

Abstract
Background:
Acute myocardial infarction (AMI) and the ensuing ischemic heart disease are approaching an epidemic state. Limited stem cell retention following intracoronary administration has reduced the clinical efficacy of this novel therapy. Polymer based cell coating is biocompatible and has been shown to be safe. Here, we assessed the therapeutic utility of gelatin-based biodegradable cell coatings on bone marrow derived cell retention in ischemic heart.
Methods:
Gelatin based cell coatings were formed from the surface-mediated photopolymerization of 3% gelatin methacrylamide and 1% PEG diacrylate. Cell coating was confirmed using a multimodality approach including flow cytometry, imaging flow cytometry (ImageStream System) and immunohistochemistry. Biocompatibility of cell coating, metabolic activity of coated cells, and the effect of cell coating on the susceptibility of cells for engulfment were assessed using in vitro models. Finally, cell adhesion to extracellular matrix was assessed in vitro using a microfluidic device. Following myocardial infarction and GFP+ BM-derived mesenchymal stem cell transplantation, flow cytometric and immunohistochemical assessment of retained cells was performed.
Results:
Coated cells are viable and metabolically active with coating degrading within 72 hours in vitro. Importantly, cell coating does not predispose bone marrow cells to aggregation or increase their susceptibility to phagocytosis. In vitro and in vivo studies demonstrated no evidence of heightened immune response or increased phagocytosis of coated cells. Cell transplantation studies following myocardial infarction proved the improved retention of coated bone marrow cells compared to uncoated cells.
Conclusion:
Gelation based polymer cell coating is biologically safe and biodegradable. Therapies employing these strategies may represent an attractive target for improving outcomes of cardiac regenerative therapies in human studies.
Keywords: Cell coating, polymer, photo-polymerization, bone marrow mesenchymal stem cells, myocardial infarction
Introduction.
Ischemic heart disease, often caused by acute myocardial infarction (AMI), is the leading cause of morbidity and mortality in the developed world [1]. While modern cardiology has achieved significant strides in revascularization and medical management, a significant portion of AMI patients progress to develop ischemic cardiomyopathy (ICM) and heart failure (HF). Bone marrow derived stem cells (BMSCs) are an attractive therapeutic target for cardiac regeneration and have been extensively investigated in animal experiments and human translational studies. However, low cell engraftment after transplantation has limited the cardiac function recovery for BMSC therapy [2]. Indeed, only a small fraction of transplanted cells remain at the injury site 24 h after injection into the heart wall [3]. Recent clinical studies [4] and meta-analyses [5] indicate the benefit of BMSC therapy is directly correlated with the quantity of cells injected, where the injection of more cells provides greater functional recovery. Clinically, the number of harvested BMSCs is limited, and larger harvest procedures are hazardous given the high-risk population in question. Additionally, ex vivo expansion to expand stem cells for further clinical use carries the risk of infection or cell phenotype changes. Therefore, efforts directed towards enhanced cell retention after transplantation are desperately needed.
Biomaterial based cardiac tissue engineering has been widely used in animal and human studies [6–8]. These studies have addressed two major limitations of current cardiac regenerative therapies, namely cytokine release to enhance the therapeutic potential of stem cells and providing mechanical support (scaffolding) to the damaged muscle through biocompatible grafts. Strategies aimed at the local sustained delivery of recombinant stromal derived factor-1 (rSDF-1) through its integration in cross-linked hyaluronic acid (HA) hydrogels resulted in enhanced bone marrow cell engraftment in the heart [9]. Furthermore, the effect of HA impregnated with an engineered SDF-1 mimetic led to significant reduction in adverse cardiac remodeling and fibrosis at 4 weeks after AMI [10]. Other studies have focused on the delivery of other cytokines such as VEGF in cell-based coatings including encapsulated mesenchymal stem cells (MSCs) using collagen and alginate polyelectrolytes [11]. Additionally, the use of biocompatible scaffolds to mitigate post-AMI adverse cardiac remodeling has demonstrated safety and therapeutic success in animal and early pre-clinical studies. Scaffolding material are either natural or synthetic [12], and in early studies natural materials appear to be more biodegradable, biocompatible, and have an advantage in recreating the native cardiac microenvironment [13]. In a previous study, D’Souza et al showed an increased cell binding to bone fragments (in vitro), when the cell membranes were modified with a bisphosphonate-containing polymer [14]. While these strategies have achieved success in proof of concept studies, they do not address the poor retention of stem cells after transplantation.
In this study, we utilized an adhesive biocompatible coating on the exterior of BM cells to improve their retention in the heart. These efforts build upon our earlier success in generating biosynthetic polymer coating on living cells. These coatings were designed using polyethylene glycol (PEG) based polymers to completely coat cells for purification purposes [15]. PEG based coatings allow essential nutrient transport thus preserving cell viability [16]. Here, we demonstrate our ability to generate gelatin-based cell coatings that are safe, biodegradable, and enhances cell retention in the ischemic myocardium without significantly impairing cell metabolism or survival.
Materials and Methods
Study Design.
Male C57BL/6 WT and C57BL/6-Tg(CAG-EGFP)131Osb/LeySopJ mice (Jackson Laboratory, BarHarbor, ME), aged 6–10 weeks, were used in this study. All procedures were conducted under the approval of the University of Kentucky IACUC in accordance with the NIH Guide for the Care and Use of Laboratory Animals (DHHS publication No. [NIH] 85–23, rev. 1996). A549 (human epithelial lung carcinoma), RAW264.7 (mouse macrophage cell line), and Jurkat (T lymphocyte) cells were obtained from ATCC.
Murine Model of Myocardial Infarction.
Mice were anesthetized with 1–3% isoflurane using an inhaled delivery system. The heart was exposed, pushed out of the thorax with a direct visual control and the left anterior descending coronary artery (LAD) was sutured and ligated at a site approximately 3 mm from its origin using a 6–0 silk suture as previously described [17, 18].
Generation of bone marrow derived mesenchymal stem cells (MSCs). Bone marrow cells were isolated and cultured at 37°C in hypoxic conditions (5% O2) as previously described [19]. Briefly, 6–8 weeks old C57BL/6-Tg (CAG-EGFP) mice (Jackson Laboratory, BarHarbor, ME) were sacrificed, femurs, tibias and hip bones were collected and then flushed with PBS supplemented with 10% FBS. Cells were cultured in complete MesenCult (StemCell Technology, Canada) supplemented with 1% penicillin-streptomycin (Life Technologies), and 1% L-Glutamine (StemCell Technology, Canada). A homogenous population of MSCs, based on morphology and cell surface marker expression, from passages 5 and 6 were used in this study. 105 cells were injected in 3 different injections (25 μL total) in the peri-infarct region.
Cell encapsulation.
