

Abstract
ONC201 is a first-in-class imipridone molecule currently in clinical trials for the treatment of multiple cancers. Despite enormous clinical potential, the mechanism of action is controversial. To investigate the mechanism of ONC201 and identify compounds with improved potency, we tested a series of novel ONC201 analogues (TR compounds) for effects on cell viability and stress responses in breast and other cancer models. The TR compounds were found to be ∼50–100 times more potent at inhibiting cell proliferation and inducing the integrated stress response protein ATF4 than ONC201. Using immobilized TR compounds, we identified the human mitochondrial caseinolytic protease P (ClpP) as a specific binding protein by mass spectrometry. Affinity chromatography/drug competition assays showed that the TR compounds bound ClpP with ∼10-fold higher affinity compared to ONC201. Importantly, we found that the peptidase activity of recombinant ClpP was strongly activated by ONC201 and the TR compounds in a dose- and time-dependent manner with the TR compounds displaying a ∼10–100 fold increase in potency over ONC201. Finally, siRNA knockdown of ClpP in SUM159 cells reduced the response to ONC201 and the TR compounds, including induction of CHOP, loss of the mitochondrial proteins (TFAM, TUFM), and the cytostatic effects of these compounds. Thus, we report that ClpP directly binds ONC201 and the related TR compounds and is an important biological target for this class of molecules. Moreover, these studies provide, for the first time, a biochemical basis for the difference in efficacy between ONC201 and the TR compounds.
ONC201 (also known as TIC10) was originally identified from an NCI chemical library screen for its ability to induce TRAIL (tumor necrosis factor-alpha-related apoptosis-inducing ligand) gene transcription in a human colon cancer cell line (HCT116).1 ONC201 has shown growth inhibitory effects in multiple cancer cell lines and antitumor activity in animal models of glioblastoma, colorectal, non-Hodgkins lymphoma, and pancreatic cancer (reviewed in ref (2)). As a result of these promising preclinical studies, its drug-like properties, its low toxicity in animals, and its penetration of the blood–brain barrier, ONC201 has rapidly advanced and is currently in 15 clinical trials (ClinicalTrials.gov identifier: NCT02863991).2,3 In 2018, ONC201 was granted Fast Track Designation for the treatment of adult recurrent H3 K27 M mutant high-grade gliomas.
Despite significant preclinical promise,3−5 no defined mechanism of action for ONC201 has been established. While TRAIL was found to be induced by ONC201 in some cell types,1,6 this was not consistently observed in all studies. Specifically, TRAIL was not increased in breast cancer cell lines even though ONC201 strongly reduced cell viability.7 ONC201 was also effective against TRAIL-resistant breast cancer cells8 and hematological malignancies independently of TRAIL.9 Although ONC201 has been reported to increase the DR5 death receptor,2 Greer et al. did not observe increased DR4 or DR5 expression after ONC201 treatment, nor did knockdown of DR4 or DR5 affect the inhibitory activity of ONC201.7
ONC201 has been reported to inhibit Akt and ERK activity, potentially providing a mechanism to explain TRAIL induction through FOX3a.2,10,11 Again, this was not observed by others despite significant effects of ONC201 on cell growth.7 Finally, it was also reported that the dopamine receptors (DRD2 and DRD3) may be targets of ONC201.12 However, direct evidence for this interaction has been sparse, and siRNA-mediated knockdown of these receptors did not reduce ONC201’s inhibitory effect.12
Recently, ONC201 was shown to kill breast cancer cells by targeting mitochondria and mitochondrial metabolism.7 Consistent with multiple previous reports,2 Lipkowitz and colleagues reported that ONC201 induced an integrated stress response (ISR) as shown by the induction of ATF4 and the C/EBP homology protein (CHOP).7,13 Although a specific mechanism was not elucidated, there was a direct correlation observed between the antiproliferative activity of ONC201, reduced oxidative phosphorylation, and the number of viable mitochondria. Moreover, ONC201 was shown to be ineffective in Rho0 cells (cells with impaired mitochondrial function due to chemical depletion of mitochondrial DNA).7
Taken together, these studies indicated the necessity to investigate and identify the potential targets for ONC201 and related compounds. The SAR of the ONC201 chemical series has been investigated though an iterative process of chemical optimization and subsequent testing in cell viability assays.11 Importantly, the initial chemical structure for the compound was determined to be incorrect by Janda and colleagues,14 spurring a series of synthetic efforts leading to the identification of novel chemical entities based on this newly discovered pharmacophore.15 Madera Therapeutics created a series of novel highly potent analogues of ONC201 and defined a new chemical series collectively known as TR compounds.16−18 To investigate potential targets for ONC201 and the related TR compounds, we applied an unbiased affinity proteomics approach to identify potential targets. In this study, we report human mitochondrial ClpP as a key protein that binds ONC201 and related compounds in a direct and specific manner. In addition, we show that pretreatment of isolated recombinant ClpP with ONC201 and the TR compounds significantly increases the activity of ClpP. We show that ClpP activation by ONC201 and related compounds in vitro correlates directly with their biological potency in cells. Finally, we demonstrate that these compounds act through ClpP activation to induce the ISR, inhibition of protein synthesis, and mitochondrial events related to inhibition of cell growth.
Methods
Chemical Compounds
ONC201 was obtained from SelleckChem (S7963). The TR compounds and D9 were supplied by Madera Therapeutics, LLC and Nanjing Gator Meditech, Ltd. ADEP was obtained from Cayman Chemical (A-54556A). All compounds were dissolved in DMSO unless otherwise stated.
Cell Culture
Human Triple Negative Breast Cancer (TNBC) cell lines SUM159 and MDA-MB-231 were a generous gift from Dr. Gary Johnson at UNC CH. SUM159 cells were cultured in Dulbecco’s modified Eagle’s medium: Nutrient Mixture F-12 (DMEM/F12, 0565-018, Thermo Fisher Scientific) supplemented with 5% fetal bovine serum (FBS, TMS-013-B, Millipore), 5 μg/mL insulin, 1 μg/mL hydrocortisone, and 1% mixture of antibiotic–antimycotic (15240062, Thermo Fisher Scientific). MDA-MB-231 cells were cultured in RPMI 1640 media (11875-093, Thermo Fisher Scientific) supplemented with 10% FBS and 1% antibiotic–antimycotic. Cells were incubated at 5% CO2 and 37°C. HeLa, A549, Panc1, PC3, and MCF-7 cells were obtained from ATCC and maintained in DMEM with 10% FBS and 1% antibiotic.
Viability Assays
Resazurin Assay
Cell viability assays were performed by plating SUM159 (1000 cells/well) on a Cellstar 96-well plate (655-180, Griener) in supplemented DMEM:F12 and allowed to incubate/adhere overnight. After adherence, the media in each well was aspirated and replaced with 50 μL of pure media and 50 μL of media containing the drug at twice the final concentration. The concentrations of drugs used are shown in the figure legends and discussed in the text. Cells treated with DMSO (vehicle only) were used as a negative control in all experiments. The final concentration of DMSO (vehicle) was kept under 1% in all experiments.
