

ABSTRACT
Despite advances in HIV therapy, there is no cure, and lifelong antiretroviral treatment is required to suppress viral replication. We hypothesized that HIV maintains the survival of latently infected CD4+ T cells through increased expression of cell survival factors including XIAP, BIRC2 and BECN1. We found that DIABLO/SMAC mimetics promote the degradation of XIAP and BIRC2 in latent HIV-infected resting memory CD4+ T cells (HIV-TCM) without activating viral transcription. Also in HIV-TCM, but not in uninfected cells, the degradation of XIAP and BIRC2 leads to the induction of macroautophagy/autophagy, and the formation of a cell death complex on phagophore membranes that consist of autophagy (ATG5, ATG7 and SQSTM1) and pro-apoptotic (FADD, RIPK1, RIPK3, CASP8) proteins. This results in autophagy-dependent apoptosis of HIV-TCM while sparing uninfected cells. These findings support the potential use of DIABLO/SMAC mimetics as part of an HIV cure strategy.
Abbreviations: ART: antiretroviral therapy; ATG2A: autophagy related 2A; ATG2B: autophagy related 2B; ATG5: autophagy related 5; ATG7: autophagy related 7; BCL2: BCL2, apoptosis regulator; BECN1: beclin 1; BIRC2: baculoviral IAP repeat containing 2; CASP8: caspase 8; CFLAR: CASP8 and FADD like apoptosis regulator; DIABLO/SMAC: diablo IAP-binding mitochondrial protein; SM: DIABLO/SMAC mimetics; DISC: death-inducing signaling complex; FADD: Fas associated via death domain; FAS: Fas cell surface death receptor; HIV: human immunodeficiency virus type 1; HIV-TCM: HIV latent resting central memory CD4+ T cells; IAP: inhibitor of apoptosis protein; RIPK1: receptor interacting serine/threonine kinase 1; RIPK3: receptor interacting serine/threonine kinase 3; RNAi: RNA interference; SQSTM1: sequestosome 1; TCM: resting central memory CD4+ T cells; TNF: tumor necrosis factor; TNFSF10: TNF superfamily member 10; XIAP: X-linked inhibitor of apoptosis.
KEYWORDS: Apoptosis, autophagy, BIRC2, caspase 8, CD4+ T cell, Fas, HIV, RIPK1, SMAC mimetics, XIAP
Although antiretroviral therapy (ART) effectively suppresses HIV replication, HIV persists in latently infected, resting memory CD4+ T cells (HIV-TCM). These latent cells contain replication-competent proviral DNA, survive for months to years, undergo proliferative renewal, and are the major obstacle to eradicating HIV. Thus, lifelong ART is required for continued viral suppression. One strategy to eradicate HIV latent viral reservoir cells is to reactivate viral transcription using a latency-reversing agent followed by clearance of these cells through a combination of viral and cell-mediated cytotoxicity. Unfortunately, reactivation from latency is insufficient to induce viral cytopathicity, and cells harboring replication-competent HIV are resistant to T cell-mediated killing, rendering the approach ineffective at both eliminating the reservoirs and preventing rebound after ART cessation. Thus, there is a need to develop alternative approaches that selectively kill HIV reservoir cells.
A crucial host defense mechanism of virally infected cells is the induction of apoptosis. Conversely, viruses survive and propagate through the prevention of apoptosis. Although the mechanism(s) by which HIV downregulates the apoptotic response in latently infected cells remains poorly understood, numerous studies indicate that HIV alters the transcriptome and proteome of these cells to inhibit apoptosis. For example, HIV inhibits cell death by upregulating the expression of anti-apoptotic proteins including BCL2, CFLAR, and the inhibitor of apoptosis proteins (IAPs) XIAP and BIRC2, rendering the infected cells less susceptible to FAS, TNFSF10 and TNF signaling. Because HIV-TCM have enhanced IAP expression, we hypothesized that IAP would be important targets for therapeutic exploitation in an HIV cure strategy. As IAPs block programmed cell death and are expressed at high levels in various human cancers, they have become attractive targets for cancer drug development. DIABLO/SMAC mimetics (SM) are small-molecule inhibitors that mimic DIABLO, an endogenous antagonist of IAPs. SM directly compete with caspases for XIAP binding, liberating caspases for apoptosis, and stimulate the E3 ubiquitin ligase activity of BIRC2 and BIRC3 promoting their autoubiquitination and proteasomal degradation with subsequent cell death. Therefore, we tested 2 SM that have been evaluated in cancer clinical trials (birinapant and GDC-0152), and embelin, a natural hydroxyl benzoquinone derived from Embelia ribes, for their effect on inducing autophagy and cell death in HIV-TCM [1].
All 3 SM tested trigger rapid and dose-dependent proteasomal degradation of IAPs in uninfected and HIV-TCM that become significant at one-tenth the concentration (10 nM birinapant, 10 nM GDC-0152 or 1 µM embelin) in the infected HIV-TCM compared to uninfected TCM. At these same concentrations, all 3 SM induce significant autophagic flux and cell death in HIV-TCM but not in TCM. At a higher SM dose (100 nM birinapant, 100 nM GDC-0152 or 10 µM embelin), there is an increase in BECN1 expression, MAP1LC3B lipidation, and CASP8 cleavage in uninfected TCM, but no induction of autophagy or biologically significant apoptosis. Conversely, we observe significant autophagy induction and apoptosis in HIV-TCM that is not TNF dependent nor secondary to toxic factors secreted into cell cultures. Importantly, there is no SM-mediated increase in HIV p24 antigen release or cell-associated HIV gag RNA indicating that the SM are selectively killing the HIV-TCM in the absence of increased viral expression.
