

Abstract
Glioblastomas are the most aggressive and lethal primary astrocytic tumors of the central nervous system. They account for 60% to 70% of all gliomas and the majority are diagnosed in Caucasian male patients at advanced age. Genetic analyses of glioblastoma show a great intra- and inter-tumor heterogeneity, which opens up a debate about its cellular origin. Different types of brain cells, including astrocytes, neural stem cells, oligodendrocyte precursor cells and glioblastoma stem cells are proposed to have a role in tumor initiation and spreading; however, data is still inconclusive. Due to short life expectancy, long-term glioblastoma survivors are defined as patients who live longer than two years post-diagnosis. Extreme survivors, living 10 years or more after diagnosis, comprise less than 1% of all patients. Molecular testing indicates genetic differences between short- and long-term survivors with glioblastoma. The most informative are IDH1/2 gene mutations and MGMT promoter methylation, which are associated with a better response to standard clinical care. Moreover, a decreased expression of the CHI3L1, FBLN4, EMP3, IGFBP2, IGFBP3, LGALS3, MAOB, PDPN, SERPING1 and TIMP1 genes has been associated with prolonged survival. In addition, emerging evidence suggests the role of different microRNAs in predicting patient survival. Other factors that may affect the survival of glioblastoma patients include clinical/demographic characteristics such as seizures at presentation, age at diagnosis, and the extent of surgical resection. Because of the small number of long-term survivors with glioblastoma, comparative studies on genetic differences between short- and long-term survivors are challenging. To improve patient management and clinical outcomes, a thorough “omics” approach is necessary for identifying differences between short- and long-term survivors with glioblastoma.
Keywords: Glioblastoma, genetics, IDH, MGMT, chromosome 1p/19q, long term survival, extreme survivors
INTRODUCTION
Diverse nature of glioblastoma
The term “glioma” refers to a type of brain tumor which originates in the parenchyma of the central nervous system (CNS), more precisely from the supportive glial cells – ependymal cells, astrocytes, and oligodendrocytes. According to the cell type they originate from or share histological features with, gliomas are divided into ependymomas, astrocytomas, and oligodendrogliomas. Of these, astrocytomas are the most common type of glial tumors. The World Health Organization (WHO) divides gliomas into four grades starting from pilocytic astrocytoma (Grade I), diffuse astrocytoma (Grade II), anaplastic astrocytoma (Grade III), and glioblastoma (Grade IV) [1,2]. Grades III and IV are considered high-grade gliomas and represent the majority of brain tumors [3]. Glioblastomas are astrocytic tumors with necrosis and microvascular proliferation. Patients suffering from this most malignant type usually succumb to the disease in 12 to 18 months after diagnosis [4]. Glioblastoma incidence is very low among all cancer types, i.e., 1 per 10 000 cases. However, with an incidence of 16% of all primary brain tumors it is the most common brain malignancy and is almost always lethal [5,6]. According to malignant progression, there are two types of glioblastomas: primary, originating de novo, and secondary, evolving from lower-grade gliomas. Based on the presence or absence of isocitrate dehydrogenase (IDH) 1 and 2 gene mutations and chromosome 1p/19q codeletion, in adults, primary glioblastomas are also defined as IDH wild-type, while secondary can either be IDH mutant and 1p/19q intact, or IDH mutant and 1p/19q codeleted [7,8]. The most frequent IDH1 mutations are at codon 132; in 90% of the cases, the mutation is R132H. Other known mutations are R132C, R132G, and R132S [9]. IDH2 mutations are less frequent and occur at codon 172, with R172K being the most common. Primary IDH wild-type glioblastomas present with mutations in the TERT, PTEN, and TP53 genes as well as with amplification of the EGFR, PDGFRA, CDK4, CDK6, MDM2, and MDM4 genes, while secondary glioblastomas show mutations in the IDH, TP53, and ATRX genes, as well as deletion of the CDKN2A [10-12]. IDH mutations are believed to be among the first changes that occur in gliomagenesis, and are most commonly accompanied by TP53 and ATRX mutations [13]. The ATRX gene enables incorporation of histone variant H3.3 into heterochromatin, which results in changes in telomere length and genomic instability [6]. A study by Reuss et al. reported the existence of three distinct glioblastoma sets: chromosome 7p gain and 10q loss with the absence of IDH mutations and the presence of nuclear ATRX expression; nuclear ATRX loss and/or IDH mutations; and nuclear ATRX loss with H3F3A mutations and without IDH mutations, which represents an adult glioblastoma subset that has similarities with pediatric glioblastoma [14]. In addition, expression profiling of glioblastoma specimens defines four different molecular subtypes. A large-scale genomic study by Verhaak et al. analyzed >200 glioblastoma samples using three gene expression platforms (Affymetrix HuEx, Affymetrix U133A, and Agilent 244K arrays) and identified four robust glioblastoma clusters or subtypes, named based on the expression of signature genes as follows: classical, mesenchymal, neural, and proneural subtype [15]. The distinct genetic events of each subtype were identified by analyzing data available from The Cancer Genome Atlas (TCGA) Research Network and are presented in Figure 1 [9,15-20].