Cells treated with appropriate concentrations of selected compounds for 72 h in 100 μL of incubation media were supplemented with 20 μL of resazurin (0.6 mM, Acros Organics 62758-13-8) and incubated for 30 min at 37°C. Cells were incubated to allow metabolism to resorufin, at which point 75 μL of each sample was transferred to a Costar black 96-well plate (CLS3915, Millipore Sigma), and the relative fluorescence of resorufin across samples was determined using a PHERAstar (BMG Labtech) with fluorescent module FI: 540-20, 590-20. The results were analyzed using GraphPad Prism 7 software which was used to generate the dose-dependence curves and to calculate IC50 values.
Total Cell Counting
Total cell counting assays were performed by plating SUM159 (50 cells/well) on a 96-well Greiner plate in supplemented DMEM:F12, allowing them to incubate/adhere overnight, and treating with drug as described above. At a predetermined time point (0, 24, 48, or 72 h), media was aspirated, and 100 μL of Hoechst stain (1 μg/mL, H3570, Thermo Fisher Scientific) was added to each well and then allowed to incubate for 30 min at 37 °C. Total cell number was then quantified using the Celigo Imaging Cytometer (Nexcelom).
Crystal Violet Assays
Crystal violet assays were performed by plating SUM159 (1000 cells/well) on a 6-well Costar plate (CLS3506, Millipore Sigma) and allowing them to incubate/adhere overnight. Media was aspirated and replaced by media containing the drug at the desired concentration. Cells were allowed to incubate for 48 h, and the media was either replaced by fresh media with drug or media without drug. Cells were then allowed to incubate until one well was 100% confluent, changing media as needed. Once confluent, cells were stained using 0.5% crystal violet in 20% methanol, allowed to incubate for 10 min at RT, then rinsed 3 times and allowed to dry overnight. Staining was quantified by dissolving crystal violet stain in Sorenson’s buffer (0.1 M sodium citrate, 50% ethanol, pH 4.2) and measuring the absorption of dissolved crystal violet at 570 nm using a BMG Polarstar Omega.
Immunoblotting
For Western blotting, SUM159 or MDA-MB-231 cells were plated and treated with compounds as described above for cell viability assays. Following treatment, cells were rinsed 3 times with 2 mL of cold PBS and lysed using RIPA buffer (no SDS) supplemented with 2 mM Na(VO3)4, 10 mM NaF, 0.0125 μM calyculin A, and complete protease inhibitor cocktail (11873580001, Roche Diagnostics). Cell lysates were clarified and immunoblotted as described earlier.19 Membranes were incubated with the following primary antibodies: [TUFM (PA5-27511), IDH2 (PA5-79436) from Invitrogen]; [ClpP (CS-14181), ATF4 (CS-11815), LONP (CS-28020), Aconitase (CS-6922s) from Cell Signaling Technologies]; [β-actin (SC-47778), CHOP (SC-575), TFAM, (SC-376672), ClpP (SC-271284) from Santa Cruz Biotechnologies] diluted 1:1000 in 5% TBST/BSA + 0.02% NaN3) overnight at 4°C, removed, washed 3 × 5 min in TBST, and placed in their respective 2° antibodies (1:10 000 dilution in 5% milk/TBST) for 1 h. Membranes washed 3 × 5 min in TBST were embedded in 2 mL of each ECL reagent and imaged using a Chemidoc MP (BioRad) or developed with film. Acquired images were processed/quantified using Image Lab software (BioRad).
Reverse Transfection
All siRNA stocks were ordered from Dharmacon at 2 nmol/siRNA and resuspended in 100 μL 1× siRNA reconstitution buffer (Dharmacon) to create a 20 μM stock. Dharmafect I was diluted 1:266 in OptiMEM media and divided into 250 μL aliquots. Individual siRNAs were added to Dharmafect aliquots for a final concentration of 125 nM, incubated at RT for 30 min, and then added to 6-well or 96-well plates. SUM159 cells were washed with PBS and trypsinized at 37 °C for 5 min. Cells were resuspended to a final concentration of 1 × 105 cells/mL (6-well) or 1 × 104 cells/mL (96-well) in 1:1 DMEM:F-12 supplemented with 5% FBS, 5 μg/mL insulin, and 1 μg/mL hydrocortisone. Cells (1.0 mL/well, 100 μL/well) were added to each well containing siRNA and incubated for 24 h.
Caspase and Apoptosis Assays
Caspase 3/7 activity was analyzed using a fluorescent peptide substrate (7-amido-4-methylcoumarin).19 The cells were plated at 8 × 105 cells per plate (6 cm plate) and treated for 24 h with 3, 30, and 300 nM TR57 and TR31; 300 nM, 3 μM, and 30 μM ONC201, as well as 0.1% DMSO and 10 nM staurosporine (IC25 on SUM159 cells determined by MTS cell viability assay (data not shown)). The samples were harvested by mechanical scraping of the cells into 400 μL of lysis buffer (50 mM HEPES (pH: 7.4), 5 mM CHAPS, and 5 mM DTT), and caspase activity was measured as described earlier.19
Affinity Capture of Protein Targets
To generate TR-80 or TR-81 affinity resins, we coupled free TR-80 or TR-81 to NHS-activated agarose (Thermo Scientific catalog #26200). Briefly, NHS-agarose beads were allowed to react with TR-80 or TR-81 at a final concentration of 5 mM in coupling buffer (PBS with 20% DMSO) for 4 h at RT. Following washing with PBS to remove unbound ligand, the beads were blocked for an additional hour with 1 M Tris-HCl, pH 7.5. Control agarose beads were generated by blocking uncoupled NHS-agarose beads with 1 M Tris-HCl, pH 7.5. Primaquine (PQ)-agarose was generated by coupling NHS-agarose with 20 mM primaquine in phosphate buffered saline, pH 8.0.
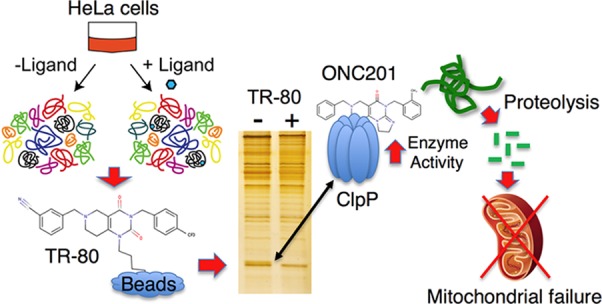
To identify TR-80 interacting proteins, approximately 50 × 106 HeLa cells were homogenized in cell lysis buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 2 mM EDTA, 0.5% NP-40) and clarified by centrifugation (15 min at 13 000g). The resultant supernatant was divided into 1 mL aliquots and mixed with ∼50 μL packed beads of control agarose, PQ-agarose, or TR-80 agarose in the presence of 2% DMSO or 1.25 mM free TR-80 and rotated for one h at RT. Beads were then collected by centrifugation and washed 3 times, 1 mL each wash, with cell lysis buffer. Beads were boiled in SDS sample buffer, resolved by SDS-PAGE using 11% polyacrylamide gels, and silver stained as described.20 A protein band, indicated by the arrow, was excised, destained, trypsin digested, and subjected to mass spectrometry for identification (see below).