Cells were washed twice with PBS by centrifugation at 500g, 3 minutes at 4 °C. 1.5 × 106 cells were collected for each polymerization trial. PBS buffer was removed, and the cell pellet was resuspended in 200 μL of 1mM Biotin sulfo NHS (Thermo Fisher Scientific, Waltham, MA) in PBS and incubated for 40 minutes on ice. Following incubation, 800 μL of PBS was added and rinsed twice by centrifuging at 500xg for 3 minutes at 4 °C. The cell pellet was then resuspended in 1 mL of PBS containing 35 μg/mL streptavidin-eosin isothiocyanate and incubated for 30 minutes on ice. Streptavidin (Thermo Fisher Scientific, Waltham, MA) and eosin-5-isothiocyanate (Sigma Aldrich, St. Louis, MO) conjugation was made in-house as described by Hansen et al. After 30 minutes of incubation, the cells were washed twice in PBS by centrifuging at 500xg for 3 minutes at 4 °C. The polymer mixture buffer was prepared using 35mM triethanol amine and 35 mM 1-vinyl-2-pyrrolidinone in PBS. This mixture was purged with ultra-pure N2 for 10 minutes. 3 wt% gelatin methacrylate (gelMA) (BioBots, Philadelphia, PA) was added to the buffer and vortexed and sonicated for 10 minutes. 1 wt% PEGDA 3500 (JenKem Technology, Plano, TX) was then added to the 3 wt% gelMA solution and vortexed. The final polymer mixture was filtered through a 0.2 μm mesh syringe filter and stored at RT. 350 μL of the polymer mixture was added to the cell pellet and gently vortexed and loaded into a chip-clip well (Whatman) with a standard microscopy slide. The chip-clip was incubated in a dark, N2 purged, sealed, zip lock bag for 3 minutes before polymerization. The cells were polymerized for 10 minutes at 30mW/cm2 under 530 nm green visible light while purging with N2. After 10 minutes, the slide was washed twice with 1ml of PBS each time.
Flow Cytometry.
Heart tissue was harvested at 7 days and placed in ice cold PBS instantly. Heart tissue was minced then digested using a collagenase B (Roche, Indianapolis, IN) and dispase II (Roche, Indianapolis, IN) solution for 30 minutes at 37°C with mixing every 5 minutes.The enzymatic reaction was stopped by dilution with Flow Buffer (PBS + 5 % normal goat serum + 0.1 % sodium azide) and the heart cell suspensions were passed through 40 μm strainers. Cells were centrifuged at 400×g for 5 min at 4 °C, then suspended in Flow Buffer. Cells were centrifuged at 400xg for 5 min at 4 °C and then resuspended in Cytofix/Cytoperm (BD Biosciences, San Jose CA) solution and incubated for 20 min at 4°C. Cells were then washed with Perm/Wash (BD Biosciences) and incubated directly for 30 minutes with APC conjugated GFP Antibody Clone 5F12.4 (eBioscience, Thermo Fisher Scientific, Waltham MA). After incubation, cells were washed twice using flow buffer and analyzed using an LSR II (Becton Dickinson) in the University of Kentucky Flow Cytometry Core. Laser calibration and compensation were carried out utilizing unstained and single fluorescent control beads (eBioscience). We utilized FlowJo v7 (FlowJo, FlowJo Ashland OR) software to generate dot plots and analyze the data.
ImageStream analyses.
All our data indicate that coated cells remain as single cells and do not aggregate. Therefore, we conducted imaging flow cytometry studies to confirm cell coating. Coated and uncoated BM-derived MSCs were stained using primary anti-collagen I antibody (Abcam) followed by secondary antibody (AF-647-Invitrogen) for analysis by ImageStream system. Hoechst33342 was added 5 min before analysis. Samples were run directly on ImageStream MKII system with 2 cameras (MilliporeSigma, Seattle, WA, USA) without any cell classifier (instrument threshold). Signals from FITC (GFP), Hoechst, AF-647 were detected by channels 2, 7, and 11, respectively; while side scatter and brightfield images were collected in channels 6 and 1, respectively.
Immunohistochemistry.
Immunohistochemical assessments were carried out on de-paraffinized and rehydrated sections as previously described [18]. Briefly, sections were exposed to heat-mediated epitope retrieval in citrate buffer, pH 6.0 (Vector Laboratories, Burlingame CA)) for 20 mins, then blocked with normal goat serum for 10 minutes at 37 °C. Slides were incubated with primary antibodies: rabbit anti-GFP (Abcam, Cambridge, United Kingdom) or rat anti-mouse CD68 (Abcam). After washing, sections were incubated with secondary antibodies conjugated to APC or FITC, respectively. The sections were finally incubated with Sudan Black B (Sigma Aldrich, St. Louis, MO) for 30 minutes. Adjacent areas in the peri-infarct and remote zones were analyzed (1 section/animal, n= 3 animals/group) at 40x magnification using Nikon Confocal Microscope A1 (Nikon, Tokyo, Japan) in the University of Kentucky Confocal Microscopy facility. Calculations were performed using the Cell Counter plugin for ImageJ, version 1.51d (NIH, Rockville, MD).
Metabolic activity assay.
Metabolic activity was assessed using MTT assay (3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide) (Thermo Fisher Scientific, Waltham, MA), which measures mitochondrial activity within the cell indicating a measure of viability and proliferation. An equal number of MSCs (20,000–50,000) from each group were incubated in triplicate in 200 μL of medium with the equal concentration of MTT for 3–4 hours at 37 ˚C. Each time point was setup in a different 96 well plate (Celltreat Scientific Products, Pepperell, MA). After incubation, the plate was centrifuged, wells were aspirated and well contents were solubilized in 200 μL of DMSO (Sigma-Aldrich, St Louis MO). Absorbance at 570 nm for each well was measured using a plate reader (Biotek, Winooski, VT). Absorbance values for each sample were averaged and then normalized to the corresponding control cultures.
Engulfment assay.
Coated and uncoated target A549 cells were labeled with MitoTracker Deep Red (Invitrogen, Thermo Fisher Scientific) according to the manufacturer’s protocol. The target cells were then cultured with effector RAW264.7 cells that had been seeded the previous day in 6 well plates. Cultures were removed with the help of a cell scraper, washed with PBS, and then subjected to flow cytometry.
Statistical Analysis.
Values are expressed as mean ± standard error of mean (SEM). We used unpaired Student t-test or analysis of variance (one-way or multiple comparisons) to estimate differences, as appropriate. We utilized two-sided Dunnett or Dunn tests for post hoc multiple comparison procedures, with control samples as the control category. Throughout the analyses, a p value less than 0.05 was considered statistically significant. All statistical analyses were performed using the Prism 7 software package (GraphPad, La Jolla, CA).
Results
Bone marrow derived MSCs remain viable after coating with gelatin methacrylate biodegradable polymer.
