

Abstract
Inflammation triggered by microbial lipopolysaccharide (LPS) through Toll‐like receptor (TLR) 4 in the presence of interferon (IFN)‐γ induces cytokine secretion in dendritic cells (DCs) tightly regulated by a defined differentiation program. This DC differentiation is characterized not only by a dynamic immune activating but also by tolerance‐inducing phenotype associated with down‐modulation of cytokines previously considered to be irreversible. CD40L on activated T cells further modifies DC differentiation. Using DNA micro‐arrays, we showed down‐regulated mRNA levels of TLR signalling molecules, whereas CD40/CD40L signalling molecules were up‐regulated at a time when LPS/IFN‐γ‐activated DCs had ceased cytokine expression. Accordingly, we demonstrated that CD40/CD40L but not TLR4 or TLR3 signalling mediated by LPS or poly (cytidylic‐inosinic) acid (poly I:C) and dsRNA re‐established the capacity for secreting interleukin (IL)‐12 in primarily LPS/IFN‐γ‐activated DCs, which have exhausted their potential for cytokine secretion. The resulting TH1 polarizing DC phenotype – which lacked accompanying secretion of the crucial immune suppressive factor IL‐10 – maintained the potential for activation of cytotoxic T lymphocytes (CTLs). We therefore conclude that immune modulation is restricted to a secondary T‐cell‐mediated stimulus at an exhausted DC state, which prevents an immune tolerant DC phenotype. These findings impact on the rational design of TLR‐activated DC‐based cancer vaccines for the induction of anti‐tumoural CTL responses.
Keywords: human dendritic cells, LPS, CD40L, IL‐12, IL‐10, TH1 polarization, cytokine exhaustion, tumour immunity
Introduction
Dendritic cells (DCs) induce, co‐ordinate and regulate adaptive immune responses [1]. Upon encountering pathogen‐associated molecular patterns (PAMP), which are recognized by pattern recognition receptors, the most prominent group of which are the Toll‐like receptors (TLR) or endogenous CD40L, DCs undergo a process of maturation [2, 3].
DC activation by TLR4 engagement with lipopolysaccharide (LPS) is commonly viewed as a rather static and irreversible terminal differentiation process. Functional capabilities are ascribed to distinct developmental phases, conventionally referred to as (i) immature DCs, which maintain tolerance against self‐antigens, or (ii) immune stimulatory mature DCs. Accumulating evidence now suggests the existence of additional functional developmental stages of DCs. Most importantly, cross‐presentation of exogenous antigens to cytotoxic T lymphocytes (CTLs) depends on a ‘semi‐mature’ status of DCs [4, 5, 6]. These DCs are characterized by active IL‐12 secretion, which is exhausted in fully matured mDCs [7]. Another only recently appreciated developmental stage is the tolerance‐inducing capacity of mDCs. Particularly, the up‐regulation of indoleamine 2,3 dioxygenase (IDO) is associated with an immune suppressive DC phenotype [8, 9].
Ligation of TLRs activates the p50/RelA NFκB subunits by the recruitment of MyD88 and the MyD88‐adapter‐like protein (MAL). Optionally, MyD88 signalling may be bypassed via TIR‐domain‐containing adapter‐inducing interferon‐β (TRIF) and the TRIF‐related adaptor molecule (TRAM) in DCs activated by the engagement of the TLR4‐MD2‐CD14 receptor complex with LPS. The TLR3 signalling route is restricted to the recruitment of TRIF for p50/RelA activation by binding of poly (cytidylic‐inosinic) acid (poly I:C) or double stranded (ds) RNA [2, 10, 11]. CD40/CD40L interaction induces the p52/RelB NFκB complex via TNF Receptor Associated Factor (TRAF) molecules [10]. The interplay of the MyD88‐dependent and ‐independent pathways in TLR4‐activated DCs triggers a specific program of target gene expression characterized by a tight regulation of cytokine secretion [7, 12, 13]. TNF‐α that signals in an autocrine fashion to DCs [14], and exogenous IFN‐γ delivered by innate immune cells, further define DC maturation and the release of immune regulatory cytokines, such as interleukin (IL)‐12 or IL‐10 [7, 15].
IL‐12 is a key cytokine supporting killer cell responses mediated by CTL through the polarization of a type 1 phenotype in T helper (TH1) cells [1]. Thereby CTL activity critically depends on antigen presentation via MHC class I molecules, a route primarily restricted to endogenously provided proteins [16]. Interaction of CD40 on DCs with CD40L on activated Th cells was identified as the critical licensing signal for cross‐presentation of exogenous antigen from DCs to CTLs, thus initiating immune responses against antigens from infected body cells or tumour cells [17, 18, 19, 20]. In contrast, IL‐10 production by phenotypically mature DCs is critical for the induction of tolerance by CD4+ T regulatory 1‐like cells [21].
In this study, we demonstrate at a molecular and cellular level that fully matured DCs that have ceased IL‐12 secretion retain the capacity to respond to CD40 ligation for up to 3 days after short exposure to LPS or poly I:C, in the presence of IFN‐γ that mimics a pro‐inflammatory situation at an infection site. As opposed to secondary TLR ligation, CD40/CD40L signalling had the potential to trigger a secondary burst of IL‐12 secretion from cytokine‐exhausted DCs. At a molecular level, endotoxin tolerance versus CD40L responsiveness correlated with a down‐modulation of molecules involved in TLR signalling and the up‐regulation of the CD40 signalling cascade. TLR4‐activated DCs, which have ceased cytokine expression engineered to express CD40L, showed a TH1 polarizing phenotype indicated by the secretion of IL‐12, but no IL‐10. This DC phenotype supported the proliferation of CD8+ CTLs, which expressed granzyme B, indicating killer cell functions. The prolonged responsiveness of DCs to CD40/CD40L signalling, enabling them for maintained TH1 immune polarization, suggests that fully matured, TLR‐activated DCs preserve some developmental flexibility. This provides evidence against a terminal differentiation state of DCs encountering TLR‐transmitted danger signals.