TR-81 Elution
To determine the specificity of protein interactions with TR-81 beads, ∼ 30 μL of TR-81 agarose was mixed with 1 mL of HeLa cell lysate generated as described above. After washing the beads to remove nonspecific interactions, the beads were incubated with 100 μL of elution buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl) containing 2% DMSO or 50 μM TR-57 for 30 min at RT. The eluant was collected, resolved on an 11% polyacrylamide gel and silver stained or immunoblotted with ClpP antibodies.
In Vitro Drug Competition Assays
To compare the relative binding ability of different compounds to ClpP, ∼200–400 μL of HeLa cell lysate (generated as described above) was mixed with vehicle (2% DMSO final) or different concentrations of ONC201 or TR compounds. After mixing for 15 min, the lysate was combined with ∼20 μL packed TR-80 beads and rotated for 1 h at RT. The supernatant was discarded, and beads were washed briefly 3 times, 1 mL each wash, with cell lysis buffer before boiling in SDS sample buffer. Samples were then resolved by SDS-PAGE and immunoblotted with ClpP antibodies. To compare the ability of the compounds to compete for ClpP binding within live cells, HeLa cells in culture were treated with DMSO (final concentration 0.1%) or various concentrations of ONC201 or TR compounds for 30 min. Cells were then snap frozen, and lysates were subsequently prepared and analyzed in a similar manner as described above.
MALDI TOF/TOF Analysis
The resultant tryptic peptides obtained from the silver stained band were desalted using a C18 Zip-Tip and applied to a MALDI target plate with α-cyano-4-hydroxycinnamic acid as the matrix. The sample was analyzed on an AB Sciex 5800 MALDI TOF/TOF mass spectrometer. MS/MS spectra were searched against a Uniprot human database using Mascot version 2.3 (Matrix Science) within ProteinPilot software version 3.0 (AB Sciex). A significance score threshold was calculated in Mascot, and an ion score above 36 was considered a significant identification (p < 0.05).
Measurement of Human ClpP Activity
Measurement of in vitro activity of recombinant human caseinolytic peptidase ClpP (Cat # MBS204060, MyBioSource) was based on monitoring the release of fluorescent coumarin from the fluorogenic substrate Ac-WLA-AMC (Cat #S330, Boston Biochem, Inc.) as described previously21−23 with minor modifications. Briefly, the activity of recombinant ClpP proteolytic subunit (1 μg/mL) was measured in assay buffer composed of 50 mM Tris, 10 mM MgCl2, 100 mM KCl, 1 mM DTT, 4 mM ATP, 0.02% Triton X-100, and 5% glycerol, pH 8.0 (HCl) using 10 μM of fluorogenic Ac-WLA-AMC substrate as described.23,24 Two different protocols were used to investigate the effects of ONC201 and the TR compounds on ClpP activity. Using the first protocol (Protocol #1), the reaction was initiated instantly by direct mixing of ClpP enzyme and substrate in the presence of indicated concentrations of compounds. Applying a second protocol (Protocol #2), the enzyme and compounds were mixed and incubated in assay buffer for 60 min before initiating the reaction by adding Ac-WLA-AMC substrate. The kinetics of the free coumarin fluorescence was monitored using black, μ-CLEAR 96-well flat bottom plates (Cat # 655090, Greiner), and the fluorescence of released coumarin was recorded at 350 nm excitation and 460 nm emission using BMR PHERAstar plate reader equipped with appropriate FI module (BMG LABTECH). The slope of the linear portion of the fluorescence signal over time used as a measure of the activity of ClpP. Measurements were carried out in triplicate and presented as the rate of fluorescence change at given concentrations of ClpP and substrate in the presence or absence of ONC201 or the TR compounds. Dose-dependence of ClpP activation with different compounds was used for determination of EC50 for each compound, and the activity of samples treated with DMSO (vehicle) measured as background and subtracted from experimental data and the activity of ClpP expressed as RFU/μg of ClpP/h.
To measure ClpP protease activity in vitro, assays were conducted using Protocol #2 as described above. Recombinant ClpP was preincubated for 1 h at 37 °C with DMSO or the indicated compounds in assay buffer and then an additional h at 37 °C in the presence of substrate. The reaction volume was 50 μL with a final concentration of 10 ng/μL ClpP, 5 μM α-casein, and 1% DMSO. Samples were boiled in SDS sample buffer, resolved by 12% SDS-PAGE, and silver stained.20
Metabolic Labeling of Nascent Proteins
Nascent proteins were metabolically labeled and quantified as described previously.25,26 Briefly, cells were incubated in methionine- and cysteine-free medium (Sigma) containing relevant concentrations of compounds tested for 15 min. 35S-labeled methionine and cysteine (125 μCi; PerkinElmer EasyTag Express Labeling Mix) were added and allowed to incorporate for 30 min. Cells were then washed twice in ice-cold PBS, scraped, and collected by centrifugation. Cell pellets were lysed in RIPA medium containing protease inhibitors (Roche), and protein concentrations were determined by the Bradford assay (Amresco). Trichloroacetic acid (TCA) was added to a final concentration of 20%, and precipitated proteins were captured on glass microfiber filters by filtration under vacuum. The filters were washed twice with 20% TCA and once with 100% ethanol and allowed to air-dry. The filters were then transferred to vials containing scintillation fluid (EcoScint), and radioactivity was quantified using a scintillation counter. The amount of radioactivity was normalized to the protein concentration for each sample.
Results and Discussion
ONC201 Analogues are Highly Potent Inhibitors of TNBC Growth
Madera Therapeutics synthesized a number of novel chemical analogues of ONC201 known as the TR compounds. TR-31, first published by Nanjing Gator Meditech, is identical to ONC212.11,16−18 TR-42, TR-65, and TR-57 were designed to address limitations of ONC201, including modest cell activity and dose-dependent pharmacokinetics. Madera also synthesized additional amine-containing analogues (TR-79, TR-80, and TR-81) for the purpose of creating affinity ligands to capture TR binding proteins (Figure 1a).
Figure 1.

ONC201 and TR analogues inhibit cell growth and induce ATF4 and CHOP activation. (a) Chemical structures of ONC201 and TR compounds. (b) Cell viability of ONC201 and TR compounds using MTS assay in SUM159 cells. IC50 values are shown in the table at left (a). Data shown as mean ± SEM, representative of N = 4 replicates. (c) Growth of SUM159 cells determined by crystal violet assay, representative of N = 3 replicates. (d) Changes in cell number determined by Hoechst stain, representative of N = 2 replicates. (e) Immunoblots of SUM159 lysates for ATF4, CHOP, and β-actin.