FIGURE 1.
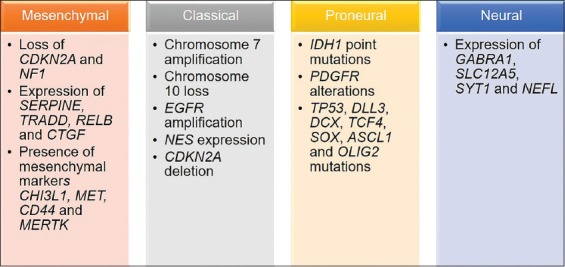
Glioblastoma subtypes. The most common genetic changes in the four glioblastoma subtypes (classical, neural, proneural, and mesenchymal), as described by Verhaak et al. [15], are given in the scheme.
Using a bioinformatics approach, Wang et al. identified 1520 differentially expressed genes in glioblastoma compared to non-tumor glial cells of epilepsy patients [21]. In their work, the signaling pathways most commonly associated with upregulated genes were the Wnt, MAPK, and ErbB pathways, while the p53 signaling cascade, extracellular matrix (ECM)-receptor interaction, and antigen processing and presentation were associated with downregulated genes. Of these, the Wnt signaling pathway plays a key role in neurogenesis and embryonic brain development, and is able to modulate self-renewal and differentiation of adult tissue stem cells [22]. Furthermore, the Wnt signaling pathway is commonly dysregulated in tumorigenesis [23]. Abnormal activation of the Wnt signaling leads to glioblastoma growth and invasion [24]. The T-cell factor/lymphoid enhancer-binding factor (TCF/LEF) family of transcription factors involved in the Wnt pathway has also been associated with glioblastoma malignancy [23]. Immunohistochemical staining of glioblastomas showed a strong expression of TCF-1 and LEF-1 in 51.6% and 71% of analyzed samples, respectively. The study by Pećina-Šlaus et al. indicated the increased expression of transcriptional factors TCF-1 and LEF-1 to be characteristic for malignant gliomas [23]. Other signaling pathways, including receptor tyrosine kinase (RTK), epidermal growth factor receptor (EGFR), platelet-derived growth factor receptor A (PDGFRA), fibroblast growth factor receptor 1 (FGFR-1), insulin-like growth factor receptor 1 (IGFR-1), nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), MET, and Sonic hedgehog (SHH) are also altered in glioblastoma [24,25]. The reason for such genetic diversity in glioblastomas is not clear. It is thought to be either the result of different cellular origin or of the same cellular origin but different response to signals from the microenvironment.
Glioblastomas are very heterogeneous in nature. They present with cellular and molecular diversity not only among tumors but also within the same tumor [26,27]. Coexistence of cells with different properties has been proven by numerous genetic studies [28-30], which suggests that glioblastomas may arise from different cell types. Various genetic changes including mutations, chromosomal aberrations, and copy number variations in both oncogenes and tumor suppressor genes have been found [7]. These changes can be either clonal (or early events) – present in all cells before the malignant transformation, or subclonal (or late events) – present in a subset of cells after the malignant transformation [31]. Such genetic diversity implies glioblastoma is not a single condition, but most likely a set of diseases.
Glioblastoma cellular origin
Glioblastomas can arise anywhere in the CNS; proneural and neural subtypes arise in or near the subventricular zone, mesenchymal and classical subtypes are distal to the subventricular zone, other gliomas arise in the superficial subcortical white matter. Proneural glioblastomas with IDH mutations are more likely to occur in the frontal lobes [5,8]. Such a correlation between tumor subtype and intracranial location can be a result of different cell origin. Glioblastoma cellular origin is controversial and still a matter of debate, with two dominant theories: dedifferentiation theory and stem cell theory (Figure 2).
FIGURE 2.
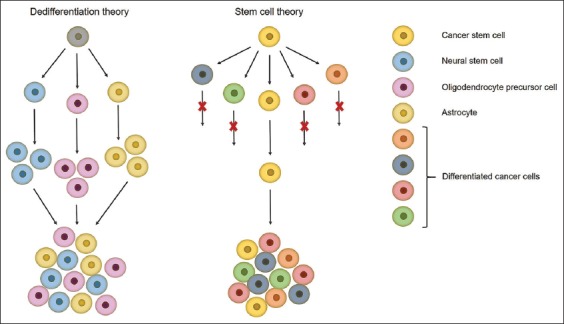
Schematic representation of the two theories about glioblastoma cellular origin. Dedifferentiation theory – all cells have tumorigenic potential but under different stimuli only some of them will contribute to tumor growth. Stem cell theory – only a fraction of cells, named cancer stem cells, are able to self-renew, initiate and regrow a tumor. Cell figures are for graphical representation only and do not show actual cell shapes.