Materials and methods
Primary cells and cell lines
Peripheral blood mononuclear cells (PBMCs) were isolated by leucocyte aphaeresis (Amicus, Baxter, Vienna, Austria) from healthy adult volunteers at the leucocyte aphaeresis facility of the St. Anna Children’s Hospital. Further monocytes were enriched from PBMCs by using an Elutra cell separator (Gambro BCT, Lakewood, CO, USA) as previously described [22].
The human neuroblastoma cell line SJ‐NB‐7 was a gift from Dr. T. Look (Memphis, TN, USA) and has been described previously [23]. SJ‐NB‐7 cells that stably express CD40L (CD40L‐SJ‐NB‐7) were generated by lenti‐viral transduction using the pLenti6/V5‐D‐TOPO vector system (Invitrogen, Carlsbad, CA, USA) followed by G418 selection. Before co‐cultivation with DCs, SJ‐NB‐7 cells and transgenic CD40L‐SJ‐NB‐7 cells were gamma‐irradiated with 6000 rad.
Generation of mature DCs
Monocyte‐derived DCs were generated in CellGro DC medium (CellGenix, Freiburg, Germany) supplemented with 1000 U/ml recombinant human GM‐CSF and 300 U/ml recombinant human IL‐4 (both from CellGenix, Freiburg, Germany) as previously described [22]. Primary stimulation was carried out on day 6 by supplementing the DC culture with either a combination of (i) 30 ng/ml lipopolysaccharide (LPS, E. coli strain O111:B4, Calbiochem, San Diego, CA, USA) and 1000 U/ml IFN‐γ (Imukin, Boehringer Ingelheim, Austria), or (ii) 25 μg/ml poly I:C (Sigma, St. Louis, MO, USA), 3000 U/ml IFN‐α (Strathmann Biotech, Munich, Germany), 25,000 U/ml IL1‐β, 100,000 U/ml TNF‐α (both from Peprotech, Rocky Hill, NJ, USA) and 1000 U/ml IFN‐γ for 6 hrs. The dose for LPS stimulation induces maximal cytokine release, which was previously titrated using DCs manufactured from a series of healthy donors [22]. After washing, the DCs were kept in culture up to 48 hrs without stimulation until secondary maturation or phenotyping. For secondary maturation, DCs were additionally stimulated by co‐cultivation of DCs with CD40L‐SJ‐NB‐7 cells in a ratio of 2:1 in the presence of 1000 U/ml IFN‐γ without removing the stimulus over the culture period. Wild‐type SJ‐NB‐7 cells plus IFN‐γ served as a control.
Differential gene expression profiling of DCs
Monocyte‐derived DCs were stimulated for 6 hrs with LPS/IFN‐γ at concentrations mentioned above. After 6 hrs, DCs were washed followed by RNA isolation using the RNeasy kit (Qiagen, Hilden, Germany) directly after 6 hrs or after 12, 24 or 48 hrs of continuous culture. Transcription values were analysed by the RZPD (German Resource Centre for Genome Research, Berlin) using the Affymetrix DNA micro‐array platform (U133 plus 2.0). DNA array data from different maturation‐time experiments and controls that were kept in culture for the same time without stimulation were normalized according to robust multichip average (RMA) normalization and compared using the CarmaWeb (Comprehensive R‐based micro‐array Analysis, Bioinformatics Graz and the Tyrolean Cancer Research Institute, Austria). Overexpression of selected genes were analysed at a linear scale over the time. For heat plotting of the TLR and CD40 pathway over the time, we used the pathway studio 5.0 software (Ariadne Genomics, Rockville, MD, USA). Single chip analyses have been submitted to Gene Expression Omnibus (GEO, Accession number: GSE11327).
Generation of transgenic DCs
Lenti‐viral particles were generated using the ViraPower™ lenti‐viral expression system using the pLenti6/V5‐D‐TOPO vector (both from Invitrogen) containing CD40L or GFP. Virus particles were 100× concentrated using an OTD Combi ultracentrifuge (Sorvall, New Castle, DE, USA), re‐suspended in CellGro DC medium and stored at –80°C until further use. Monocyte‐derived DCs were stimulated with LPS/IFN‐γ either for 6 hrs to generate IL‐12 secreting DCs or for 6 hrs followed by a continuous culture up to 48 hrs without stimulation in order to generate cytokine‐exhausted DCs. 106 cells of both, 6 and 48 hrs matured DCs, were then transduced with 250 μl 100× concentrated lenti‐virus in combination with 6 μg/ml polybrene (Sigma‐Aldrich) in culture medium supplemented with IL‐4, GM‐CSF and IFN‐γ in concentrations mentioned above.
Quantification of IL‐10, IL‐12 and kynurenine
IL‐12 and IL‐10 was analysed in the supernatants 48 hrs after primary or secondary stimulation or lenti‐viral transduction following a previously described ELISA protocol [6] using the following antibodies and recombinant proteins: anti‐IL‐12 p70 and anti‐IL‐10 capture antibodies, biotinylated anti‐IL‐12 p40/p70 and anti‐IL‐10 detection antibodies, recombinant IL‐12 and IL‐10 (all from BD Pharmingen, San Diego, CA, USA). Further supernatants of DCs were analysed for kynurenine by HPLC as previously described [24].
Flow cytometry of DC membrane molecules
The immune phenotypic maturation status of DCs was determined after 48 hrs of primary or secondary stimulation or lenti‐viral transduction using the following antibodies: anti‐CD80‐PE (Immunotech, Beckman Coulter, Fullerton, CA, USA), anti‐CD86‐ Allophycocyanin (APC), anti‐CD1a‐FITC, anti‐CD14‐Peridinin chlorophyll protein (PerCP)‐Cy5.5, anti‐CD83‐APC and anti‐CD45‐PerCP (all from BD Pharmingen), anti‐MHC I‐PE, and anti‐MHC II‐FITC (both from Dako Cytomation, Carpinteria, CA, USA). The expression density of membrane molecules involved in the TLR4 and CD40 signalling pathways were analysed on 48 hrs LPS/IFN‐γ‐matured DCs using anti‐TLR4‐PE (Abcam, Cambridge, UK), anti‐CD14‐PerCP‐Cy5.5 (BD Pharmingen) and anti‐CD40‐FITC (BD Pharmingen). Ectopic CD40L (and GFP) expression was measured after 48 hrs of viral transduction using anti‐CD40L‐PE (BD Pharmingen). Cells were analysed using a FACScalibur flow cytometer (Becton Dickinson, Mountain View, CA, USA). Data analysis was performed using CellQuest software (Becton Dickinson). The appropriate isotype control antibodies were included in the analysis.