We compared ONC201 and the TR compounds for effects on growth inhibition of two established models of TNBC (MDA-MB-231, SUM159 cells). Cells were incubated with ONC201 or the TR compounds for 24–72 h, and cell viability was measured using MTS assay. The TR compounds were all significantly more potent compared to ONC201, with IC50 values ∼100-fold lower than ONC201 in these cells (Figure 1b). Growth inhibition was confirmed by crystal violet colony formation assays (Figure 1c) and cell counting experiments (Figure 1d).
ONC201 and analogues have been shown to induce an ISR in multiple cancer cell lines.1,7,9,27 CHOP and ATF4 proteins are common markers of the ISR response and ER stress,28 and we compared the ability of ONC201 and the TR compounds to induce CHOP and ATF4. Incubation of SUM159 or MDA-MB-231 cells with ONC201 or TR-57 strongly induced dose- and time-dependent increases in ATF4 as measured by immunoblotting. The amount of ONC201 or TR-57 required to increase ATF4 closely reflected the IC50 values for growth inhibition in these cells. While the effects on CHOP were more modest and peaked earlier than ATF4 (Figure 1e), these data confirmed the activation of the ISR by these compounds. Under the same conditions, we observed increased Erk and Akt phosphorylation (Figure S1).
ONC201 was shown to increase PARP cleavage and apoptosis in some cancer models2,29 but not others.7 We tested the effects of ONC201 and select TR analogues on caspase activity in SUM159 cells using a fluorogenic Ac-DEVD-AMC substrate. No significant increase in caspase activity was detected by either fluorogenic assays or immunoblotting for PARP cleavage, even at the highest concentrations of ONC201 or TR-57 tested. By contrast, staurosporine, an established apoptosis inducer, increased Ac-DEVD-AMC cleavage as expected (Figure S1). Cell counting experiments confirmed that these compounds did not reduce the total number of cells below the initial value, even after 72 h incubation (Figure 1d). Thus, these compounds, at the concentrations tested, are having cytostatic effects but not increasing caspase-dependent cell death in SUM159 cells.
Affinity Chromatography Identifies the Mitochondrial Protease ClpP as a Target of ONC201 and the TR Compounds
To identify protein targets of ONC201, we coupled modified ONC201 analogues (TR-79, TR-80, and TR-81 (Figure 1a)) to agarose beads. Addition of the spacer arm and primary amine did not significantly reduce the potency of TR-79 to inhibit TNBC growth (Figure 1b). To control for nonspecific interactions, we compared our TR-80 affinity column protein binding results to agarose alone or agarose coupled to an unrelated drug, PQ. HeLa cell lysates were mixed with control beads, PQ-beads, or TR-80 beads in the absence or presence of excess free TR-80. Following incubation of beads with lysate, the beads were washed and boiled in sample buffer to release bound proteins, and the samples were resolved by SDS-PAGE and silver stained. A distinct protein band of ∼24 kDa was observed that did not bind the control beads or the PQ-beads and only bound TR-80 beads in the absence of free TR-80. The protein, indicated by the arrow (Figure 2a), was excised, trypsin digested, and subjected to MALDI TOF/TOF mass spectrometry. MASCOT analysis identified this protein as the human mitochondrial protease, ClpP (Methods and Table S1).
Figure 2.

Identification of ClpP as an ONC201 and TR compound binding protein. (a) HeLa cell lysate was mixed with the indicated beads in the absence or presence of free TR-80. A silver stained gel is shown, and the band indicated by the arrow was excised and subjected to mass spectrometry for identification. (b) Replicate samples from panel a were immunoblotted with ClpP antibodies. (c, d) TR-81 beads were charged with HeLa cell lysate and eluted with 50 μM TR-57. A portion of the eluant was silver stained (c) or immunoblotted (d) for ClpP. (e) Pure, recombinant human ClpP was mixed with the indicated beads in the absence or presence of 50 μM TR-57 and immunoblotted for ClpP. (f, g) HeLa cell lysates were mixed with TR-81 beads in the presence of the indicated compounds and immunoblotted for ClpP. (h) HeLa cells were treated with the indicated compounds for 30 min, and lysates were mixed with TR-81 beads and immunoblotted for ClpP.
To confirm the mass spectrometry results, the experiment was repeated as above except the samples were immunoblotted with anti-ClpP antibodies. ClpP was only detected bound to TR-80 beads in the absence of free TR-80 (Figure 2b). Because total cell lysate was applied to TR-81 beads, we sought to determine if ClpP binding to TR-81 was direct or mediated by other proteins. To this end, TR-81 beads were charged with HeLa cell lysates, washed to remove nonspecific proteins, and eluted with 50 μM free TR-57. The eluant was resolved by SDS-PAGE and silver stained or immunoblotted with ClpP antibodies. A single protein band at 24 kDa was observed upon silver staining of the eluate (Figure 2c), and immunoblotting confirmed the protein to be ClpP (Figure 2d). Because ClpP was the only protein detected in the eluate (Figure 2c), it suggests that ClpP binds directly to TR-81 and also that TR-81 is highly specific for ClpP. Confirmation of direct binding between TR-81 and ClpP was shown by incubation of pure recombinant human ClpP with TR-81 beads (Figure 2e).
To compare the relative ability of ONC201 or TR compounds to bind ClpP in vitro, we used TR-81 beads in a competition assay. Increasing concentrations of ONC201, TR-31 (ONC212),11 or TR-57 were mixed with HeLa cell lysates or added directly to HeLa cells in culture. These lysates were applied to TR-81 beads and the amount of ClpP bound assessed by immunoblotting. ONC201, TR-57, and TR-31 all reduced ClpP binding to TR-81 beads in a dose-dependent manner whether mixed with HeLa cell lysates (Figure 2f,g) or added to cell culture media (Figure 2h). Importantly, both TR-57 and TR-31 exhibited a ∼10-fold greater potency over ONC201 in reducing ClpP binding whether applied to cell lysates or cell cultures. To determine if TR-81 bound ClpP from other cancer cell lines, we generated cell lysates from breast cancer (SUM159, MCF7), lung (A549), prostate (PC3), or pancreatic (PANC1) cells and performed similar experiments. ClpP bound immobilized TR-81 from all cell lines tested, and this binding could be prevented with TR-57 (Figure S2), indicating that the interaction between TR compounds and ClpP was not restricted to HeLa cells but was observed from multiple different cancer cell types.
ONC201 and TR Compounds Activate ClpP Peptidase and Protease Activity
We next tested the effects of these compounds on ClpP enzymatic activity. Using purified, recombinant human ClpP protein and a select ClpP peptide fluorogenic substrate (Ac-WLA-AMC), we measured the peptidase activity of ClpP in the presence or absence of ONC201 and TR compounds. As shown in Figure 3a, incubation of ClpP with ONC201 or TR-57 strongly increased ClpP peptidase activity in a dose- and time-dependent manner. We then compared the effects of other TR compounds on ClpP activity. TR-27, 65, 66, and 79 all activated ClpP in a dose-dependent manner similarly to that observed with TR-57 and ONC201. All of the TR compounds were observed to be more potent activators of ClpP than ONC201. We also tested the effects of the recently reported ClpP activator D9.30 D9 also increased ClpP activity albeit at much higher concentrations (EC50 ∼ 110 μM), compared to that observed for ONC201 or the TR compounds (Figure S4).