Our prior studies documented our ability to generate biosynthetic polymer coating on living cells. These coatings were designed using polyethylene glycol (PEG) based polymers to completely coat cells for purification purposes [15]. PEG based coatings allow essential nutrient transport thus preserving cell viability [16]. In this study, we elected to use gelatin methacrylate biodegradable coating to achieve improved cell adhesion and retention in cardiac tissue. Initially, we wanted to see if our protocol would coat bone marrow cells with a gelatin-based polymer. To evaluate our cell coating technique on a homogeneous population of cells, we coated BM-MSCs using same coating polymer and protocol. Successful coating on BM-MSCs surface was observed when stained with anti-collagen antibodies and analyzed using imaging flow cytometer (Fig 1A). A thin layer of gelatin coating was demonstrated on BM-MSCs surface after cell surface polymerization process. Cells remained intact and viable as shown by Hoechst staining. We further confirmed the coating using histochemistry by integrating picosirius red in the polymer to identify coated BMCs cells (Fig 1B). Gelatin methacrylate coated BMCs were viable as assessed by histological and flow cytometric techniques (Fig. 1B). On average, we achieved ~40–50% coating and 85% of coated cells appeared to be viable based on calcein viability staining.
Figure 1. BM-derived MSCs remained viable after coating with polymer.
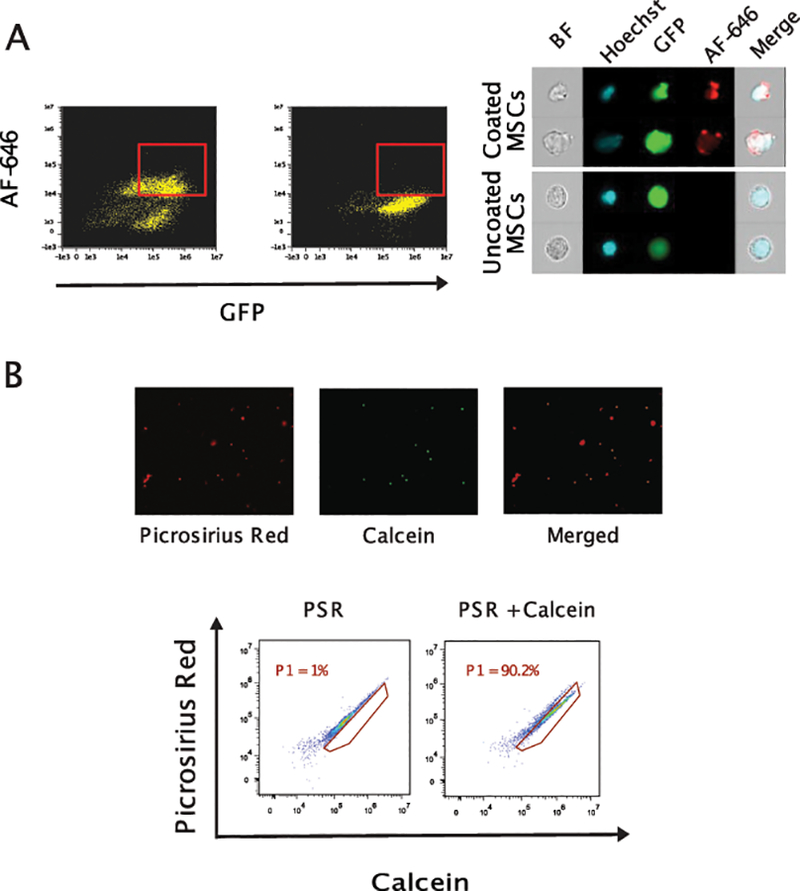
Coated BM-MSCs were visualized by imaging flow cytometer-ImageStream System. Coated and uncoated BM-MSCs were fixed, stained with antibodies against collagen I antibody (AF-647) for the presence of cell surface gelatin coating, and acquired on an ImageStream flow cytometer. (Panel A) Representative FACS plots and images demonstrating nucleated coated GFP+ BM-MSCs with Hoechst33342 and Alexa-Fluor 647 positive staining. (Panel B) Coated cells remained alive after coating process as stained double positively with picosirius red (red for gelatin coating) and calcein (green for viability) as assessed by microscopy (upper panel) and flow cytometry (lower panel).
Coated bone marrow derived cells do not aggregate.
Cell coating and enhanced transplanted cell retention can be of great benefit in stem cell-based studies, especially in cardiac applications. However, cell-cell aggregation can be detrimental in certain delivery methods such as intracoronary infusion where aggregated cells can block microvasculature leading to myocardial infarction. We conducted further studies to rule out the possibility that monomers on two adjacent cells do crosslink resulting in cell-cell aggregates. To determine what impact, if any, the cell coating had on cell-cell aggregates, we quantified the number of aggregates in coated BM-MSCs vs. monomer incubated or uncoated cells. Using flow cytometry, we analyzed cell aggregates using the forward scatter height and area. Our analyses did not show a significant difference in cell-cell aggregates between coated and uncoated cells (Fig. 2). Therefore, our data shows that neither the process of polymerization nor the coating itself had a significant impact on cell aggregation.
Figure 2. Coating did not predispose cells to aggregate.
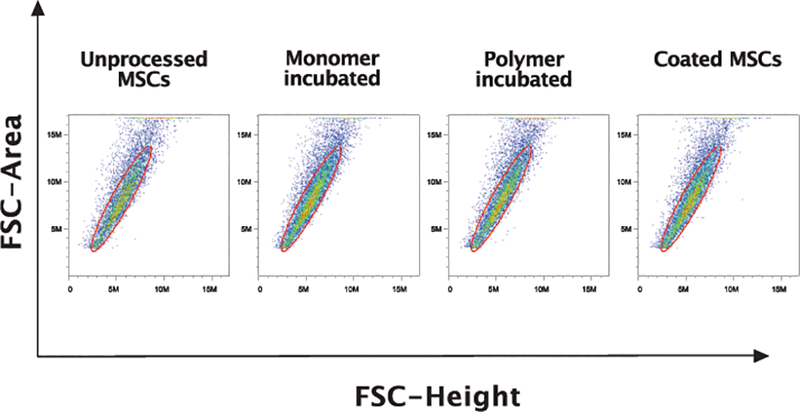
Coated and uncoated control BM-MSCs were analyzed for cell-cell aggregates using flow cytometry by examining the forward scatter area vs. height (FSC) plots. Events falling outside the diagonal of forward scatter area versus height were considered to be aggregates. On forward scatter plots, where the area under the curve is plotted against the maximum height of the curve for each event, single cells fall along the diagonal, indicated by the drawn gate. We did not observe changes in cell aggregates with our coating strategy.
Gelatin based cell coatings are degradable.
In clinical applications, a cell coating is ideally degradable allowing stem cells to exert their beneficial effects after the resolution of inflammation. This is particularly important during the early inflammatory phase that peaks at 72 hours after MI [20–22]. Hence, we designed our gelatin methacrylate coating to degrade naturally, by metalloproteinases (MMPs), within 72 hours. While bone marrow cells express low levels of MMP activity, the ischemic heart expresses high levels of MMPs [23]. To demonstrate that coated cells are capable of degrading their gelatin coating, we coated A549 cells, human lung carcinoma cell line, that have high level of MMP secretion capability (A549 cells) and cultured them for three days in RPMI + 10% FBS medium under normal culture conditions. Cell coating was confirmed using Picosirius red which was assessed by flow cytometry (Fig. 3). Within one day of culturing, there was a reduction in the amount of coating on cells which further decreased by Day 3. Additionally, in our in vivo studies, we did not observe any coating in our immunohistochemistry studies (data not shown). Taken together, our findings suggest that the gelatin coating is transient and can be degraded to release cells to exert their therapeutic effect after transplantation.