The first proposed mechanism of glioblastoma origin is the “dedifferentiation theory or stochastic model” which states that all cells are equipotent, but under different genetic or epigenetic stimuli only some of them will contribute to tumor growth [32,33]. In the past, it was thought that the adult brain does not regenerate, and astrocytes were considered to be the only dividing cells in the adult brain. In addition to this, the glial fibrillary acidic protein (GFAP) astrocytic marker was frequently found in glioma tissues, which led to the conclusion that astrocytes are the cells of origin of gliomas, as reported by Jiang and Uhrbom [1]. This theory was questioned after the discovery of self-renewing and multipotent neural stem cells (NSCs) in the late 1990s, which have been successfully isolated from the subventricular zone, hippocampus, and dentate gyrus of the adult mammalian brain [34-36]. NSCs express the surface markers nestin, SRY (sex determining region Y)-box 2 (SOX2), prominin-1 (CD133), and glial fibrillary acidic protein (GFAP) [8]. Gliomas arise near the subventricular zone which is consistent with the location of NSCs in the mammalian brain, and this is why NSCs were proposed as the cells of origin of gliomas. Furthermore, the EGFR and AKT/PKB signaling pathways, which are important for NSC proliferation and differentiation, are commonly altered in gliomas. Lastly, oligodendrocyte precursor cells (OPCs) were proposed as the cells of origin of gliomas, for several reasons: they are the major dividing cells in the adult brain, they can give rise to oligodendrocytes and they are present in the subventricular zone, white and gray brain matter [1]. This hypothesis was supported by frequent alterations in the PDGFRA signaling pathway in gliomas – a pathway that is important for normal oligodendrocyte development. Moreover, the OPC markers neuron-glial antigen 2 (NG2), oligodendrocyte transcription factor 2 (OLIG2), and PDGFR are found in gliomas, which indicates a direct link between the tumors and OPCs. The regenerative potential of astrocytes, NSCs, and OPCs makes them plausible candidates for cells of origin of gliomas [37]. Still, despite all evidence pointing in the direction of these cells, glioblastoma origin remains unsolved.
The second mechanism is the so-called “hierarchical model or stem cell theory” which argues that tumors contain a subset of cells, named cancer stem cells (CSCs), able to proliferate, give rise to and reseed a tumor. CSCs are described as cells able to self-renew and generate more differentiated tumor cells, which is believed to be accomplished by asymmetric division where one daughter cell retains stem cell properties while the other differentiates into different types of tumor cells [38]. CSCs have been proven to be more resistant to genotoxic treatments, which seems to be the cause of tumor recurrence [34,38]. Their resistance to radiation is a result of a higher DNA repair rate, as shown by Bao et al. [39]. The authors compared early DNA damage checkpoint responses in CD133+ and CD133− cells and found that ionizing radiation initiates activating phosphorylation of ATM, Rad17, checkpoint kinase 1 (Chk1), and Chk2 checkpoint proteins in these cells. Moreover, the activating phosphorylation was significantly higher in CD133+ compared to CD133− cells, indicating that CD133+ cells are characterized by greater checkpoint activation to DNA damage. The authors also showed that CD133+ cells repair DNA damage more efficiently than CD133− cells. This ability to repair DNA damage more rapidly suggests CD133+ cells are able to survive radiation and reseed a tumor [39]. The great self-renewal ability of CSCs favors the accumulation of mutations, which ultimately leads to tumor formation and progression [22]. However, the existence of CSCs is difficult to prove because of a lack of specific biomarkers. For the identification of CSCs, markers associated with immature cells and normal stem cells are used. CSCs expressing CD133 were isolated from glioblastomas, so CD133 is considered as a potential biomarker of CSCs [36,40,41]. However, it was later shown that CD133− cells can also give rise to a glioma [42-44]. Moreover, the cell surface marker cluster of differentiation 15 (CD15) has also been proposed as a glioma stem cell marker [44,45]. Still, the “gold standard” for the identification of CSCs is their ability to give rise to a phenotypically identical tumor as the primary malignancy, in immunocompromised mice.
In the case of glioblastomas, it is highly likely that multiple cell lineages are simultaneously present in the tumor. This also suggests that various cell types are responsible for glioma initiation and development. It is, thus, important to continue investigating glioma cellular origin and how specific cell types can contribute to glioma formation, progression, and recurrence [1].