Figure 3.

Activation of ClpP by ONC201 and the TR compounds. (a) Purified, recombinant human ClpP was incubated with the indicated concentrations of each compound, and ClpP peptidase activity was measured. (b) Recombinant human ClpP was preincubated for 1 h at 37 °C with the indicated compounds and then for an additional hour at 37 °C in the presence of casein. Reaction products were resolved by SDS-PAGE and silver stained.
ONC201 and TR-57 were tested for their effects on ClpP protease activity. We preincubated recombinant ClpP with DMSO, ONC201, or TR-57 and then assayed for casein proteolysis. ONC201 and TR-57 increased ClpP activity toward casein proteolysis with an observed half maximal dose of ∼1.25 μM and ∼200 nM for ONC201 and TR-57, respectively (Figure 3b). These results demonstrate that ONC201 and the TR compounds increase ClpP activity toward unstructured proteins (casein) and peptide substrates.
ClpP Knockdown Reduces the Effects of ONC201 and TR-57 on the ISR and Cell Growth
To investigate if ClpP was a biological target for ONC201 and the TR compounds, siRNA knockdown of ClpP was performed in SUM159 cells. ClpP knockdown was verified by immunoblotting, and ClpP expression was almost completely eliminated by this treatment (Figure 4a). Next, the effect of ClpP knockdown on CHOP/ATF4 induction by ONC201 and TR-57 was examined. Incubation of WT SUM159 cells with ONC201 or TR-57, increased the amount of CHOP protein after 24 h. By contrast, no increase in CHOP was observed after ONC201/TR-57 treatment of the ClpP knockdown cells (Figure 4a). Similar results were observed with ATF4 (Figure S3).
Figure 4.

Effects of ONC201 and TR-57 are prevented by ClpP knockdown in SUM159 cells. (a) Immunoblot of lysates from SUM159 cells transfected with a nontargeting siRNA (siCTRL) or siRNA targeting ClpP (siClpP) and treated with either 10 μM ONC201, 150 nM TR-57, or DMSO for 24 h. Blots shown are representative of three biological replicates. (b) Total protein synthesis measured in SUM159 cells transfected with siCTRL or siClpP after treatment with ONC201, TR-57, cycloheximide, or DMSO. (c) Cell viability plots for SUM159 cells transfected with siCTRL (black circles) or siClpP (white squares) using Hoechst stain for total cell count after 48-h treatment with ONC201 or TR-57. Graphs shown in (b) and (c) are representative of 2 biological replicates. Error bars represent standard error of the mean (SEM) from replicates.
The mitochondrial transcription factor A (TFAM) was recently shown to be reduced by ONC201 treatment.7 From a global proteomics analysis, we observed that the mitochondrial protein elongation factor Tu (TUFM) and other mitochondrial proteins were strongly reduced by ONC201 and TR-57 treatment of SUM159 cells (E.M.J. Fennell, unpublished observations). The effects of ONC201 and TR-57 on the reduction of TFAM and TUFM were compared by immunoblotting control or ClpP knockdown cells. Interestingly, the ONC201/TR-57-stimulated reduction of these proteins was prevented in the ClpP knockdown cells (Figure 4a). Moreover, the less potent ClpP activator D9, also decreased TFAM and TUFM in a ClpP-dependent manner (Figure S3).
ONC201 has been shown to activate the protein synthesis inhibitory eIF2a kinases.27 Using 35S-methionine incorporation into proteins,25,26 we measured the effects of ONC201 and TR-57 on protein synthesis in SUM159 cells. Both compounds significantly inhibited total protein synthesis (>50%) after 24 h (Figure 4b) with TR-57 much more potent than ONC201. Knockdown of ClpP strongly inhibited this response, consistent with activation of ClpP being required. Thus, these results support the importance of ClpP activation by ONC201 or TR-57 as essential for the inhibition of protein synthesis by these compounds.
Lastly, the effects of ClpP knockdown on growth inhibition by ONC201 and TR-57 were examined. ClpP knockdown cells were incubated with or without ONC201 or TR-57, and cell proliferation was measured. The results of these studies demonstrated that the growth inhibitory effects of ONC201 or TR-57 were significantly reduced in the ClpP knockdown cells as compared to WT cells. Thus, these studies demonstrate that the cytostatic effects of ONC201 and TR-57 are in part dependent on ClpP, providing further evidence that this protein is an important target for these compounds (Figure 4c).
In conclusion, the rapid clinical advance of ONC201 has preceded a detailed understanding of the molecular mechanism of drug action.3 We now demonstrate that highly potent chemical agents related to ONC201 may be prepared and that the mitochondrial protein ClpP is a novel target for ONC201 and the related TR compounds. Our data provide evidence for direct binding and activation of the peptide and protease activity of ClpP. Furthermore, through knockdown experiments, we demonstrate that ClpP is essential for the activation of the ISR and subsequent events, including protein synthesis inhibition and ultimately mitochondrial changes initiated by these compounds (Figure 5).
Figure 5.
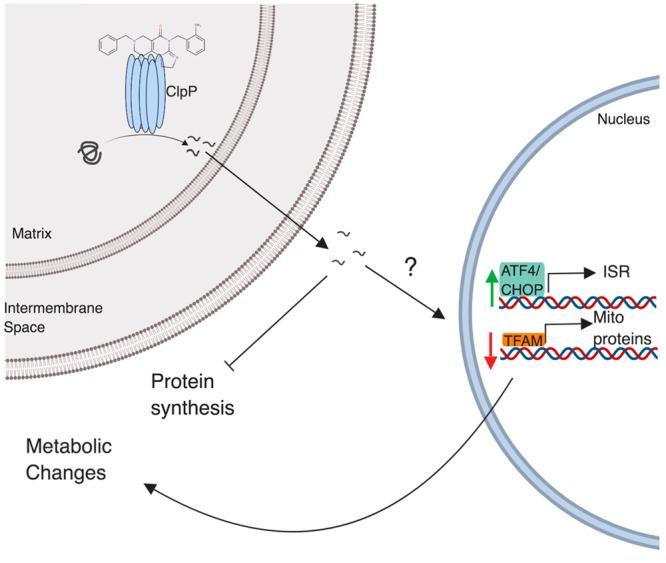
Model for the regulation of ClpP and the ISR by ONC201 and the TR compounds. ClpP, a mitochondrial protease, is activated by ONC201 and TR analogues to degrade mitochondrial proteins. Protein fragments are exported from the mitochondria and activate the unfolded protein response (ATF4, CHOP), decrease TFAM activity, and inhibit protein synthesis. ClpP-mediated changes in ATF4, CHOP, and TFAM lead to alterations in metabolism.