As autophagy and apoptosis are closely linked, we further examined the role of autophagy in the apparent selective hypersensitivity of HIV-TCM to apoptosis. We initially ascertained that the degradation of both XIAP and BIRC2 is autophagy independent. To investigate how autophagy affects SM-mediated cell death in HIV-TCM, we inhibited the autophagy conjugation cascade with a combination of RNA interference (RNAi) and pharmacological inhibition. We observed that inhibition of early stages of autophagy (wortmannin or RNAi for ULK1, ATG5 or ATG7) prevents cell death. Conversely, neither inhibition of mid stages (simultaneous RNAi for ATG2A and ATG2B [which causes the accumulation of immature unclosed autophagosomal/phagophore structures]) nor late stages of autophagy (using chloroquine or bafilomycin A1) protects cells from SM-mediated apoptosis. These observations led us to hypothesize that components of the autophagy machinery were mediating cell death by serving as a scaffold for efficient death-inducing signaling complex (DISC) formation rather than by turnover of cellular components by autophagy (Figure 1). In support of this hypothesis, characterization of the DISC consisting of pro-apoptotic (FADD, RIPK1, RIPK3, CASP8) and autophagy (ATG5, ATG7 and SQSTM1) proteins and localization of the DISC to phagophores was confirmed through a series of co-immunoprecipitation and subcellular fractionation experiments using ATG5-, ATG7-, and ATG2A/B-silenced cells. Moreover, localization of the DISC to the phagophore membrane requires SQSTM1 binding to RIPK1, as RNAi for SQSTM1 is sufficient to completely inhibit SM-mediated DISC formation, RIPK1 cleavage, and apoptosis.
Figure 1.
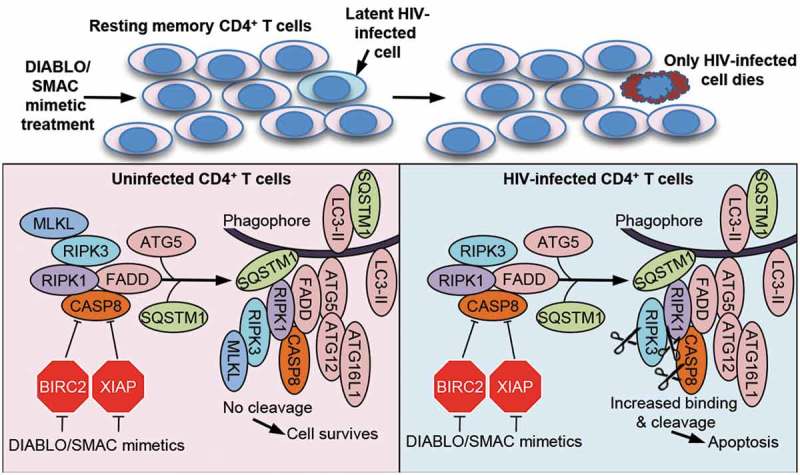
SMAC mimetics promote apoptosis in HIV-TCM via the autophagy-dependent formation of a CASP8-activating platform on phagophore membranes. Treatment of a heterogeneous population of uninfected and latent HIV-infected TCM with DIABLO/SMAC mimetics induces the degradation of XIAP and BIRC2. This triggers the induction of autophagy and the formation of a cell death complex consisting of pro-apoptotic (FADD, RIPK1, RIPK3, CASP8) and autophagy (ATG5, ATG7 and SQSTM1) proteins on unclosed autophagosomal/phagophore structures in HIV-TCM, but not uninfected TCM resulting in the selective apoptosis of only the infected HIV-TCM, while sparing uninfected bystander cells in the absence of viral activation.
Although SM induces apoptosis in resting HIV-TCM, it will be important to evaluate their impact on dividing CD4+ T cells, as well as other cell (sub)types, especially in the presence of proinflammatory cytokines. Importantly, we observed minimal apoptosis in uninfected cells and our preliminary research suggests a similar selective potency in macrophages. In addition, the ability of SM to penetrate tissue sites such as gut-associated lymphoid tissue and the central nervous system must also be determined. In summary, we showed that SM mediate the selective induction of HIV-TCM apoptosis through the assembly of a ripoptosome-like DISC involving autophagy machinery, but not the degradative role of autophagy, and without viral reactivation while sparing uninfected cells. These findings support the use of SM in future strategies designed to eradicate HIV.
Funding Statement
This work was supported by the National Institute of Neurological Disorders and Stroke [NS084912 and NS104015], the California HIV/AIDS Research Program [ID12-SD-255], and the International Maternal Pediatric Adolescent AIDS Clinical Trials Network (IMPAACT). Overall support for IMPAACT was provided by the National Institute of Allergy and Infectious Diseases [UM1AI068632 (IMPAACT LOC), UM1AI068616 (IMPAACT SDMC) and UM1AI106716 (IMPAACT LC)], with co-funding from the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD) and the National Institute of Mental Health (NIMH). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript .
Disclosure statement
No potential conflict of interest was reported by the authors.
Reference
- [1].Campbell GR, Bruckman RS, Chu YL, et al. SMAC mimetics induce autophagy-dependent apoptosis of HIV-1-infected resting memory CD4+ T cells. Cell Host Microbe. 2018. November 14;24(15):689–702.e7. PMID: 30344003. [DOI] [PMC free article] [PubMed] [Google Scholar]