CLINICAL MANAGEMENT OF GLIOBLASTOMA AND MOLECULAR BIOMARKERS
Late diagnosis of glioblastoma is a result of unspecific symptoms such as headache, confusion, memory loss and personality changes, which can also be accompanied by problems in motor function and speech [46]. Diagnosis is performed with computed tomography and magnetic resonance imaging or magnetic resonance spectroscopy and confirmed with molecular techniques including immunohistochemistry, Sanger sequencing, fluorescence in situ hybridization, and microsatellite analysis [47]. Standard of care consists of maximal surgical resection followed by radiation and temozolomide chemotherapy [48,49]. However, even with such an aggressive treatment in 75% to 90% of the glioblastoma cases, the tumor recurs within 7 to 10 months after surgery. Only 9% of glioblastoma patients are still alive two years post diagnosis and these are considered long-term survivors [50]. Major issues in glioblastoma management are its intracranial location, fast growth, and infiltrative nature that leads to incomplete surgical resection and development of therapy resistance [37]. Furthermore, chemotherapy offers limited options due to poor drug penetration through the blood–brain barrier [51]. Identifying the cell of origin of glioblastoma is of great importance for patient care. If treatment could be tailored to target a specific subset of cells in every patient, the effectiveness of clinical care would be greatly improved (Figure 3) [5]. Besides, targeting specific cell types would lead to the design of novel drugs with minimal toxicity to other non-malignant cells [8].
FIGURE 3.
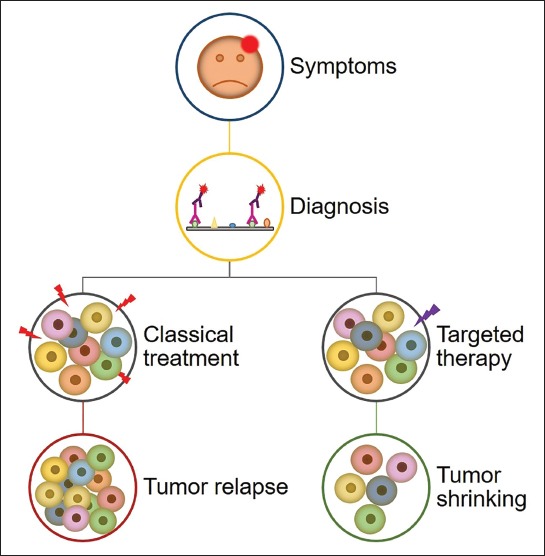
Conventional vs. targeted therapy in glioblastoma. A schematic representation of appearance of symptoms and glioblastoma diagnosis by immunohistochemistry, followed by two possible treatment approaches: conventional therapy is aiming at the majority of cells, which in most cases results in tumor recurrence; and targeted therapy that targets a specific cell type or cell property that will ultimately lead to tumor shrinking.
So far, the only confirmed molecular biomarkers for glioblastoma are O6-methylguanine-DNA methyltransferase (MGMT) promoter methylation, IDH1/2 mutations, and loss of heterozygosity in chromosome 1p/19q [3,14,51,52]. The alkylating agent temozolomide causes DNA damage by adding alkyl groups to guanine O6 position. This change is effectively repaired by the DNA repair protein MGMT which restores guanine from O6-methylguanine and reverses the effect of chemotherapy. Nevertheless, when MGMT promoter is methylated, the protein expression is decreased, which leads to better sensitivity of tumor to temozolomide chemotherapy [53]. The survival of patients with MGMT promoter methylation is significantly higher (21.7 months) compared to patients with non-methylated promoter (15.3 months) [51]. IDH mutations are correlated with excessive genome methylation resulting in glioma-specific CpG island methylator phenotype (G-CIMP). IDH mutations are also commonly associated with MGMT promoter methylation as they occur in 79% of G-CIMP and 46% of non-G-CIMP [3,51]. Moreover, IDH wild-type glioblastomas are often found in brain areas that are difficult to access surgically. IDH mutations alone are not related to long-term survival, but when paired with MGMT promoter methylation they are considered a significant prognostic factor. Finally, chromosome 1p/19q codeletion is considered beneficial for elderly patients receiving procarbazine (P), lomustine (C or CCNU), vincistrine (V) – PCV chemotherapy. The combination of whole arm 1p/19q codeletion and IDH mutations seems to be favorable. In patients who receive temozolomide chemotherapy the presence of 1p/19q codeletion was linked to a longer duration of response to chemotherapy [54]. In contrast, chemoresistant tumors were found to contain both copies of chromosome 1p. In addition, patients with chromosomes 1p and 19q have 5.7 times higher risk of recurrence compared to patients with allele loss on these chromosomes [52].
LONG-TERM GLIOBLASTOMA SURVIVORS
Gliomas can occur at any age, but the majority arise in older patients. Primary glioblastomas are more common among Caucasian men in advanced age, while lower-grade gliomas and secondary glioblastomas are more common in younger adults (45 years and younger) [55]. Gender differences are attributed to hormonal changes and genetic features of patients [8]. Due to frequent disease recurrence, only 3% to 5% of glioblastoma patients live longer than three years after diagnosis [56]. Prognosis depends on patients’ age at diagnosis, with younger patients having a better outcome. This can be partially explained with better overall health, but it can also be a result of different molecular and genetic alterations in younger compared to older patients [5]. Although glioblastomas from short- and long-term survivors are histologically the same, their biological and molecular characteristics are remarkably different [57].