While multiple mechanisms of ONC201 action have been proposed, our results support the importance of ClpP activation to the ISR and subsequent mitochondrial events. This includes ClpP-dependent reduction of the mitochondrial proteins TFAM and TUFM in cells treated with ONC201 or the TR compounds. In this way, our findings are in agreement with Greer et al., who identified reduced TFAM and mitochondrial metabolism as part of the ONC201 response. Interestingly, they observed TFAM protein levels decreased prior to a change in RNA levels.7 Our data showing inhibition of protein synthesis by ONC201 and TR-57, in a ClpP-dependent manner, further supports this observation (Figure 4).
The ATP-dependent CLP protease (ClpP) is a highly conserved serine protease found in species ranging from bacteria to humans.31−33 In humans, ClpP is localized to the mitochondrial matrix and is important for regulating responses to cell stresses, including heat shock, nutrient deprivation, and other cellular insults.34,35 ClpP has been linked to the mitochondrial unfolded protein response (UPR), where it is involved in the regulation of protein homeostasis.36 ClpP is overexpressed in some leukemias, and human ClpP has recently been recognized as a potential target for cancer chemotherapy.37,38 Activators of ClpP, including the acyldepsipeptides (ADEPs) and other compounds, are currently being investigated as novel anticancer or antimicrobial treatments.30,38,39
The mechanism of ClpP activation by ONC201 and TR compounds remains to be established. In cells, the ClpP complex is composed of two components, the ATP-binding chaperone protein (ClpX) and the barrel-shaped ClpP peptidase.40 Other known activators of human and bacterial ClpP (i.e., ADEPs) are believed to displace ClpX and open the central substrate cavity. Elegant structural studies demonstrated that opening this cavity may increase the peptidase activity of ClpP independently of the protein unfolding activity of ClpX.41 Wong and demonstrated that the ADEPs bound to a hydrophobic cleft between the monomers in the heptamer structure of purified ClpP.39
Our data are consistent with the effects of ONC201 and the TR compounds being mediated through direct binding to ClpP. This thesis is supported by affinity column binding assays and biochemical assays where we used isolated, purified ClpP in the absence of ClpX to assess the effects on enzyme activity. While we only tested activity on peptides and a protein lacking significant structure (casein), the effects on intact proteins remains to be determined. However, our proteomics analysis after ONC201 or TR-57 treatment indicates a significant reduction in multiple mitochondrial proteins, suggesting that ClpP activation in cells is leading to the degradation of intact mitochondrial proteins (E.M.J. Fennell, unpublished observations).
In all studies, we observed that the TR compounds were more potent binders and activators of ClpP than ONC201 or the recently reported compound D9. While we were unable to prevent binding of ClpP to TR-81 beads with ADEP or D9 (Figure S2), whether or not ONC201 or the TR compounds bind to the same pocket as these compounds or compete with ClpX binding in intact cells remains to be determined. Importantly, the potent effects of the TR compounds on ClpP directly paralleled the effects of these compound on ISR induction, protein synthesis, and growth inhibition.
The mechanisms by which ONC201 and the TR compounds inhibit cell growth or induce cell death also remains to be determined. The ADEPs affect mitochondrial morphology and inhibit oxidative phosphorylation after extended exposure. We confirmed that ONC201 and the TR compounds induce changes in mitochondrial morphology, inhibition of oxidative phosphorylation, and increased lactic acid formation as reported by others (E. Holmuhamedov, unpublished observations). While we observed primarily cytostatic responses in our studies, these results may be cell type-dependent, as others have observed apoptotic responses. We also did not observe a complete elimination of the cytostatic effects of these compounds in ClpP knockdown cells, suggesting that additional mechanisms of action may also contribute to the growth inhibitory effects of these compounds.
In summary, ONC201 as an unoptimized screening hit has displayed remarkable clinical activity in a range of difficult-to-treat cancers. We propose that the clinical response observed by treatment with ONC201 underscores the importance of a common biological mechanism of action that explains its broad efficacy against multiple cancer types. Our studies demonstrate that ClpP is the common target for both ONC201 and the TR compounds, and activation of ClpP results in modulation of the integrated stress response pathway. Thus, the TR compounds, based on the pioneering work of Janda and colleagues,14 are novel and highly potent activators of ClpP and represent the future direction of ClpP activators as anticancer agents.
Acknowledgments
We would like to gratefully acknowledge D.J. Graves for helpful comments and advice on the manuscript. We would also like to acknowledge the UNC Michael Hooker Proteomics Facility. L.J.A.-C was supported by the NIH 5 T32 GM007040. This work was supported by a Lineberger University Cancer Research Grant (UNC) to L.M.G. and M.P.L. and support from the NIHP30 CA016086, R01 CA199064 (L.M.G.). N.J.M. was supported by funding from NIH R01 AI103311. This work was also supported by Russian Science Foundation, Grant #19-75-20145 (E.H.). P.R.G. received funding from the New York-Presbyterian Brooklyn Methodist Hospital.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschembio.9b00222.
Supplemental Figures 1–4, Table S1, and Supplemental Methods, which describe: chemical methods of synthesis for TR79, 80, 81; the signaling effects of ONC201 and TR-57 in SUM159 cells (Figure S1); the ability of TR-57 but not D9 and ADEP to compete for ClpP binding (Figure S2); prevention by ClpP knockdown of ATF4 induction by ONC201 and TR-57 (Figure S3); prevention of the effects of D9 by ClpP knockdown in SUM159 cells (Figure S4); identification of ClpP through PID (MASCOT) analysis (Table S1) (PDF)
Author Contributions
¶ P.R.G., L.J.A.-C., and E.M.J.F. contributed equally.
The authors declare the following competing financial interest(s): Dr. Edwin Iwanowicz has ownership rights in Madera Therapeutics, LLC which in turn has rights to chemical matter described in this paper.