Allogeneic mixed leucocyte reaction
DCs were primarily maturated with LPS, a poly I:C cocktail, irradiated CD40L‐SJ‐NB‐7 cells or SJ‐NB‐7 control cells in the presence of IFN‐γ at the concentrations given above for 6 hrs and continuously cultivated up to 48 hrs without stimulation in order to generate cytokine‐exhausted DCs. Those 48 hrs matured DCs were further co‐cultivated with irradiated CD40L‐SJ‐NB‐7 or SJ‐NB‐7 control cells also in the presence of IFN‐γ. Differently stimulated DCs were washed and co‐cultivated with allogeneic responder lymphocytes. DCs (10,000, 2000 or 400) were placed in triplicates (100 μl per well) on a 96‐well round bottom plate along with 105 responder cells in 100 μl AIM‐V medium (Invitrogen) supplemented with 2% human plasma (Octapharma, Vienna, Austria). For a positive reference, 105 responder cells were stimulated in 100 μl medium with Staphylococcal enterotoxin A/B (Toxin Technologies Inc., Sarasota, FL, USA) at 100 ng/ml final concentration. On day 4 of the co‐culture, cells were incubated for another 18 hrs with 1 μCi of tritium thymidine solution (NEN Life Science Products, Boston, MA, USA). Finally, the cells were harvested with a Skatron harvesting device (Skatron, Lier, Norway) and the incorporated tritium thymidine was counted on a Trilux β‐plate reader (Wallac Oy, Turku, Finland). In another MLR system, DCs were stimulated with LPS/IFN‐γ at concentrations given above for 6 hrs or for 6 hrs with additional cultivation up to 48 hrs without stimulation. Both, 6 and 48 hrs matured DCs, were then treated with CD40L or GFP containing lenti‐viral particles. Six hours after viral treatment, an alloMLR was assessed using responder T cells that were isolated from PBMCs by magnetic bead sorting using a pan T‐cell isolation kit (Miltenyi Biotec, Bergisch Gladbach, Germany) and labelled with 5 μM CFSE (Invitrogen). As controls we used 6 or 48 hrs LPS/IFN‐γ matured DCs without lenti‐viral treatment. For the MLR, 105 responder cells together with stimulating DCs at a 1:3, 1:9 or 1:27 DC:T cell ratio were cultivated in 200 μl AIM‐V/ 2% human plasma on a 96‐well round bottom plate. On day 6, the T cells were harvested and fixed with cytofix/cytoperm (BD Pharmigen). CD8+ T cells were analysed for CFSE dilution, and CD25 or granzyme B expression with a FACS LSRII flow cytometer (Becton Dickinson) using the following antibodies: anti‐CD3‐PE‐Texas‐RED, anti‐CD8‐APC‐Cy7 BD, anti‐granzyme B‐Alexa Fluor 700 (all from BD Pharmingen) and anti‐CD25‐Alexa Fluor 647 (Serotec, Oxford, UK).
Results
Molecules of the CD40 signalling cascade are strongly induced in TLR4‐activated cytokine‐exhausted DCs
In order to mimic pro‐inflammation at an infection site followed by DC migration into the lymph node, we briefly activated monocyte‐derived DCs for 6 hrs with LPS/IFN‐γ and analysed differential mRNA expression levels of key molecules in the TLR and CD40 signalling cascade over a time course of 48 hrs. Following an early induction of TLR4 after 6 hrs, expression of the whole TLR4‐MD2‐CD14 receptor complex together with MyD88‐MAL adaptor molecules was strongly decreased after 48 hrs (Fig. 1A and B). This correlated with increased mRNA expression levels of cytokines associated with inflammation, immune activation and regulation, reaching peak expression during the first 24 hrs followed by a strong decline at 48 hrs after DC activation (Fig. 1B). The TLR3‐TRIF cascade and the TRIF‐TRAM adaptor complex, which is engaged upon TLR4 signalling independently of MyD88, were slightly induced over 48 hrs after LPS/IFN‐γ stimulation. This was also the case for the RelA/NF‐κB subunit that is essential for TLR signalling. In contrast, CD40, whose up‐regulated expression on DCs is a well‐established response to the encounter of a maturation signal, and the TRAF adaptor molecules were highly up‐regulated 48 hrs after TLR4 induction. In addition, the RelB/NF‐κB subunit engaged upon CD40 signalling was also induced, reaching maximum mRNA expression after 48 hrs (Fig. 1A and B).
Figure 1.

Molecules of the CD40 signalling cascade are highly induced 48 hrs after TLR4 activation at the time of cytokine exhaustion. (A) Immature DCs were stimulated with LPS/IFN‐γ for 6 hrs and compared to un‐stimulated DCs by DNA micro‐array analysis. In addition, 12, 24 and 48 hrs matured DCs receiving a 6‐hr LPS/IFN‐γ stimulus were also compared to un‐stimulated DCs. Key molecules of the TLR and CD40 signalling cascade are analysed by heat plotting over the time. Up‐regulation is indicated by red and down‐regulation by different green colouring. (B) Detailed illustration of differentially regulated genes encoding secreted proteins or proteins involved in TLR or CD40/CD40L signalling. mRNA fold expression is depicted over 48 hrs after LPS/IFN‐γ activation at a linear scale. *Down‐regulation compared to immature DCs. (C) Quality control by analysing the activation status of LPS/IFN‐γ‐treated DCs in (A). IL‐10, IL‐12 and the IDO metabolite kynurenine were measured as depicted over 48 hrs of maturation. These data were reproducible using two different donors. (D) Expression density of TLR4, CD14 and CD40 after 48 hrs of LPS/IFN‐γ activation (black histogram) compared to un‐stimulated immature DCs (dashed histogram) and isotype control stained immature DCs (filled black histogram). One out of two independent experiments using two different donors is depicted.