In general, gliomas are very aggressive tumors. Astrocytomas are associated with worse prognosis, while oligodendrogliomas have been related to better outcomes [5]. Long-term survivors comprise approximately 10% of all glioblastoma patients [58]. Still, only a small number of patients show strong response to therapy and extremely long-term survival of 10 years or more. Tykocki and Eltayeb performed literature analysis on clinical studies containing information for extreme survivors, i.e., glioblastoma patients surviving 10 years or longer [59]. According to their systematic review, 0.71% of all glioblastoma patients have a survival longer than 10 years. Moreover, the authors found a relationship between age at diagnosis and overall survival (OS), for every 4.7 years younger age at diagnosis the OS was one year longer after 10 years of survival. In general, the 10-year OS of glioblastoma patients varies among different age groups: 0–14, 15–39, and 40+ years groups have 14.9%, 13.6%, and 1.6% of long-term survivors, respectively. The most common clinical symptom of glioblastoma, epileptic seizures, is another factor associated with the survival of patients. Seizures can occur either at initial diagnosis or result from recurrence of the disease. Although they reduce the patient quality of life, seizures as a symptom are positively correlated to longer survival in glioblastoma patients [60-62]. However, in cases where there is a longer delay between epileptic seizures as a symptom and surgical resection of tumor, this correlation does not seem to be significant [62,63].
Regarding the association between genetic mutations and survival, reports show that glioblastomas with IDH1/2 mutations and MGMT promoter methylation are more responsive to surgical resection and temozolomide chemotherapy, and have better prognosis [57,64-66]. Hartmann et al. compared tumor samples from 33 patients with primary glioblastoma who lived longer than 60 months to patients whose OS was not longer than 36 months [67]. They showed more frequent MGMT promoter methylation (66.7% vs. 33.6%) and less frequent IDH1/2 mutations (63.6% vs. 96.4% for IDH1 and 96.9% vs. 99.2% for IDH2) in the long-term survival group. In their study, 1p/19q codeletions were not common and EGFR amplifications were absent. Li et al. analyzed three microarray datasets containing eight controls, 58 long-term and 135 short-term survivors, and found that the FBLN4, IGFBP2, and CHI3L1 genes are negatively associated with glioblastoma survival [68]. Expression levels of these three genes were significantly increased in short-term vs. long-term survivors and normal controls. The authors concluded that increased FBLN4, IGFBP2, and CHI3L1 mRNA expression levels are associated with decreased survival probability. Similarly, Gerber et al. found that decreased mRNA expression levels of the CHI3L1, EMP3, IGFBP2, IGFBP3, LGALS3, MAOB, PDPN, SERPING1, and TIMP1 genes are associated with longer survival [66]. Moreover, they showed high MGMT promoter methylation in long-term survivors. Although glioblastoma samples in Gerber’s study could not be classified into a single subtype as described by the Verhaak classification [15], several genes, such as the CHI3L1, EMP3, PDPN and TIMP1 genes, were linked to the mesenchymal subtype [66]. The study by Michaelsen et al. reported that the MGMT, IFNG, CXLC9, LGALS4, and CD34 genes are prognostic factors in glioblastoma [65]. The authors further validated bioinformatically the potential use of MGMT, IFNG, CXLC9, LGALS4 and CD34 as prognostic factors using data available from AVAglio study, and confirmed high expression of CD34 in the long-term survival group [65,69]. When CD34 expression was analyzed in relation to clinical and pathological features of patients (age, corticosteroid use, and MGMT promoter methylation) it showed association with prolonged OS. This was also the case for MGMT mRNA expression [65]. Moreover, an interesting observation is the involvement of alpha thalassemia/mental retardation syndrome X-linked (ATRX) gene in longer survival of glioblastoma patients [6,70]. Using a gene-targeted next-generation sequencing panel, Cantero et al. found better prognosis for glioblastoma patients whose tumors present with ATRX or DAXX mutations in the absence of IDH or H3F3A mutations [70]. Similar findings were published by Chaurasia et al. who reported significantly higher OS and progression-free survival (PFS) in glioblastoma patients with alpha thalassemia/mental retardation syndrome X-linked (ATRX) and IDH1 mutant protein expression (ATRX- and IDH+, respectively) [6]. Moreover, their immunohistochemical examination of formalin-fixed paraffin-embedded tissue samples showed that ATRX-/IDH+ patients had the longest OS (42.71 months) and PFS (42.2 months), while ATRX+/IDH- patients had the lowest OS (20.7 months) and PFS (16.8 months). Increasing evidence shows that microRNAs (miRNAs) have a prognostic value in long-term survival of glioblastoma patients [22,71-73]. Hermansen et al. reported differences in miRNA profiles of short- and long-term glioblastoma survivors (40 patients total). They identified four miRNAs (miR-107, hsa-miR-548, miR-3125, and miR-331-3p) that were associated with shorter survival. However, when only glioblastoma patients with known MGMT methylation status were analyzed, low miR sum score was found to be an independent negative prognostic factor [74]. On the other hand, Yuan et al. [57] found increased levels of let-7g-5p, miR-139-5p, miR-17-5p, and miR-9-3p in tumor tissue samples from long-term glioblastoma survivors independent of their MGMT methylation status. Patients with high expression levels of these miRNAs lived 88 days longer than patients with low expression levels (439 vs. 351 days, respectively) [57].