Figure 3. Cell coating began to degrade in vitro within 24 hr.
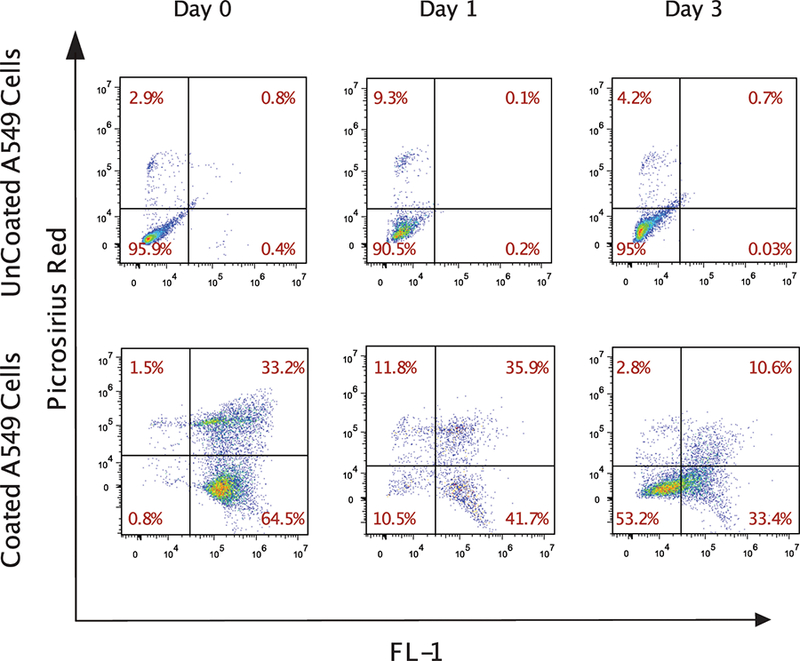
Coated and uncoated control A549 lung carcinoma cells were cultured for up to three days. Coated cells were identified with Picosirius red. The fluorescence in the FL-1 channel of the coated cells was due to the presence of eosin, which was used to initiate polymerization. The plots demonstrate the progressive loss of coating on cultured cells over time.
Coated bone marrow derived cells remain metabolically active in vitro.
Our prior studies demonstrated that a PEG based biosynthetic cell coating is compatible with nutrient transfer. It was then necessary to determine if cells coated with the gelatin-based biodegradable material remained metabolically active. For these studies, coated and uncoated BM-MSCs were cultured until passage 5 in mesencult medium under hypoxic conditions. Immediately after coating and on daily time intervals afterwards, cells were harvested and their metabolic activity was measured by the MTT assay. Initially, coated cells expressed lower metabolic activity as compared to uncoated controls. However, the MTT assay showed equivalent activity between coated and uncoated cells over time (Fig. 4). This lag could be explained, at least in part, by the delayed attachment of coated cells which required an additional one to one and half days to adhere to the surface of the plate compared to uncoated cells.
Figure 4. Coated BM-MSCs are metabolically active.
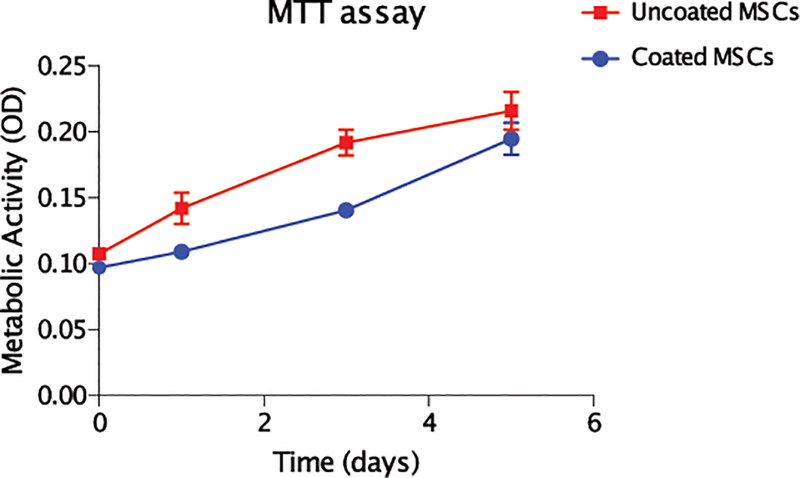
Coated and control uncoated cells were assessed for up to five days in vitro. Each day, cultures of cells were examined for metabolic activity by the MTT assay. After an initial lag phase, the metabolic activity of coated cells was comparable to that of the control uncoated cells.
Coated bone marrow derived cells do not elicit an immune response.
While cell coating could provide cell protection from inflammatory conditions, the possibility of coating triggering an immune response needs to be excluded. We used a cell tracking system where all cells (coated and uncoated) were incubated with Mitotracker Deep Red. For this assay, we utilized a well-defined in vitro system with target A549 cells cultured with mouse RAW264.7 macrophage effector cells at a ratio of 5:1 for 15 minutes. Effector cells that had engulfed target cells acquired Deep Red from the target cells and then expressed a low level of Deep Red (Fig 5A). We observed no significant difference in mean fluorescence intensity for the Mitotracker deep red dye in effector cells (RAW264.7 macrophage) when incubated with coated or uncoated cells. This pattern was observed for up to 3 hours of co-culture (Fig. 5B). On the other hand, we had no effector cells expressing Deep Red dye when effector cells were cultured without target cells confirming the specificity of our approach (data not shown).
Figure 5. Coating does not predispose coated cells to be engulfed by macrophages.