The activation status of LPS/IFN‐γ‐activated DCs used for transcriptional profiling was further analysed by IL‐10 or IL‐12 secretion over the time also indicating cytokine exhaustion after 48 hrs by reaching a plateau of accumulating cytokines in the supernatants (Fig. 1C). Effective activation of DCs was additionally shown by the increase of kynurenine, the immune suppressive metabolite of IDO, which was highly induced upon LPS/IFN‐γ stimulation (data not shown).
The above results indicated that 48 hrs after LPS/IFN‐γ stimulation, at which time the DCs have ceased cytokine production, DCs maintain expression of molecules critically needed for responding to CD40 ligation. In contrast, molecules of the TLR signalling pathway were down‐modulated after 48 hrs. We observed a five‐fold decreased expression density of the surface molecule TLR4 and undetectable levels of the CD14 co‐receptor, whereas CD40 expression density, as expected, was increased after 48 hrs of LPS/IFN‐γ maturation (Fig. 1D).
CD40/CD40L signalling enables IL‐12 secretion in TLR‐activated, cytokine‐exhausted DCs
Having demonstrated that molecules of the CD40 signalling cascade are induced in cytokine‐exhausted DCs, we analysed IL‐12 secretion from 48 hrs primarily LPS/IFN‐γ matured DCs that received a secondary CD40L stimulus. After primary engagement of TLR4 (with LPS) or TLR3 (with poly I:C), both in the presence of IFN‐γ, DCs released considerable amounts of IL‐12 (Fig. 2A). Independent of the initial stimulus, exposure of 48 hrs LPS/IFN‐γ or poly I:C/IFN‐γ pre‐matured DCs to CD40L‐SJ‐NB‐7/IFN‐γ had the capacity to trigger a secondary burst of IL‐12 secretion. The CD40L‐SJ‐NB‐7 cells homogenously expressed CD40L two log steps above background (data not shown). Wild‐type SJ‐NB‐7 cells plus IFN‐γ did not trigger IL‐12 release; similarly, a TLR‐mediated maturation stimulus did not have the capacity to induce secondary IL‐12 secretion. We observed a similar IL‐12 secretion profile in DCs re‐stimulated 24 or 72 hrs after primary maturation LPS/IFN‐γ or poly I:C/IFN‐γ when secondarily exposed to CD40L‐SJ‐NB‐7 in the presence of IFN‐γ (data not shown).
Figure 2.
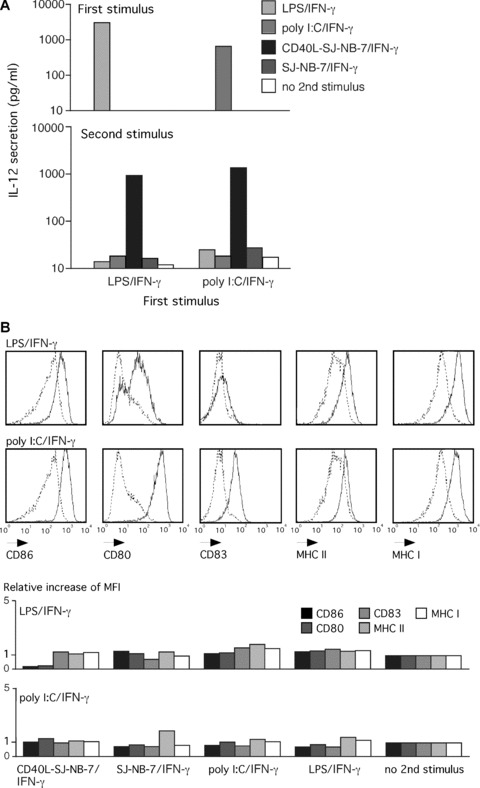
CD40L re‐induces IL‐12 secretion in TLR‐activated DCs that have exhausted their capacity for cytokine secretion. (A) Upper panel: IL‐12 secreted from DCs after a 6‐hr exposure to the indicated stimuli and the adequate co‐factors (see main text). Accumulation of IL‐12 after 48 hrs of activation was measured. Lower panel: IL‐12 secretion after a second round of stimulation analysed from 48 hrs pre‐matured DCs in (A) re‐stimulated for 48 hrs with the indicated stimuli (colour code). One representative experiment out of five is given. (B) Immune phenotype of DCs analysed in (A) measured by flow cytometry after 48 hrs of primary maturation with the indicated maturation stimuli. LPS/IFN‐γ and poly I:C/IFN‐γ stimulated DCs (black histogram) are compared to immature DCs (dashed histogram). (C) Relative increase of the expression density (mean fluorescence intensity, MFI) of depicted surface maturation markers on 48 hrs pre‐matured DCs analysed in (A) stimulated with the indicated stimuli (top left) followed by a 48‐hr re‐stimulation with the stimuli given at the bottom of the graph in comparison to primarily stimulated DCs.
Furthermore, DCs primarily stimulated with LPS/IFN‐γ or poly I:C/IFN‐γ showed a typical mature membrane molecule expression profile characterized by an up‐regulated expression density of the membrane molecules CD86, CD80, CD83, MHC class I, and II after 48 hrs (Fig. 2B). No significant further increase in the expression density of the already highly induced maturation markers could be triggered upon re‐stimulation with CD40L‐SJ‐NB‐7/IFN‐γ, LPS/IFN‐γ or poly I:C/IFN‐γ.