A case report described a single patient with 20-year survival after glioblastoma diagnosis [75]. The 45-year-old Caucasian man was diagnosed with glioblastoma at the age of 25 years. After initial treatment (surgery and radiation) there were two tumor recurrences, two and 20 years after initial diagnosis. Molecular testing was performed only in tumor specimens from the second recurrence and revealed MGMT promoter methylation, PTEN and TP53 expression positive, EGFR and protein kinase AKT expression negative. Although this isolated case is not enough to draw a general conclusion, the molecular profile of the patient could give information about better response to treatment and potentially long-term survival of glioblastoma patients who appear with the described genetic profile.
In general, younger age is a predictor of better OS. Patients diagnosed with glioblastoma below the age of 40 years have a greater chance for longer survival, especially for a survival of more than 10 years after diagnosis [76,77]. Another factor affecting the prognosis of glioblastoma patients is the extent of surgical resection, e.g., the 12-month PFS is increased by 50% after complete tumor resection. However, there are also reports of patients who underwent incomplete resection or biopsy only but who survived for more than a decade [59]. In addition to younger age and good performance status at diagnosis, adjuvant chemotherapy is considered beneficial for glioblastoma patients [58]. Epileptic seizures are also favorable, probably due to a higher chance of an early stage disease diagnosis, when the tumor is smaller, and a total resection of tumor. Regarding the genetic factors, patients with MGMT promoter methylation and IDH1/2 mutations are more common among long-term survivors.
CONCLUSION
Glioblastoma is a significant medical problem of modern society because of its high mortality rate. Even with aggressive clinical care, long-term glioblastoma survivors comprise <15% of all cases. Further extensive research and multidisciplinary “omics” approach is needed to understand the natural causes of glioblastoma occurrence. Clinical features such as age at diagnosis, presence of seizures as an initial symptom, and extent of surgical resection are known factors that contribute to patients’ life expectancy. However, for better patient management, genetic, epigenetic, transcriptomic and proteomic information should supplement the early clinical symptoms. So far, IDH mutations and MGMT promoter methylation are the most important molecular factors for determining long-term survival of glioblastoma patients. Revealing the factors that contribute to patients’ longevity is important for precise diagnosis and correct clinical management of the disease. Due to the small number of patients with long life expectancy, comparative studies about genetic differences between short- and long-term survivors are challenging, making this phenomenon poorly understood. However, identification of such differences is crucial for establishing the mechanism of glioblastoma pathology.
ACKNOWLEDGMENTS
The author would like to thank the Interreg EC Project 2014-2020, Ref. No. 146, Acronym: TRANS-GLIOMA, and the Research Programme Grant P1-0390 from the Slovenian Research Agency (ARRS) for funding. The funders had no role in the decision to publish or preparation of the manuscript.
DECLARATION OF INTERESTS
The author declares no conflict of interests.