Supplementary Material
References
- Allen J. E.; Krigsfeld G.; Mayes P. A.; Patel L.; Dicker D. T.; Patel A. S.; Dolloff N. G.; Messaris E.; Scata K. A.; Wang W.; Zhou J. Y.; Wu G. S.; El-Deiry W. S. (2013) Dual inactivation of Akt and ERK by TIC10 signals Foxo3a nuclear translocation, TRAIL gene induction, and potent antitumor effects. Sci. Transl. Med. 5, 171ra17. 10.1126/scitranslmed.3004828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen J. E.; Kline C. L.; Prabhu V. V.; Wagner J.; Ishizawa J.; Madhukar N.; Lev A.; Baumeister M.; Zhou L.; Lulla A.; Stogniew M.; Schalop L.; Benes C.; Kaufman H. L.; Pottorf R. S.; Nallaganchu B. R.; Olson G. L.; Al-Mulla F.; Duvic M.; Wu G. S.; Dicker D. T.; Talekar M. K.; Lim B.; Elemento O.; Oster W.; Bertino J.; Flaherty K.; Wang M. L.; Borthakur G.; Andreeff M.; Stein M.; El-Deiry W. S. (2016) Discovery and clinical introduction of first-in-class imipridone ONC201. Oncotarget 7, 74380–74392. 10.18632/oncotarget.11814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrillaga-Romany I.; Chi A. S.; Allen J. E.; Oster W.; Wen P. Y.; Batchelor T. T. (2017) A phase 2 study of the first imipridone ONC201, a selective DRD2 antagonist for oncology, administered every three weeks in recurrent glioblastoma. Oncotarget 8, 79298–79304. 10.18632/oncotarget.17837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein M. N.; Bertino J. R.; Kaufman H. L.; Mayer T.; Moss R.; Silk A.; Chan N.; Malhotra J.; Rodriguez L.; Aisner J.; Aiken R. D.; Haffty B. G.; DiPaola R. S.; Saunders T.; Zloza A.; Damare S.; Beckett Y.; Yu B.; Najmi S.; Gabel C.; Dickerson S.; Zheng L.; El-Deiry W. S.; Allen J. E.; Stogniew M.; Oster W.; Mehnert J. M. (2017) First-in-Human Clinical Trial of Oral ONC201 in Patients with Refractory Solid Tumors. Clin. Cancer Res. 23, 4163–4169. 10.1158/1078-0432.CCR-16-2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner J.; Kline C. L.; Zhou L.; Campbell K. S.; MacFarlane A. W.; Olszanski A. J.; Cai K. Q.; Hensley H. H.; Ross E. A.; Ralff M. D.; Zloza A.; Chesson C. B.; Newman J. H.; Kaufman H.; Bertino J.; Stein M.; El-Deiry W. S. (2018) Dose intensification of TRAIL-inducing ONC201 inhibits metastasis and promotes intratumoral NK cell recruitment. J. Clin. Invest. 128, 2325–2338. 10.1172/JCI96711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen J. E.; Krigsfeld G.; Patel L.; Mayes P. A.; Dicker D. T.; Wu G. S.; El-Deiry W. S. (2015) Identification of TRAIL-inducing compounds highlights small molecule ONC201/TIC10 as a unique anti-cancer agent that activates the TRAIL pathway. Mol. Cancer 14, 99. 10.1186/s12943-015-0346-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer Y. E.; Porat-Shliom N.; Nagashima K.; Stuelten C.; Crooks D.; Koparde V. N.; Gilbert S. F.; Islam C.; Ubaldini A.; Ji Y.; Gattinoni L.; Soheilian F.; Wang X.; Hafner M.; Shetty J.; Tran B.; Jailwala P.; Cam M.; Lang M.; Voeller D.; Reinhold W. C.; Rajapakse V.; Pommier Y.; Weigert R.; Linehan W. M.; Lipkowitz S. (2018) ONC201 kills breast cancer cells in vitro by targeting mitochondria. Oncotarget 9, 18454–18479. 10.18632/oncotarget.24862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan X.; Kho D.; Xu J.; Gajan A.; Wu K.; Wu G. S. (2017) ONC201 activates ER stress to inhibit the growth of triple-negative breast cancer cells. Oncotarget 8, 21626–21638. 10.18632/oncotarget.15451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizawa J.; Kojima K.; Chachad D.; Ruvolo P.; Ruvolo V.; Jacamo R. O.; Borthakur G.; Mu H.; Zeng Z.; Tabe Y.; Allen J. E.; Wang Z.; Ma W.; Lee H. C.; Orlowski R.; Sarbassov D.; Lorenzi P. L.; Huang X.; Neelapu S. S.; McDonnell T.; Miranda R. N.; Wang M.; Kantarjian H.; Konopleva M.; Davis R. E.; Andreeff M. (2016) ATF4 induction through an atypical integrated stress response to ONC201 triggers p53-independent apoptosis in hematological malignancies. Sci. Signaling 9, ra17. 10.1126/scisignal.aac4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y.; Zhou J.; Li Z.; Jiang Y.; Zhou Y. (2016) Small Molecular TRAIL Inducer ONC201 Induces Death in Lung Cancer Cells: A Preclinical Study. PLoS One 11, e0162133. 10.1371/journal.pone.0162133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner J.; Kline C. L.; Ralff M. D.; Lev A.; Lulla A.; Zhou L.; Olson G. L.; Nallaganchu B. R.; Benes C. H.; Allen J. E.; Prabhu V. V.; Stogniew M.; Oster W.; El-Deiry W. S. (2017) Preclinical evaluation of the imipridone family, analogs of clinical stage anti-cancer small molecule ONC201, reveals potent anti-cancer effects of ONC212. Cell Cycle 16, 1790–1799. 10.1080/15384101.2017.1325046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kline C. L. B.; Ralff M. D.; Lulla A. R.; Wagner J. M.; Abbosh P. H.; Dicker D. T.; Allen J. E.; El-Deiry W. S. (2018) Role of Dopamine Receptors in the Anticancer Activity of ONC201. Neoplasia 20, 80–91. 10.1016/j.neo.2017.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su N.; Kilberg M. S. (2008) C/EBP homology protein (CHOP) interacts with activating transcription factor 4 (ATF4) and negatively regulates the stress-dependent induction of the asparagine synthetase gene. J. Biol. Chem. 283, 35106–35117. 10.1074/jbc.M806874200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob N. T.; Lockner J. W.; Kravchenko V. V.; Janda K. D. (2014) Pharmacophore reassignment for induction of the immunosurveillance cytokine TRAIL. Angew. Chem., Int. Ed. 53, 6628–6631. 10.1002/anie.201402133. [DOI] [PubMed] [Google Scholar]
- Ma Z.; Gao G.; Fang K.; Sun H. (2019) Development of Novel Anticancer Agents with a Scaffold of Tetrahydropyrido[4,3-d]pyrimidine-2,4-dione. ACS Med. Chem. Lett. 10, 191–195. 10.1021/acsmedchemlett.8b00531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu R. L. Y.Imidazo-Pyrimidine Compounds, and Preparation Methods and Application Thereof. Chinese Patent 1048609482015, 100.
- Iwanowicz E. J.Protein Kinase Regulators. Patent Application WO2018/0319872018, 92.
- Iwanowicz E. J.Protein Kinase Regulators. Patent Application WO2018/0319902018, 81.