CD40 ligation maintains the lymphocyte stimulatory capacity of cytokine‐exhausted TLR‐activated DCs
Because 48 hrs cytokine‐exhausted DCs have the ability to maintain their responsiveness to CD40/CD40L signalling, we assessed the immune stimulatory capacity of 48 hrs pre‐matured DCs re‐stimulated with CD40L in a co‐culture with allogeneic lymphocytes (Fig. 3). We compared lymphocytes stimulated with cytokine‐exhausted DCs 48 hrs after primary activation that received a secondary CD40L stimulus delivered by CD40L‐SJ‐NB‐7/IFN‐γ compared to SJ‐NB‐7/IFN‐γ control stimulation. A secondary exposure of 48 hrs LPS/IFN‐γ or poly I:C/IFN‐γ matured DCs to CD40L‐SJ‐NB‐7 cells in the presence of IFN‐γ led to a high capacity for lymphocyte activation. In contrast, the immune stimulatory capacity of 48 hrs matured DCs exposed to SJ‐NB‐7 control cells was poor. Successful primary maturation shown by IL‐12 release and immune phenotyping of surface markers of DCs used for the MLR was analysed as shown in Fig. 2 (data not shown).
Figure 3.
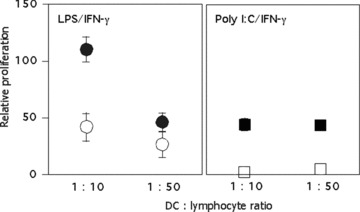
CD40 ligation induces strong stimulatory capacity in cytokine‐exhausted TLR‐activated DCs. Allogeneic MLR using DCs 48 hrs after receiving a primary maturation stimulus as indicated at the top of each panel (LPS/IFN‐γ; circles, poly I:C/IFN‐γ; squares) followed by a 6‐hr co‐cultivation with CD40L‐SJ‐NB‐7 cells (black symbols) or control SJ‐NB‐7 cells (white symbols), both in the presence of IFN‐γ. Proliferation was assessed by tritium thymidine incorporation at the indicated DC:lymphocyte ratios. Experiments from three different donors are combined using an internal SEA/SEB proliferation control defined as 100; unstimulated T cells were normalized to 0; the proliferation ± S.D. relative to the SEA/SEB (100) and un‐stimulated (0) controls is shown.
CD40L ectopically expressed on cytokine‐exhausted TLR4‐activated DCs sustains stimulatory potential for CTLs
In our allogeneic test system described above, we delivered the CD40L stimulus using irradiated transgenic SJ‐NB‐7 cells, which might interfere with the DC‐T cell interaction. In order to avoid the presence of a cell line, reduce the complexity of the system and introduce a concept that might be used in a clinical setting, we engineered DCs 6 or 48 hrs after LPS/IFN‐γ activation to express CD40L using a recombinant lenti‐virus (lv‐CD40L) thus continuously exposing DCs to CD40/CD40L signalling. Lenti‐viral GFP transfer was used in control DCs (Fig. 4A). Six hours LPS/IFN‐γ matured DCs showed increased IL‐12 and IL‐10 secretion upon CD40L expression (Fig. 4C). The lv‐GFP control DCs also enhanced secretion of IL‐12 and IL‐10 due to synergistic effects of TLR4 (LPS) and TLR3 (lenti‐viral dsRNA) engagement. In contrast, IL‐12 secretion was restored upon lv‐CD40L engineering of DCs 48 hrs after LPS/IFN‐γ activation, but not upon lv‐GFP. Of note, lv‐CD40L and lv‐GFP did not trigger IL‐10 release from any cytokine‐exhausted DCs. Engineering with lenti‐viral constructs did not alter the expression of the maturation markers CD86, CD80 and CD83 (Fig. 4B).
Figure 4.
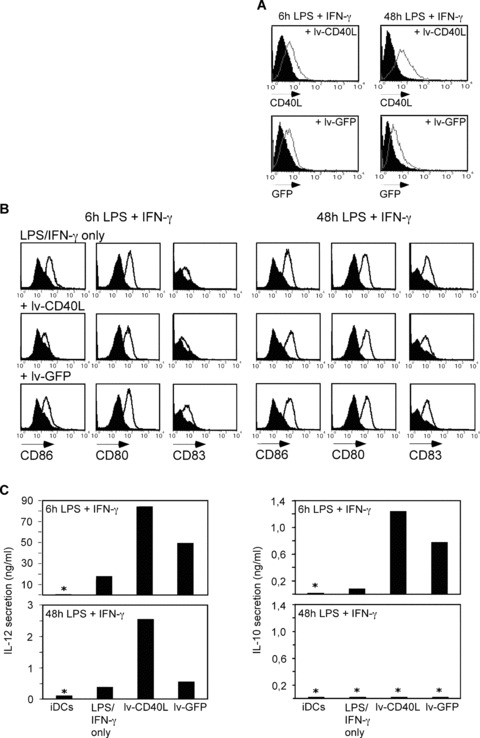
CD40L ectopically expressed on cytokine‐exhausted TLR4‐activated DCs re‐induces IL‐12 secretion. (A) CD40L or GFP expression after lenti‐viral gene transfer into 6 or 48 hrs pre‐matured DCs that received a 6‐hr LPS/IFN‐γ stimulus. Expression density was measured by flow cytometry 48 hrs after transduction with the lenti‐viral constructs lv‐CD40L or lv‐GFP (grey histograms) compared to untreated DCs (filled black histogram). (B) Immune phenotype of DCs described in (A) measured 48 hrs after viral transduction. Shown is one representative out of five experiments with three different donors. (C) IL‐12 and IL‐10 secretion from DCs described in (A) measured 48 hrs after lenti‐viral transduction. *Below the detection limit of the ELISA.
We further analysed the potential of CD40L expressing TLR4 pre‐activated DCs to stimulate potentially cytolytic CD8+ T lymphocytes in an alloMLR (Fig. 5). Expression of CD40L on 48 hrs LPS/IFN‐γ matured DCs further increase the expression density of CD25 on CD8+ T lymphocytes. There was no difference in CD25 expression on CD4+ T cells (data not shown). Granzyme B expression in CD8+ T lymphocytes was also induced. GFP expressing control DCs did not enhance T‐cell stimulation compared to un‐treated 48 hrs LPS/IFN‐γ matured DCs. In contrast, 6 hrs LPS/IFN‐γ‐activated DCs did not trigger CD25 expression on T lymphocytes. Only a slight induction of granzyme B was observed when T lymphocytes were stimulated with lv‐CD40L expressing DCs compared to lv‐GFP or untreated 6 hrs LPS/IFN‐γ matured DCs. These data provide further evidence for the strong capacity of CD40L‐engineered DCs to maintain the capacity for triggering cytolytic immune responses 48 hrs after LPS/IFN‐γ treatment when DCs enter a phase of cytokine exhaustion.
