [DOI] [PubMed] [Google Scholar]
- 145. Barclay W, Shinohara ML. Inflammasome activation in multiple sclerosis and experimental autoimmune encephalomyelitis (EAE). Brain Pathol 2017; 27:213–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Chung Y, Chang SH, Martinez GJ, Yang XO, Nurieva R, Kang HS et al Critical regulation of early Th17 cell differentiation by interleukin‐1 signaling. Immunity 2009; 30:576–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Mufazalov IA, Schelmbauer C, Regen T, Kuschmann J, Wanke F, Gabriel LA et al IL‐1 signaling is critical for expansion but not generation of autoreactive GM‐CSF+ Th17 cells. EMBO J 2017; 36:102–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Matsuki T, Nakae S, Sudo K, Horai R, Iwakura Y. Abnormal T cell activation caused by the imbalance of the IL‐1/IL‐1R antagonist system is responsible for the development of experimental autoimmune encephalomyelitis. Int Immunol 2006; 18:399–407. [DOI] [PubMed] [Google Scholar]
- 149. El‐Behi M, Ciric B, Dai H, Yan Y, Cullimore M, Safavi F et al The encephalitogenicity of T(H)17 cells is dependent on IL‐1‐ and IL‐23‐induced production of the cytokine GM‐CSF. Nat Immunol 2011; 12:568–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Gris D, Ye Z, Iocca HA, Wen H, Craven RR, Gris P et al NLRP3 plays a critical role in the development of experimental autoimmune encephalomyelitis by mediating Th1 and Th17 responses. J Immunol 2010; 185:974–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Aksentijevich I, Putnam CD, Remmers EF, Mueller JL, Le J, Kolodner RD et al The clinical continuum of cryopyrinopathies: novel CIAS1 mutations in North American patients and a new cryopyrin model. Arthritis Rheum 2007; 56:1273–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Fu SP, Li SN, Wang JF, Li S, Xie SS, Xue WJ et al BHBA suppresses LPS‐induced inflammation in BV‐2 cells by inhibiting NF‐kappaB activation. Mediators Inflamm 2014; 2014:983 401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med 2003; 9:669–76. [DOI] [PubMed] [Google Scholar]
- 154. Li Y, Liu Y, Wang C, Xia WR, Zheng YJ, Yang J et al Succinate induces synovial angiogenesis in rheumatoid arthritis through metabolic remodeling and HIF‐1alpha/VEGF axis. Free Radic Biol Med 2018; 126:1–14. [DOI] [PubMed] [Google Scholar]
- 155. Karouzakis E, Neidhart M, Gay RE, Gay S. Molecular and cellular basis of rheumatoid joint destruction. Immunol Lett 2006; 106:8–13. [DOI] [PubMed] [Google Scholar]
- 156. Yoshida K, Kikutani H. Genetic and immunological basis of autoimmune diabetes in the NOD mouse. Rev Immunogenet 2000; 2:140–6. [PubMed] [Google Scholar]
- 157. Carlos D, Costa FR, Pereira CA, Rocha FA, Yaochite JN, Oliveira GG et al Mitochondrial DNA activates the NLRP3 inflammasome and predisposes to type 1 diabetes in murine model. Front Immunol 2017; 8:164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Liu H, Xu R, Kong Q, Liu J, Yu Z, Zhao C. Downregulated NLRP3 and NLRP1 inflammasomes signaling pathways in the development and progression of type 1 diabetes mellitus. Biomed Pharmacother 2017; 94:619–26. [DOI] [PubMed] [Google Scholar]
- 159. Hu B, Elinav E, Huber S, Booth CJ, Strowig T, Jin C et al Inflammation‐induced tumorigenesis in the colon is regulated by caspase‐1 and NLRC4. Proc Natl Acad Sci USA 2010; 107:21 635–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Zhai Z, Liu W, Kaur M, Luo Y, Domenico J, Samson JM et al NLRP1 promotes tumor growth by enhancing inflammasome activation and suppressing apoptosis in metastatic melanoma. Oncogene 2017; 36:3820–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Dupaul‐Chicoine J, Arabzadeh A, Dagenais M, Douglas T, Champagne C, Morizot A et al The Nlrp3 inflammasome suppresses colorectal cancer metastatic growth in the liver by promoting natural killer cell tumoricidal activity. Immunity 2015; 43:751–63. [DOI] [PubMed] [Google Scholar]
- 162. Guo B, Fu S, Zhang J, Liu B, Li Z. Targeting inflammasome/IL‐1 pathways for cancer immunotherapy. Sci Rep 2016; 6:36 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Small W Jr, Bacon MA, Bajaj A, Chuang LT, Fisher BJ, Harkenrider MM et al Cervical cancer: a global health crisis. Cancer 2017; 123:2404–12. [DOI] [PubMed] [Google Scholar]
- 164. Rouble AN, Storey KB. Characterization of the SIRT family of NAD+‐dependent protein deacetylases in the context of a mammalian model of hibernation, the thirteen‐lined ground squirrel. Cryobiology 2015; 71:334–43. [DOI] [PubMed] [Google Scholar]
- 165. Nagini S. Breast cancer: current molecular therapeutic targets and new players. Anti‐Cancer Agents Medl Chem 2017; 17:152–63. [DOI] [PubMed] [Google Scholar]
- 166. Engin A. Obesity‐associated breast cancer: analysis of risk factors. Adv Exp Med Biol 2017; 960:571–606. [DOI] [PubMed] [Google Scholar]
- 167. Kolb R, Phan L. Obesity‐associated NLRC4 inflammasome activation drives breast cancer progression. Oncogene 2016; 7:13 007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Chen J, Pitmon E, Wang K. Microbiome, inflammation and colorectal cancer. Semin Immunol 2017; 32:43–53. [DOI] [PubMed] [Google Scholar]
- 169. Hou JK, Abraham B, El‐Serag H. Dietary intake and risk of developing inflammatory bowel disease: a systematic review of the literature. Am J Gastroenterol 2011; 106:563–73. [DOI] [PubMed] [Google Scholar]
- 170. Giovannucci E, Michaud D. The role of obesity and related metabolic disturbances in cancers of the colon, prostate, and pancreas. Gastroenterology 2007; 132:2208–25. [DOI] [PubMed] [Google Scholar]
- 171. Zhao S, Gong Z, Zhou J, Tian C, Gao Y, Xu C et al Deoxycholic acid triggers NLRP3 inflammasome activation and aggravates DSS‐induced colitis in mice. Front Immunol 2016; 7:536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Liu L, Dong W, Wang S, Zhang Y, Liu T, Xie R et al Deoxycholic acid disrupts the intestinal mucosal barrier and promotes intestinal tumorigenesis. Food Function 2018; 9:5588–97. [DOI] [PubMed] [Google Scholar]
- 173. Yuan F, Kolb R, Pandey G, Li W, Sun L, Liu F et al Involvement of the NLRC4‐inflammasome in diabetic nephropathy. PLoS ONE 2016; 11:e0164135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Wilms H, Sievers J, Rickert U, Rostami‐Yazdi M, Mrowietz U, Lucius R. Dimethylfumarate inhibits microglial and astrocytic inflammation by suppressing the synthesis of nitric oxide, IL‐1beta, TNF‐alpha and IL‐6 in an in‐vitro model of brain inflammation. J Neuroinflamm 2010; 7:30–7. [DOI] [PMC free article] [PubMed] [Google Scholar]