- Okumu D. O.; East M. P.; Levine M.; Herring L. E.; Zhang R.; Gilbert T. S. K.; Litchfield D. W.; Zhang Y.; Graves L. M. (2017) BIRC6 mediates imatinib resistance independently of Mcl-1. PLoS One 12, e0177871. 10.1371/journal.pone.0177871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevchenko A.; Wilm M.; Vorm O.; Mann M. (1996) Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal. Chem. 68, 850–858. 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- Thompson M. W.; Maurizi M. R. (1994) Activity and specificity of Escherichia coli ClpAP protease in cleaving model peptide substrates. J. Biol. Chem. 269, 18201–18208. [PubMed] [Google Scholar]
- Thompson M. W.; Singh S. K.; Maurizi M. R. (1994) Processive degradation of proteins by the ATP-dependent Clp protease from Escherichia coli. Requirement for the multiple array of active sites in ClpP but not ATP hydrolysis. J. Biol. Chem. 269, 18209–18215. [PubMed] [Google Scholar]
- Woo K. M.; Chung W. J.; Ha D. B.; Goldberg A. L.; Chung C. H. (1989) Protease Ti from Escherichia coli requires ATP hydrolysis for protein breakdown but not for hydrolysis of small peptides. J. Biol. Chem. 264, 2088–2091. [PubMed] [Google Scholar]
- Maurizi M. R.; Thompson M. W.; Singh S. K.; Kim S. H. (1994) Endopeptidase Clp: ATP-dependent Clp protease from Escherichia coli. Methods Enzymol. 244, 314–331. 10.1016/0076-6879(94)44025-5. [DOI] [PubMed] [Google Scholar]
- Lenarcic E. M.; Ziehr B.; De Leon G.; Mitchell D.; Moorman N. J. (2014) Differential role for host translation factors in host and viral protein synthesis during human cytomegalovirus infection. J. Virol 88, 1473–1483. 10.1128/JVI.02321-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziehr B.; Vincent H. A.; Moorman N. J. (2016) Human Cytomegalovirus pTRS1 and pIRS1 Antagonize Protein Kinase R To Facilitate Virus Replication. J. Virol. 90, 3839–3848. 10.1128/JVI.02714-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kline C. L.; Van den Heuvel A. P.; Allen J. E.; Prabhu V. V.; Dicker D. T.; El-Deiry W. S. (2016) ONC201 kills solid tumor cells by triggering an integrated stress response dependent on ATF4 activation by specific eIF2alpha kinases. Sci. Signaling 9, ra18. 10.1126/scisignal.aac4374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quiros P. M.; Prado M. A.; Zamboni N.; D’Amico D.; Williams R. W.; Finley D.; Gygi S. P.; Auwerx J. (2017) Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J. Cell Biol. 216, 2027–2045. 10.1083/jcb.201702058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni X.; Zhang X.; Hu C. H.; Langridge T.; Tarapore R. S.; Allen J. E.; Oster W.; Duvic M. (2017) ONC201 selectively induces apoptosis in cutaneous T-cell lymphoma cells via activating pro-apoptotic integrated stress response and inactivating JAK/STAT and NF-kappaB pathways. Oncotarget 8, 61761–61776. 10.18632/oncotarget.18688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl M.; Korotkov V. S.; Balogh D.; Kick L. M.; Gersch M.; Pahl A.; Kielkowski P.; Richter K.; Schneider S.; Sieber S. A. (2018) Selective Activation of Human Caseinolytic Protease P (ClpP). Angew. Chem., Int. Ed. 57, 14602–14607. 10.1002/anie.201808189. [DOI] [PubMed] [Google Scholar]
- Maurizi M. R.; Clark W. P.; Kim S. H.; Gottesman S. (1990) Clp P represents a unique family of serine proteases. J. Biol. Chem. 265, 12546–12552. [PubMed] [Google Scholar]
- Baker T. A.; Sauer R. T. (2012) ClpXP, an ATP-powered unfolding and protein-degradation machine. Biochim. Biophys. Acta, Mol. Cell Res. 1823, 15–28. 10.1016/j.bbamcr.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexopoulos J. A.; Guarne A.; Ortega J. (2012) ClpP: a structurally dynamic protease regulated by AAA+ proteins. J. Struct. Biol. 179, 202–210. 10.1016/j.jsb.2012.05.003. [DOI] [PubMed] [Google Scholar]
- Kang S. G.; Ortega J.; Singh S. K.; Wang N.; Huang N. N.; Steven A. C.; Maurizi M. R. (2002) Functional proteolytic complexes of the human mitochondrial ATP-dependent protease, hClpXP. J. Biol. Chem. 277, 21095–21102. 10.1074/jbc.M201642200. [DOI] [PubMed] [Google Scholar]
- Matsushima Y.; Kaguni L. S. (2012) Matrix proteases in mitochondrial DNA function. Biochim. Biophys. Acta, Gene Regul. Mech. 1819, 1080–1087. 10.1016/j.bbagrm.2011.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes C. M.; Petrova K.; Benedetti C.; Yang Y.; Ron D. (2007) ClpP mediates activation of a mitochondrial unfolded protein response in C. elegans. Dev. Cell 13, 467–480. 10.1016/j.devcel.2007.07.016. [DOI] [PubMed] [Google Scholar]
- Leung E.; Datti A.; Cossette M.; Goodreid J.; McCaw S. E.; Mah M.; Nakhamchik A.; Ogata K.; El Bakkouri M.; Cheng Y. Q.; Wodak S. J.; Eger B. T.; Pai E. F.; Liu J.; Gray-Owen S.; Batey R. A.; Houry W. A. (2011) Activators of cylindrical proteases as antimicrobials: identification and development of small molecule activators of ClpP protease. Chem. Biol. 18, 1167–1178. 10.1016/j.chembiol.2011.07.023. [DOI] [PubMed] [Google Scholar]
- Bhandari V.; Wong K. S.; Zhou J. L.; Mabanglo M. F.; Batey R. A.; Houry W. A. (2018) The Role of ClpP Protease in Bacterial Pathogenesis and Human Diseases. ACS Chem. Biol. 13, 1413–1425. 10.1021/acschembio.8b00124. [DOI] [PubMed] [Google Scholar]
- Wong K. S.; Mabanglo M. F.; Seraphim T. V.; Mollica A.; Mao Y. Q.; Rizzolo K.; Leung E.; Moutaoufik M. T.; Hoell L.; Phanse S.; Goodreid J.; Barbosa L. R. S.; Ramos C. H. I.; Babu M.; Mennella V.; Batey R. A.; Schimmer A. D.; Houry W. A. (2018) Acyldepsipeptide Analogs Dysregulate Human Mitochondrial ClpP Protease Activity and Cause Apoptotic Cell Death. Cell Chem. Biol. 25, 1017–1030 e9. 10.1016/j.chembiol.2018.05.014. [DOI] [PubMed] [Google Scholar]
- Kang S. G.; Dimitrova M. N.; Ortega J.; Ginsburg A.; Maurizi M. R. (2005) Human mitochondrial ClpP is a stable heptamer that assembles into a tetradecamer in the presence of ClpX. J. Biol. Chem. 280, 35424–35432. 10.1074/jbc.M507240200. [DOI] [PubMed] [Google Scholar]
- Li D. H.; Chung Y. S.; Gloyd M.; Joseph E.; Ghirlando R.; Wright G. D.; Cheng Y. Q.; Maurizi M. R.; Guarne A.; Ortega J. (2010) Acyldepsipeptide antibiotics induce the formation of a structured axial channel in ClpP: A model for the ClpX/ClpA-bound state of ClpP. Chem. Biol. 17, 959–69. 10.1016/j.chembiol.2010.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.