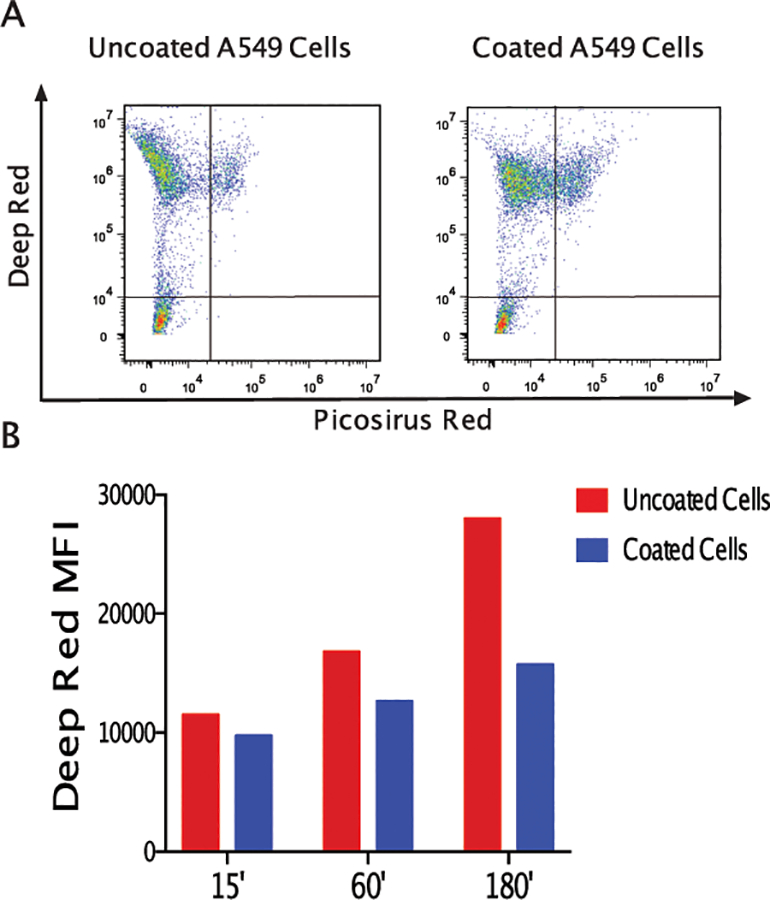
All A549 target cells were labeled with Deep Red while coated cells were identified by Picosirius Red. Target cells were cultured with unlabeled RAW264.7 macrophage effector cells for 15 minutes. Effector cells that had engulfed target cells were found in the lower left region (Panel A). Fewer professional phagocytes were present when coated A549 cells were co-cultured (right plot) than uncoated cells (left plot). The brightness of deep red, an indication of engulfment, was plotted for cultures indicating that coating may protect target cells from engulfment as detailed in the quantitative assessment of plots (Panel B).
We further confirmed that cell coating does not elicit an immune response in the heart using our in vivo studies. We stained heart sections 7 days after MI and cell transplantation against CD68, a macrophages cell surface marker. The number of CD68+ macrophages was similar in the hearts of mice injected with coated bone marrow cells compared to control uncoated cells (Fig. 6) implying that the coating did not enhance the inflammatory response seen upon MI.
Figure 6. The coating on the cells did not induce a localized inflammatory response.
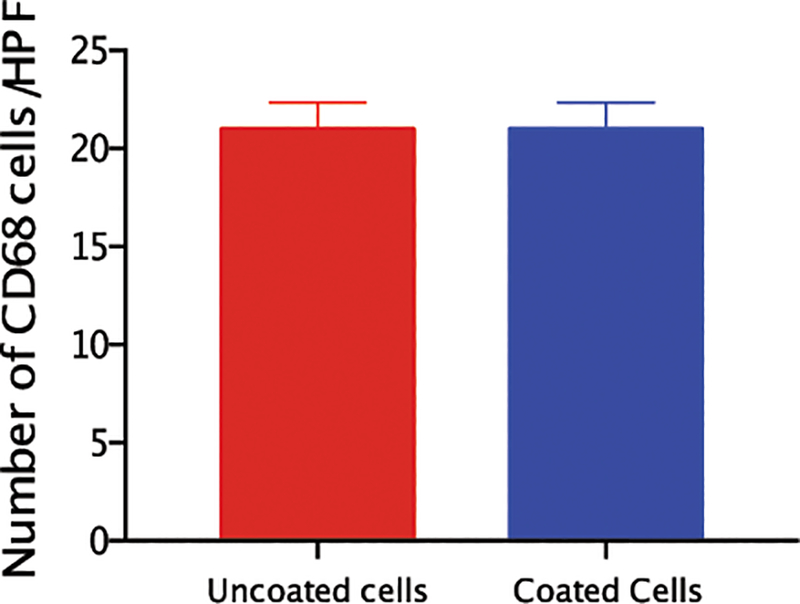
Paraffin imbedded sections were stained for CD68 and quantified as number of cells per power field were quantified. Quantitative analysis did not show significant difference between mice treated with coated or uncoated cells (N = 3 mice/group).
Polymer coated cells demonstrate enhanced retention in the heart following transplantation.
We next investigated cell retention in the heart after acute myocardial infarction. Immediately after WT BL/6 mice were subjected to MI using the LAD permanent model, 1.25×105 BM-MSCs from GFP mice were injected in two separate peri-infract locations. The use of GFP donor cells allows us to track the retention and fate of transplanted cells. Seven days later, we examined the number of BM-MSCs retained in the heart using immunohistochemistry and flow cytometry. Hearts were isolated, digested to single cell suspension then stained against GFP. We observed significantly higher numbers of GFP+ MSCs in the coated arm compared to uncoated cells (uncoated = 10.8 ± 3.4 vs. coated = 27 ± 3.8 cells/mg heart tissue, P = 0.03) (Fig. 7A and B). Interestingly, coated bone marrow MSCs had a higher expression of GFP on flow cytometry, supporting their viability. Our immunohistochemistry data confirmed these findings with higher numbers of GFP cells in mice transplanted with coated cells compared to those transplanted with uncoated cells (uncoated = 21.1 ± 5.4 vs. coated = 39.2 ± 3.3 cells/high power field, P < 0.01). GFP+ cells were observed primarily in the peri-infarct region (Fig. 7C and D).
Figure 7. Coated cells were more efficiently retained in the heart.