Figure 5.
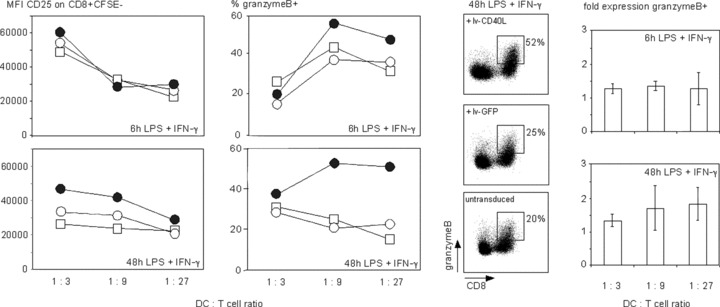
CD40L ectopically expressed on cytokine‐exhausted TLR4‐activated DCs triggers activation of CD8+ T lymphocytes. Allogeneic MLR using DCs that were matured for 6 or 48 hrs upon receiving a 6‐hr LPS/IFN‐γ stimulus as indicated (bottom right of each panel) followed by a 6‐hr treatment with the lenti‐virus lv‐CD40L (black circles) or lv‐GFP (white circles) in comparison to only primary stimulated DCs (squares). T‐cell activation was measured by the mean fluorescence intensity (MFI) of CD25 expressed on proliferating CFSE negative CD8+ T cells. Granzyme B expressing cells are given as the percentage in CD8+ T cells. The dot plots show CD8+ granzyme B+ T cells at a DC:T cell ratio = 1:9. Shown is one representative experiment of three using three different donors. The bar diagram combines the data of three different donors, which shows fold expression ± S.D. of granzyme B expressing CD8+ T cells stimulated with lv‐CD40L DCs normalized to lv‐GFP DCs; 6 hrs LPS/IFN‐γ pre‐matured DCs; P= 0,01; 48 hrs LPS/IFN‐γ pre‐matured DCs, P= 0,004. CD40L or GFP expression as well as the phenotype of the DCs used for the depicted alloMLRs is shown in Fig. 4.
Discussion
It is generally assumed that DCs follow a pre‐determined differentiation program upon encountering danger signals [7, 12, 13]. Prior to maturation, DCs contribute to the maintenance of tolerance against auto‐antigens, which is their default function. Exposure to a PAMP, inflammatory cytokines, or the T‐cell‐mediated CD40L signal causes DCs to acquire a strong immune stimulatory capacity. Twenty‐four hours after initiation of maturation, DCs supposedly start assuming an immune suppressive phenotype that is thought to be accompanied by the exhaustion of the DC’s capacity for cytokine secretion, most critically IL‐12 [7, 8, 9]. In contrast to this presumption, we hypothesized that in the final immune suppressive differentiation phase DCs might retain some developmental plasticity, which would allow them to respond to secondary immune regulatory signals. This was confirmed by demonstrating that DCs, after encountering a TLR‐mediated stimulus, could be prevented from assuming an immune suppressive phenotype by secondary CD40/DC40L signalling at a time of cytokine exhaustion. In contrast, secondary TLR‐mediated signals did not have the capacity for supporting an immune response.
The developmental process of DCs starts with monocytes entering peripheral tissues [25]. There the monocytes differentiate into what is commonly referred to as immature DCs. The main function of iDCs is to maintain peripheral tolerance and to act as sentinels for microbial invasion. Phenotypically, they are characterized by a low expression density of T‐lymphocyte co‐stimulatory molecules such as the members of the B7 family CD80 and CD86, lack of cytokine secretion and an overall poor capacity for T lymphocyte stimulation. Encountering of TLR ligands such as endotoxin (LPS), which is a TLR4 ligand, initiates DC differentiation providing the DCs with an enhanced ability for T‐cell activation by up‐regulated expression of co‐stimulatory membrane molecules and cytokine secretion. In vivo DC maturation and T‐cell activation is initiated within a narrow time window. We induced DC maturation by a brief exposure to LPS plus IFN‐γ for 6 hrs, which is sufficient to initiate the entire differentiation program necessary for stimulating T lymphocytes [22]. Employing such an experimental setting, we devised an in vivo 2‐step maturation hypothesis. TLR‐activated DCs are expected to withdraw from peripheral exposure to microbial danger signals (LPS) due to migration into secondary lymphoid organs. In the lymph node, a second differentiation step controlled by CD40L signalling from activated T cells, which is crucial for antigen specific CTL activation, may take place [18, 19, 20, 26]. In all our experiments with TLR‐activated DCs, we added IFN‐γ, which synergistically strengthened the TLR‐mediated DC differentiation process by an increase of cytokine secretion without kinetic changes [7]. DNA chip array analyses of IFN‐γ stimulated DCs also demonstrated that interferon regulatory factor (IRF) ‐1, IRF3 and IRF7, which assume an important role in TLR signalling, are induced (data not shown) [11].
The release of inflammatory and immune regulatory cytokines, most importantly IL‐12 and IL‐10, confers the DC’s ability for regulating T lymphocyte activity. However, within 2 days after encountering a PAMP, the aptitude of DCs for cytokine secretion is terminated (Fig. 1), a status referred to as DC ‘exhaustion’[7]. During this phase of the DC differentiation process, immune suppressive factors are switched on [8, 9, 27]. Consequentially, it appears reasonable to assume that DC differentiation does not necessarily branch off into subsets but rather, during the various phases of their life cycle, DCs pass through several phenotypic stages, suggesting a dynamic differentiation process. Indeed, we observed an immediate induction of IL‐12 followed by a delay of 6 hrs by IL‐10 mRNA expression (Fig. 1), which correlated with the secretion kinetic of the cytokines. In contrast, the formation of kynurenines, the immune suppressive metabolites of IDO‐mediated tryptophan degradation, was delayed. Together this suggests an early immune activating followed by an immune suppressive phenotype of TLR4‐activated DCs (Fig. 1C). Further evidence supporting this hypothesis comes from the observation of late production of IFN‐γ (Fig. 1), which is assumed to enhance IDO activity via an autocrine feedback loop [28].