REFERENCES
- 1.Jiang Y, Uhrbom L. On the origin of glioma. Ups J Med Sci. 2012;117(2):113–21. doi: 10.3109/03009734.2012.658976. https://doi.org/10.3109/03009734.2012.658976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 world health organization classification of tumors of the central nervous system:A summary. Acta Neuropathol. 2016;131(6):803–20. doi: 10.1007/s00401-016-1545-1. https://doi.org/10.1007/s00401-016-1545-1. [DOI] [PubMed] [Google Scholar]
- 3.Zhang C, Bao Z, Zhang W, Jiang T. Progress on molecular biomarkers and classification of malignant gliomas. Front Med. 2013;7(2):150–6. doi: 10.1007/s11684-013-0267-1. https://doi.org/10.1007/s11684-013-0267-1. [DOI] [PubMed] [Google Scholar]
- 4.Nørøxe DS, Poulsen HS, Lassen U. Hallmarks of glioblastoma:A systematic review. ESMO Open. 2016;1(6):e000144. doi: 10.1136/esmoopen-2016-000144. http://dx.doi.org/10.1136/esmoopen-2016-000144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zong H, Verhaak RG, Canoll P. The cellular origin for malignant glioma and prospects for clinical advancements. Expert Rev Mol Diagn. 2012;12(4):383–94. doi: 10.1586/erm.12.30. https://doi.org/10.1586/erm.12.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chaurasia A, Park SH, Seo JW, Park CK. Immunohistochemical analysis of ATRX, IDH1 and p53 in glioblastoma and their correlations with patient survival. J Korean Med Sci. 2016;31(8):1208–14. doi: 10.3346/jkms.2016.31.8.1208. https://doi.org/10.3346/jkms.2016.31.8.1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barthel FP, Wesseling P, Verhaak RGW. Reconstructing the molecular life history of gliomas. Acta Neuropathol. 2018;13(5):649–70. doi: 10.1007/s00401-018-1842-y. https://doi.org/10.1007/s00401-018-1842-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cahill D, Turcan S. Origin of gliomas. Semin Neurol. 2018;38(1):5–10. doi: 10.1055/s-0037-1620238. https://doi.org/10.1055/s-0037-1620238. [DOI] [PubMed] [Google Scholar]
- 9.Agnihotri S, Aldape KD, Zadeh G. Isocitrate dehydrogenase status and molecular subclasses of glioma and glioblastoma. Neurosurg Focus. 2014;37(6):E13. doi: 10.3171/2014.9.FOCUS14505. https://doi.org/10.3171/2014.9.FOCUS14505. [DOI] [PubMed] [Google Scholar]
- 10.Ohgaki H, Dessen P, Jourde B, Horstmann S, Nishikawa T, Di Patre PL, et al. Genetic pathways to glioblastoma:A population-based study. Cancer Res. 2004;64(19):6892–9. doi: 10.1158/0008-5472.CAN-04-1337. https://doi.org/10.1158/0008-5472.CAN-04-1337. [DOI] [PubMed] [Google Scholar]
- 11.Ohgaki H, Kleihues P. Genetic pathways to primary and secondary glioblastoma. Am J Pathol. 2007;170(5):1445–53. doi: 10.2353/ajpath.2007.070011. https://doi.org/10.2353/ajpath.2007.070011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Olar A, Aldape KD. Using the molecular classification of glioblastoma to inform personalized treatment. J Pathol. 2014;232(2):165–77. doi: 10.1002/path.4282. https://doi.org/10.1002/path.4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jovčevska I. Sequencing the next generation of glioblastomas. Crit Rev Clin Lab Sci. 2018;55(4):264–82. doi: 10.1080/10408363.2018.1462759. https://doi.org/10.1080/10408363.2018.1462759. [DOI] [PubMed] [Google Scholar]
- 14.Reuss DE, Sahm F, Schrimpf D, Wiestler B, Capper D, Koelsche C, et al. ATRX and IDH1-R132H immunohistochemistry with subsequent copy number analysis and IDH sequencing as a basis for an “integrated”diagnostic approach for adult astrocytoma, oligodendroglioma and glioblastoma. Acta Neuropathol. 2015;129(1):133–46. doi: 10.1007/s00401-014-1370-3. https://doi.org/10.1007/s00401-014-1370-3. [DOI] [PubMed] [Google Scholar]
- 15.Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17(1):98–110. doi: 10.1016/j.ccr.2009.12.020. https://doi.org/10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Friedmann-Morvinski D. Glioblastoma heterogeneity and cancer cell plasticity. Crit Rev Oncog. 2014;19(5):327–36. doi: 10.1615/critrevoncog.2014011777. https://doi.org/10.1615/CritRevOncog.2014011777. [DOI] [PubMed] [Google Scholar]
- 17.Li R, Li H, Yan W, Yang P, Bao Z, Zhang C, et al. Genetic and clinical characteristics of primary and secondary glioblastoma is associated with differential molecular subtype distribution. Oncotarget. 2015;6(9):7318–24. doi: 10.18632/oncotarget.3440. https://doi.org/10.18632/oncotarget.3440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Parker NR, Hudson AL, Khong P, Parkinson JF, Dwight T, Ikin RJ, et al. Intratumoral heterogeneity identified at the epigenetic, genetic and transcriptional level in glioblastoma. Sci Rep. 2016;6:22477. doi: 10.1038/srep22477. https://doi.org/10.1038/srep22477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mooney KL, Choy W, Sidhu S, Pelargos P, Bui TT, Voth B, et al. The role of CD44 in glioblastoma multiforme. J Clin Neurosci. 2016;34:1–5. doi: 10.1016/j.jocn.2016.05.012. https://doi.org/10.1016/j.jocn.2016.05.012. [DOI] [PubMed] [Google Scholar]