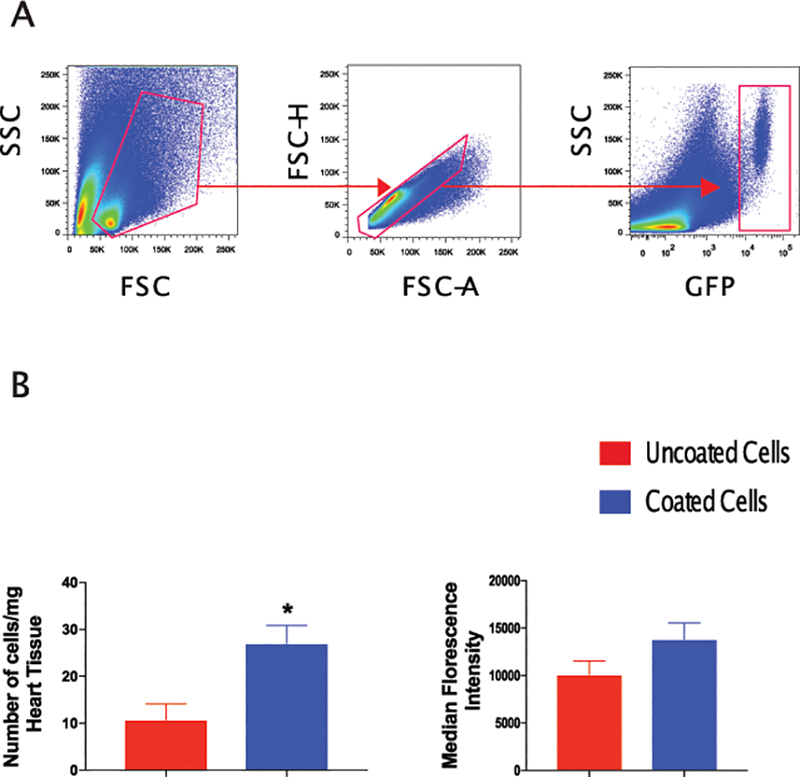
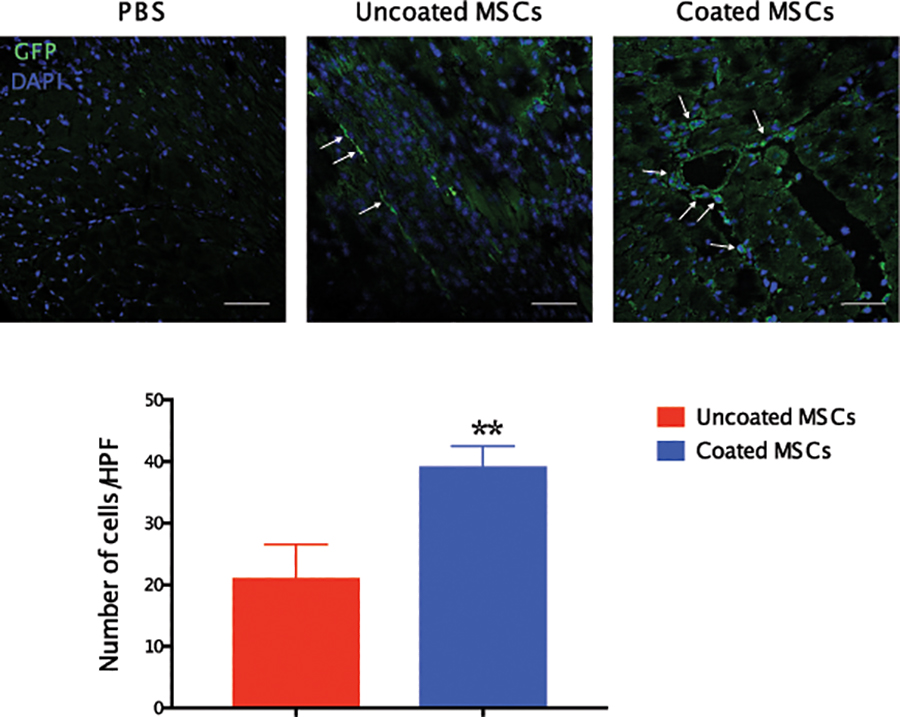
Flow cytometry analyses of digested heart tissue demonstrate higher percentage of GFP+ cells in mice treated with coated cells comparted to mice transplanted with uncoated cells. Panel A shows the gating strategy for GFP+ cells in the heart as assessed 7 days after MI and transplantation. Panel B illustrates a quantitative analysis of GFP+ cells in digested heart tissue of mice treated with coated and uncoated bone marrow cells showing higher percentage of GFP+ cells and trend towards higher GFP mean fluorescence intensity in mice treated with coated MSCs. Immunohistochemical analysis demonstrated higher numbers of retained GFP+ bone marrow cells in mice treated with coated cells (Panel C). Quantitatively, mice treated with coated cells exhibited higher numbers for GFP+ cells per high power field as compared to mice treated with uncoated cells (Panel D) (N = 4 mice/group; *P<0.05, **P<0.01).
Discussion
There is a great need for effective therapies to treat ischemic heart disease as an alternative to heart transplantation, which suffers significant limitations. The use of bone marrow derived stem cells for cardiac regeneration has gained traction in animal research and translational human studies [24–29]. However, attempts at achieving myocardial regeneration after ischemic tissue loss have achieved limited clinical success. Stem cell based regenerative therapies in cardiac applications are hampered by the low engraftment of transplanted cells [3]. In this study, we demonstrate a novel approach to enhance the engraftment of transplanted BM cells following transplantation in the ischemic myocardium. Gelatin-based polymer coatings are biologically compatible with BM cells achieving excellent coating without affecting cell survival. BM mesenchymal stem cells coated with polymer showed enhanced retention in cardiac tissue after MI without evidence of increased inflammatory or phagocytosis response. This is the first study to examine the retention of stem cell engraftment for myocardial regenerative therapies using biologically compatible individual cell polymer coating and could provide the basis for successful and reproducible regenerative cardiac studies.
Current BM based regenerative studies rely mostly on unselected BMCs which include non-stem cell populations believed to reduce the beneficial effects of the therapy. On the other hand, utilizing selected BM stem cell populations (BMSCs) such as CD34 and CD133 cells by depleting inflammatory cells is more scientifically promising due to their ability to enhance cardiac recovery; their resistance to apoptosis; and their angiogenic potential [30, 31]. The majority of benefit from cardiac transplantation of stem cells originates from enhanced angiogenesis [24, 32, 33]. However, due to the limited yield of selected BMSCs and the low retention after transplantation in cardiac tissue [29], stem cell regenerative therapy has faced limited benefit and widespread scrutiny. Human and animal studies suggest limited engraftment of stem cells after their transplantation in acute and chronic ischemic heart disease [3, 34]. In acute ischemic injury, SDF-1 and homing chemokines are expressed in the heart [35–37]. However, upregulated metalloproteinases and proteases lead to their degradation and therefore reduction in the homing signal for transplanted cells [38, 39]. Indeed, studies on mice lacking the expression of CXCR4 (SDF-1 receptor) demonstrated similar homing of transplanted BM stem cells compared to WT mice after MI [40]. In this study, we adopt a novel approach to address this limitation. Gelatin based individual cell polymer coating has been shown to be biocompatible in different biological applications. Our prior studies have demonstrated the successful use of biocompatible polymer coating for cell selection and other regenerative applications [15, 16, 41, 42].
The use of synthetic cell coatings has gained momentum in cellular therapy. Hydrogel architectures have been utilized in multiple clinical applications including as vehicles for drug delivery and as scaffolds for cell grafts to achieve better cell survival and cardiac muscle support [43–45]. Collagen is one of the main proteins in mammalian extracellular matrix (ECM) and has multiple integral functions including structural support. Small collagen derived peptides, such as gelatin, have collagen specific affinity and saturable binding can increase cell retention upon transplant [46]. In fact, studies have shown that gelatin exhibit specific bindings to collagen mimetic peptides which contains naturally occurring sequences identical to those found in collagen [47, 48]. Moreover, the use of a gelatin-based cell coating is attractive because of its biocompatibility, adhesiveness, and porous nature that maintain the metabolism and survival of coated cells. Cardiac tissue engineering studies utilizing gelatin and collagen scaffolds, even in the absence of stem cell incorporation, showed cardiac recovery, angiogenesis and cell-cell electrical coupling [49–51]. These characteristics made collagen-derivatives ideal candidates for our cell coating approach. In our studies, gelatin-based cell coating did not affect cell viability or metabolic activity over 5 days in vitro. Gelatin-coated cells were more abundant in the myocardium after transplantation in the heart following myocardial infarction. Our ongoing studies are examining the therapeutic utility of cell coating in a myocardial infarction model.