DCs upon encountering an initial PAMP‐associated danger signal in an inflammatory area still need to remain susceptible to modulatory signals when reaching the lymph node. Beside the observation that macrophages become resistant to LPS after TLR4 engagement [12, 29], which may be essential to prevent autoimmune responses, it appears reasonable to assume that PAMPs, except under septic conditions, are not present in the lymph node. Thus, retained responsiveness to TLR signalling was not expected. In line with this expectation, we observed that the expression of molecules involved in the TLR4 signalling pathway in DCs is gradually down‐modulated upon encountering an LPS signal in the presence of pro‐inflammatory IFN‐γ. Thus, the only signal with the capacity to modulate DC function that is likely to be present in the lymph node would be derived from T lymphocytes, which points towards CD40L as a major candidate. Similar to TLR engagement, CD40L signalling from activated T cells causes DC activation characterized by the release of IL‐12, which is, like TLR‐mediated DC differentiation, strongly dependent on IFN‐γ co‐signalling [30]. Indeed, expression of the entire CD40 signalling cascade was up‐regulated after 2 days of LPS/IFN‐γ activation. This was confirmed in experiments that demonstrated that a CD40L‐mediated signal in the presence of IFN‐γ had the potential to revert DC exhaustion after 48 hrs of TLR‐mediated maturation resulting in renewed IL‐12 secretion and strong T lymphocyte stimulatory activity. This observation depended on membrane‐associated CD40L expressed from SJ‐NB‐7 cells or ectopically expressed from DCs by lenti‐viral gene transfer. Soluble recombinant CD40L in our hands does not have the potential to trigger the release of considerable amounts of IL‐12 from human DCs [31]. Re‐induction of IL‐12 secretion in TLR pre‐activated DCs was also rapidly down‐modulated when the number of CD40L expressing SJ‐NB‐7 cells was reduced from a 1:2 to a 1:5 SJ‐NB‐7:DC ratio. This may explain the observation of Langenkamp et al. that DCs did not respond to secondary CD40/CD40L signalling in DCs that had stopped producing cytokines, as the ration of DCs to CD40L transgenic cells was lower compared to our experiments. Furthermore, IL‐12 release strongly depended on the presence of IFN‐γ (data not shown) [7]. An issue that needs to be taken into consideration when stimulating DCs with a transgenic cell line is that feedback stimuli other than CD40L derived from the transgenic cells might synergize with CD40/CD40L signalling. In order to attribute the enhanced stimulatory capacity of DCs to CD40 ligation alone, we established a test system based on lenti‐viral gene transfer of CD40L into DCs for delivering the CD40L signal via a crosstalk of CD40L expressing DCs. Of particular interest, CD40 ligation induces a second burst of IL‐12 but not IL‐10 release indicating that such DCs are re‐enabled for TH1 polarization and the support of CTL activity. These findings also correlate with observations in the murine system, where IL‐10 is important in the DC exhaustion process that can be rescued by CD40 ligation [32]. In contrast to CD40/CD40L signalling, secondary encounter of a TLR‐mediated signal such as LPS, poly I:C or dsRNA from a lenti‐viral vector could not revert cytokine exhaustion, suggesting a general TLR unresponsiveness at this DC maturation state.
In summary, this study demonstrates that TLR‐activated DCs retain the developmental plasticity for responding to CD40/CD40L signalling in the presence of IFN‐γ, thus maintaining the capacity for TH1 polarization and the support of CTL‐mediated immune responses. Our findings may be of particular relevance for the rational design of DC‐based cancer vaccines. In order to present soluble tumour antigens to CTLs, DCs must have the capacity for cross‐presentation, which may critically depend on a secondary CD40L‐mediated signal most likely derived from activated TH1 cells expressing CD40L and secreting IFN‐γ. Furthermore, secondary exposure of fully matured DCs to CD40/CD40L signalling appears to have the capacity to prevent DCs from assuming an immune suppressive phenotype, another critical aspect for DC cancer vaccination.
Acknowledgements
We are grateful to R. Panzer, A. Heitger and B. Jürgens for critically reviewing our manuscript and for helpful discussion, and to S. Piechot for proof reading. This work was supported by grants from the Vienna Business Agency and the Austrian Research Promotion Fund (project number 810109). Trimed Biotech GmbH is a subsidiary of AOP Orphan Pharmaceuticals AG that also provides private equity funding for Trimed. A.M. Dohnal and T. Felzmann are employees of Trimed and thus declare competing financial interest.