Our prior studies demonstrated the safety and feasibility of depositing 100 nm thick PEG-based cell coating polymers on the surface of cells [15, 16, 41, 42]. The aim of this study is to investigate the effectiveness of the biodegradable gelatin-based cell coating polymer with a goal of enhancing cell adhesion to cardiac tissue and cell retention at sites of myocardial infarction. This approach offers multiple advantages to current therapies. Several studies have demonstrated that the benefit of bone marrow-derived stem cells in cardiac applications is proportional to their transplanted numbers [5, 52]. This is particularly important in clinical trials where purified stem cell populations are rare and difficult to isolate. Higher volume bone marrow harvest to isolate larger numbers of stem cells is clinically hazardous in heart failure patients while smaller numbers may not be clinically beneficial. Therefore, by increasing cell retention, we could achieve similar beneficial effects with fewer stem cells or more benefit with the current numbers of stem cells used in clinical application. Our results confirm the increased number of coated BM-MSCs in the peri-infarct region when transplanted after MI. We however do not know if the higher numbers of BM-MSCs in the heart after MI in the coated arm is due to higher cell retention or the protection of cells from the surrounding deleterious microenvironment. Nonetheless, the clinical applicability of these results is huge with multiple studies correlating the benefit of stem cells in the heart to the number of cells transplanted and retained in the heart. Future studies examining the efficacy of coated BM-MSCs in cardiac regeneration could establish this new approach and provide proof of concept for clinical trials.
In conclusion, we demonstrate successful encapsulation of bone marrow derived cells using gelatin based biodegradable coating. The coating is compatible with cell viability and does not elicit an immune response. Coated bone marrow cells achieve higher numbers in the ischemic myocardium after transplantation. This methodology is capable of clinical translation and can improve the success and reproducibility of cell therapy studies in general and cardiac regeneration studies in particular.
ACKNOWLEDGMENTS
Dr. Abdel-Latif is supported by the University of Kentucky COBRE Early Career Program (P20 GM103527) and the NIH Grant R01 HL124266. This work was partially supported by R01 HL127682 and the National Science Foundation under Award CBET-1351531.
Footnotes
Conflict of interest: None.
References.
- 1.Mozaffarian D, et al. , Heart disease and stroke statistics−-2015 update: a report from the American Heart Association. Circulation, 2015. 131(4): p. e29–322. [DOI] [PubMed] [Google Scholar]
- 2.Asahara T, Kawamoto A, and Masuda H, Concise review: Circulating endothelial progenitor cells for vascular medicine. Stem Cells, 2011. 29(11): p. 1650–5. [DOI] [PubMed] [Google Scholar]
- 3.Hofmann M, et al. , Monitoring of bone marrow cell homing into the infarcted human myocardium. Circulation, 2005. 111(17): p. 2198–202. [DOI] [PubMed] [Google Scholar]
- 4.Quyyumi A, et al. , One year follow-up results from PRESERVE-AMI: a randomized, double-blind, placebo controlled clinical trial of intracoronary infusion of autologous CD34+ cells in patients with left ventricular dysfunction post STEMI. J Am Coll Cardiol, 2015. 55(10): p. A1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Afzal MR, et al. , Adult Bone Marrow Cell Therapy for Ischemic Heart Disease: Evidence and Insights From Randomized Controlled Trials. Circ Res, 2015. 117(6): p. 558–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kavanagh DP, Robinson J, and Kalia N, Mesenchymal stem cell priming: fine-tuning adhesion and function. Stem Cell Rev, 2014. 10(4): p. 587–99. [DOI] [PubMed] [Google Scholar]
- 7.Zhou Y, et al. , Effects of Human Fibroblast-Derived Extracellular Matrix on Mesenchymal Stem Cells. Stem Cell Rev, 2016. 12(5): p. 560–572. [DOI] [PubMed] [Google Scholar]
- 8.Arnaoutova I, et al. , Basement membrane matrix (BME) has multiple uses with stem cells. Stem Cell Rev, 2012. 8(1): p. 163–9. [DOI] [PubMed] [Google Scholar]
- 9.Purcell BP, et al. , Synergistic effects of SDF-1alpha chemokine and hyaluronic acid release from degradable hydrogels on directing bone marrow derived cell homing to the myocardium. Biomaterials, 2012. 33(31): p. 7849–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.MacArthur JW Jr., et al. , Sustained release of engineered stromal cell-derived factor 1-alpha from injectable hydrogels effectively recruits endothelial progenitor cells and preserves ventricular function after myocardial infarction. Circulation, 2013. 128(11 Suppl 1): p. S79–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu G, et al. , A VEGF delivery system targeting MI improves angiogenesis and cardiac function based on the tropism of MSCs and layer-by-layer self-assembly. Biomaterials, 2017. 127: p. 117–131. [DOI] [PubMed] [Google Scholar]
- 12.Nash ME, et al. , Thermoresponsive substrates used for the expansion of human mesenchymal stem cells and the preservation of immunophenotype. Stem Cell Rev, 2013. 9(2): p. 148–57. [DOI] [PubMed] [Google Scholar]
- 13.Sarig U and Machluf M, Engineering cell platforms for myocardial regeneration. Expert Opin Biol Ther, 2011. 11(8): p. 1055–77. [DOI] [PubMed] [Google Scholar]
- 14.D’Souza S, et al. , Engineering of cell membranes with a bisphosphonate-containing polymer using ATRP synthesis for bone targeting. Biomaterials, 2014. 35(35): p. 9447–58. [DOI] [PubMed] [Google Scholar]
- 15.Romero G, et al. , Protective Polymer Coatings for High-Throughput, High-Purity Cellular Isolation. ACS Appl Mater Interfaces, 2015. 7(32): p. 17598–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lilly JL, et al. , Characterization of molecular transport in ultrathin hydrogel coatings for cellular immunoprotection. Biomacromolecules, 2015. 16(2): p. 541–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gao E, et al. , A novel and efficient model of coronary artery ligation and myocardial infarction in the mouse. Circ Res, 2010. 107(12): p. 1445–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Klyachkin YM, et al. , Pharmacological Elevation of Circulating Bioactive Phosphosphingolipids Enhances Myocardial Recovery After Acute Infarction. Stem Cells Transl Med, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Caroti CM, et al. , A Novel Technique for Accelerated Culture of Murine Mesenchymal Stem Cells that Allows for Sustained Multipotency. Sci Rep, 2017. 7(1): p. 13334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nahrendorf M, et al. , The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med, 2007. 204(12): p. 3037–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van der Laan AM, et al. , Monocyte subset accumulation in the human heart following acute myocardial infarction and the role of the spleen as monocyte reservoir. Eur Heart J, 2014. 35(6): p. 376–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Epelman S, Liu PP, and Mann DL, Role of innate and adaptive immune mechanisms in cardiac injury and repair. Nat Rev Immunol, 2015. 15(2): p. 117–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peterson JT, et al. , Evolution of matrix metalloprotease and tissue inhibitor expression during heart failure progression in the infarcted rat. Cardiovasc Res, 2000. 46(2): p. 307–15. [DOI] [PubMed] [Google Scholar]
- 24.Guerin CL, et al. , Human very Small Embryonic-like Cells Support Vascular Maturation and Therapeutic Revascularization Induced by Endothelial Progenitor Cells. Stem Cell Rev, 2017. 13(4): p. 552–560. [DOI] [PubMed] [Google Scholar]
- 25.Kaushik A and Bhartiya D, Pluripotent Very Small Embryonic-Like Stem Cells in Adult Testes - An Alternate Premise to Explain Testicular Germ Cell Tumors. Stem Cell Rev, 2018. 14(6): p. 793–800. [DOI] [PubMed] [Google Scholar]
- 26.Shaikh A, et al. , Mouse Bone Marrow VSELs Exhibit Differentiation into Three Embryonic Germ Lineages and Germ & Hematopoietic Cells in Culture. Stem Cell Rev, 2017. 13(2): p. 202–216. [DOI] [PubMed] [Google Scholar]
- 27.Ratajczak MZ, et al. , Pivotal role of paracrine effects in stem cell therapies in regenerative medicine: can we translate stem cell-secreted paracrine factors and microvesicles into better therapeutic strategies? Leukemia, 2012. 26(6): p. 1166–73. [DOI] [PubMed] [Google Scholar]
- 28.Wojakowski W, et al. , Very small embryonic-like stem cells in cardiovascular repair. Pharmacol Ther, 2011. 129(1): p. 21–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zuba-Surma EK, et al. , Very small embryonic-like stem cells: biology and therapeutic potential for heart repair. Antioxid Redox Signal, 2011. 15(7): p. 1821–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Losordo DW, et al. , Intramyocardial, autologous CD34+ cell therapy for refractory angina. Circ Res, 2011. 109(4): p. 428–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zuba-Surma EK, et al. , Transplantation of expanded bone marrow-derived very small embryonic-like stem cells (VSEL-SCs) improves left ventricular function and remodelling after myocardial infarction. J Cell Mol Med, 2011. 15(6): p. 1319–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Blandinieres A, et al. , Endothelial Colony-Forming Cells Do Not Participate to Fibrogenesis in a Bleomycin-Induced Pulmonary Fibrosis Model in Nude Mice. Stem Cell Rev, 2018. 14(6): p. 812–822. [DOI] [PubMed] [Google Scholar]
- 33.d’Audigier C, et al. , Egfl7 Represses the Vasculogenic Potential of Human Endothelial Progenitor Cells. Stem Cell Rev, 2018. 14(1): p. 82–91. [DOI] [PubMed] [Google Scholar]
- 34.Brenner W, et al. , 111In-labeled CD34+ hematopoietic progenitor cells in a rat myocardial infarction model. J Nucl Med, 2004. 45(3): p. 512–8. [PubMed] [Google Scholar]
- 35.Abbott JD, et al. , Stromal cell-derived factor-1alpha plays a critical role in stem cell recruitment to the heart after myocardial infarction but is not sufficient to induce homing in the absence of injury. Circulation, 2004. 110(21): p. 3300–5. [DOI] [PubMed] [Google Scholar]
- 36.Kucia M, et al. , CXCR4-SDF-1 signalling, locomotion, chemotaxis and adhesion. J Mol Histol, 2004. 35(3): p. 233–45. [DOI] [PubMed] [Google Scholar]
- 37.Marquez-Curtis LA, et al. , The ins and outs of hematopoietic stem cells: studies to improve transplantation outcomes. Stem cell reviews, 2011. 7(3): p. 590–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McQuibban GA, et al. , Matrix metalloproteinase activity inactivates the CXC chemokine stromal cell-derived factor-1. J Biol Chem, 2001. 276(47): p. 43503–8. [DOI] [PubMed] [Google Scholar]
- 39.McQuibban GA, et al. , Matrix metalloproteinase processing of monocyte chemoattractant proteins generates CC chemokine receptor antagonists with anti-inflammatory properties in vivo. Blood, 2002. 100(4): p. 1160–7. [PubMed] [Google Scholar]
- 40.Agarwal U, et al. , Role of cardiac myocyte CXCR4 expression in development and left ventricular remodeling after acute myocardial infarction. Circ Res, 2010. 107(5): p. 667–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lilly JL and Berron BJ, The Role of Surface Receptor Density in Surface-Initiated Polymerizations for Cancer Cell Isolation. Langmuir, 2016. 32(22): p. 5681–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lilly JL, et al. , Interfacial polymerization for colorimetric labeling of protein expression in cells. PLoS One, 2014. 9(12): p. e115630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fedorovich NE, et al. , Hydrogels as extracellular matrices for skeletal tissue engineering: state-of-the-art and novel application in organ printing. Tissue Eng, 2007. 13(8): p. 1905–25. [DOI] [PubMed] [Google Scholar]
- 44.Hamidi M, Azadi A, and Rafiei P, Hydrogel nanoparticles in drug delivery. Adv Drug Deliv Rev, 2008. 60(15): p. 1638–49. [DOI] [PubMed] [Google Scholar]
- 45.Lin CC and Anseth KS, PEG hydrogels for the controlled release of biomolecules in regenerative medicine. Pharm Res, 2009. 26(3): p. 631–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Takagi J, Asai H, and Saito Y, A collagen/gelatin-binding decapeptide derived from bovine propolypeptide of von Willebrand factor. Biochemistry, 1992. 31(36): p. 8530–4. [DOI] [PubMed] [Google Scholar]
- 47.San BH, et al. , Nanoparticle Assembly and Gelatin Binding Mediated by Triple Helical Collagen Mimetic Peptide. ACS Appl Mater Interfaces, 2016. 8(31): p. 19907–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chan TR, et al. , Collagen-gelatin mixtures as wound model, and substrates for VEGF-mimetic peptide binding and endothelial cell activation. Acta Biomater, 2015. 15: p. 164–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Blackburn NJ, et al. , Timing underpins the benefits associated with injectable collagen biomaterial therapy for the treatment of myocardial infarction. Biomaterials, 2015. 39: p. 182–92. [DOI] [PubMed] [Google Scholar]
- 50.Holladay CA, et al. , Recovery of cardiac function mediated by MSC and interleukin-10 plasmid functionalised scaffold. Biomaterials, 2012. 33(5): p. 1303–14. [DOI] [PubMed] [Google Scholar]
- 51.Serpooshan V, et al. , The effect of bioengineered acellular collagen patch on cardiac remodeling and ventricular function post myocardial infarction. Biomaterials, 2013. 34(36): p. 9048–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Quyyumi AA, et al. , PreSERVE-AMI: A Randomized, Double-Blind, Placebo-Controlled Clinical Trial of Intracoronary Administration of Autologous CD34+ Cells in Patients With Left Ventricular Dysfunction Post STEMI. Circ Res, 2017. 120(2): p. 324–331. [DOI] [PMC free article] [PubMed] [Google Scholar]