References
- 1. Steinman RM, Hemmi H. Dendritic cells: translating innate to adaptive immunity. Curr Top Microbiol Immunol. 2006; 311: 17–58. [DOI] [PubMed] [Google Scholar]
- 2. Akira S, Takeda K. Toll‐like receptor signalling. Nat Rev Immunol. 2004; 4: 499–511. [DOI] [PubMed] [Google Scholar]
- 3. Matzinger P. Tolerance, danger, and the extended family. Ann Rev Immunol. 1994; 12: 991–1045. [DOI] [PubMed] [Google Scholar]
- 4. Felzmann T, Huttner KG, Breuer SK, et al . Semi‐mature IL‐12 secreting dendritic cells present exogenous antigen to trigger cytolytic immune responses. Cancer Immunol Immunother. 2005; 54: 769–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hüttner KG, Breuer SK, Paul P, et al . Generation of potent anti‐tumor immunity in mice by interleukin‐12‐secreting dendritic cells. Cancer Immunol Immunother. 2005; 54: 67–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dohnal A, Witt V, Hugel H, et al . Phase I study of tumor Ag‐loaded IL‐12 secreting semi‐mature DC for the treatment of pediatric cancer. Cytotherapy. 2007: 1–16. [DOI] [PubMed] [Google Scholar]
- 7. Langenkamp A, Messi M, Lanzavecchia A, et al . Kinetics of dendritic cell activation: impact on priming of TH1, TH2 and nonpolarized T cells. Nat Immunol. 2000; 1: 3116. [DOI] [PubMed] [Google Scholar]
- 8. Mellor AL, Munn DH. IDO expression by dendritic cells: tolerance and tryptophan catabolism. Nat Rev Immunol. 2004; 4: 762–74. [DOI] [PubMed] [Google Scholar]
- 9. Hill M, Tanguy‐Royer S, Royer P, et al . IDO expands human CD4+CD25high regulatory T cells by promoting maturation of LPS‐treated dendritic cells. Eur J Immunol. 2007; 37: 3054–62. [DOI] [PubMed] [Google Scholar]
- 10. Bonizzi G, Karin M. The two NF‐kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 2004; 25: 280–8. [DOI] [PubMed] [Google Scholar]
- 11. O’Neill LA, Bowie AG. The family of five: TIR‐domain‐containing adaptors in Toll‐like receptor signalling. Nat Rev Immunol. 2007; 7: 353–64. [DOI] [PubMed] [Google Scholar]
- 12. Foster SL, Hargreaves DC, Medzhitov R. Gene‐specific control of inflammation by TLR‐induced chromatin modifications. Nature. 2007; 447: 972–8. [DOI] [PubMed] [Google Scholar]
- 13. Huang Q, Liu D, Majewski P, et al . The plasticity of dendritic cell responses to pathogens and their components. Science. 2001; 294: 870–5. [DOI] [PubMed] [Google Scholar]
- 14. Covert MW, Leung TH, Gaston JE, et al . Achieving stability of lipopolysaccharide‐induced NF‐kappaB activation. Science. 2005; 309: 1854–7. [DOI] [PubMed] [Google Scholar]
- 15. Trinchieri G. Interleukin‐12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol. 2003; 3: 133–46. [DOI] [PubMed] [Google Scholar]
- 16. Rock KL, York IA, Goldberg AL. Post‐proteasomal antigen processing for major histocompatibility complex class I presentation. Nat Immunol. 2004; 5: 670–7. [DOI] [PubMed] [Google Scholar]
- 17. Lanzavecchia A. Immunology. Licence to kill. Nature. 1998; 393: 413–4. [DOI] [PubMed] [Google Scholar]
- 18. Bennett SR, Carbone FR, Karamalis F, et al . Help for cytotoxic‐T‐cell responses is mediated by CD40 signalling. Nature. 1998; 393: 478–80. [DOI] [PubMed] [Google Scholar]
- 19. Ridge JP, Di‐Rosa F, Matzinger P. A conditioned dendritic cell can be a temporal bridge between a CD4+ T‐helper and a T‐killer cell. Nature. 1998; 393: 474–8. [DOI] [PubMed] [Google Scholar]
- 20. Schoenberger SP, Toes RE, van‐der‐Voort EI, et al . T‐cell help for cytotoxic T lymphocytes is mediated by CD40‐CD40L interactions. Nature. 1998; 393: 480–3. [DOI] [PubMed] [Google Scholar]
- 21. Akbari O, DeKruyff RH, Umetsu DT. Pulmonary dendritic cells producing IL‐10 mediate tolerance induced by respiratory exposure to antigen. Nat Immunol. 2001; 2: 725–31. [DOI] [PubMed] [Google Scholar]
- 22. Dohnal AM, Graffi S, Witt V, et al . Comparative evaluation of techniques for the manufacturing of dendritic cell‐based cancer vaccines. J Cell Mol Med. 2009; 13: 125–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shapiro DN, Valentine MB, Rowe ST, et al . Detection of N‐myc gene amplification by fluorescence in situ hybridization. Diagnostic utility for neuroblastoma. Am J Pathol. 1993; 142: 1339–46. [PMC free article] [PubMed] [Google Scholar]
- 24. Laich A, Neurauter G, Widner B, et al . More rapid method for simultaneous measurement of tryptophan and kynurenine by HPLC. Clin Chem. 2002; 48: 579–81. [PubMed] [Google Scholar]
- 25. Shortman K, Naik SH. Steady‐state and inflammatory dendritic‐cell development. Nat Rev Immunol. 2007; 7: 19–30. [DOI] [PubMed] [Google Scholar]
- 26. Macagno A, Napolitani G, Lanzavecchia A, et al . Duration, combination and timing: the signal integration model of dendritic cell activation. Trends Immunol. 2007; 28: 227–33. [DOI] [PubMed] [Google Scholar]
- 27. Logue EC, Sha WC. CD28‐B7 bidirectional signaling: a two‐way street to activation. Nat Immunol. 2004; 5: 1103–5. [DOI] [PubMed] [Google Scholar]
- 28. Puccetti P. On watching the watchers: IDO and type I/II IFN. Eur J Immunol. 2007; 37: 876–9. [DOI] [PubMed] [Google Scholar]
- 29. West MA, Heagy W. Endotoxin tolerance: a review. Crit Care Med. 2002; 30: S64–73. [PubMed] [Google Scholar]
- 30. Kalinski P, Schuitemaker JHN, Hilkens CMU, et al . Final maturation of dendritic cells is associated with impaired responsiveness to IFN‐{gamma} and to bacterial IL‐12 inducers: decreased ability of mature dendritic cells to produce IL‐12 during the interaction with Th cells. J Immunol. 1999; 162: 3231–6. [PubMed] [Google Scholar]
- 31. Felzmann T, Buchberger M, Lehner M, et al . Functional maturation of dendritic cells by exposure to CD40L transgenic tumor cells, fibroblasts or keratinocytes. Cancer Lett. 2001; 168: 145–54. [DOI] [PubMed] [Google Scholar]
- 32. Kajino K, Nakamura I, Bamba H, et al . Involvement of IL‐10 in exhaustion of myeloid dendritic cells and rescue by CD40 stimulation. Immunology. 2007; 120: 28–37. [DOI] [PMC free article] [PubMed] [Google Scholar]