- 20.Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455(7216):1061–8. doi: 10.1038/nature07385. https://doi.org/10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang R, Wei J, Li Z, Tian Y, Du C. Bioinformatical analysis of gene expression signatures of different glioma subtypes. Oncol Lett. 2018;15(3):2807–14. doi: 10.3892/ol.2017.7660. https://doi.org/10.3892/ol.2017.7660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zuccarini M, Giuliani P, Ziberi S, Carluccio M, Iorio PD, Caciagli F, et al. The role of Wnt signal in glioblastoma development and progression:A Possible new pharmacological target for the therapy of this tumor. Genes (Basel) 2018;9(2):E105. doi: 10.3390/genes9020105. https://doi.org/10.3390/genes9020105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pećina-Šlaus N, Kafka A, Tomas D, Marković L, Okštajner PK, Sukser V, et al. Wnt signaling transcription factors TCF-1 and LEF-1 are upregulated in malignant astrocytic brain tumors. Histol Histopathol. 2014;29(12):1557–64. doi: 10.14670/HH-29.1557. https://doi.org/10.14670/HH-29.1557. [DOI] [PubMed] [Google Scholar]
- 24.Lee Y, Lee JK, Ahn SH, Lee J, Nam DH. WNT signaling in glioblastoma and therapeutic opportunities. Lab Invest. 2016;96(2):137–50. doi: 10.1038/labinvest.2015.140. https://doi.org/10.1038/labinvest.2015.140. [DOI] [PubMed] [Google Scholar]
- 25.Chowdhury FA, Hossain MK, Mostofa AG, Akbor MM, Bin Sayeed MS. Therapeutic potential of thymoquinone in glioblastoma treatment:Targeting major gliomagenesis signaling pathways. Biomed Res Int. 2018;2018:4010629. doi: 10.1155/2018/4010629. https://doi.org/10.1155/2018/4010629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aum DJ, Kim DH, Beaumont TL, Leuthardt EC, Dunn GP, Kim AH, et al. Molecular and cellular heterogeneity:The hallmark of glioblastoma. Neurosurg Focus. 2014;37(6):E11. doi: 10.3171/2014.9.FOCUS14521. https://doi.org/10.3171/2014.9.FOCUS14521. [DOI] [PubMed] [Google Scholar]
- 27.Soeda A, Hara A, Kunisada T, Yoshimura S, Iwama T, Park DM. The evidence of glioblastoma heterogeneity. Sci Rep. 2015;5:7979. doi: 10.1038/srep07979. https://doi.org/10.1038/srep07979;https://doi.org/10.1038/srep09630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eder K, Kalman B. Molecular heterogeneity of glioblastoma and its clinical relevance. Pathol Oncol Res. 2014;20(4):777–87. doi: 10.1007/s12253-014-9833-3. https://doi.org/10.1007/s12253-014-9833-3. [DOI] [PubMed] [Google Scholar]
- 29.Meyer M, Reimand J, Lan X, Head R, Zhu X, Kushida M, et al. Single cell-derived clonal analysis of human glioblastoma links functional and genomic heterogeneity. Proc Natl Acad Sci U S A. 2015;112(3):851–6. doi: 10.1073/pnas.1320611111. https://doi.org/10.1073/pnas.1320611111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344(6190):1396–401. doi: 10.1126/science.1254257. https://doi.org/10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim H, Zheng S, Amini SS, Virk SM, Mikkelsen T, Brat DJ, et al. Whole-genome and multisector exome sequencing of primary and post-treatment glioblastoma reveals patterns of tumor evolution. Genome Res. 2015;25(3):316–27. doi: 10.1101/gr.180612.114. https://doi.org/10.1101/gr.180612.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Driessens G, Beck B, Caauwe A, Simons BD, Blanpain C. Defining the mode of tumour growth by clonal analysis. Nature. 2012;488(7412):527–30. doi: 10.1038/nature11344. https://doi.org/10.1038/nature11344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vermeulen L, Sprick MR, Kemper K, Stassi G, Medema JP. Cancer stem cells - old concepts, new insights. Cell Death Differ. 2008;15(6):947–58. doi: 10.1038/cdd.2008.20. https://doi.org/10.1038/cdd.2008.20. [DOI] [PubMed] [Google Scholar]
- 34.Borovski T, De Sousa E, Melo F, Vermeulen L, Medema JP. Cancer stem cell niche:The place to be. Cancer Res. 2011;71(3):634–9. doi: 10.1158/0008-5472.CAN-10-3220. https://doi.org/10.1158/0008-5472.CAN-10-3220. [DOI] [PubMed] [Google Scholar]
- 35.Eriksson PS, Perfilieva E, Björk-Eriksson T, Alborn AM, Nordborg C, Peterson DA, et al. Neurogenesis in the adult human hippocampus. Nat Med. 1998;4(11):1313–7. doi: 10.1038/3305. https://doi.org/10.1038/3305. [DOI] [PubMed] [Google Scholar]
- 36.Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, et al. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63(18):5821–8. [PubMed] [Google Scholar]
- 37.Zong H, Parada LF, Baker SJ. Cell of origin for malignant gliomas and its implication in therapeutic development. Cold Spring Harb Perspect Biol. 2015;7(5):a020610. doi: 10.1101/cshperspect.a020610. https://doi.org/10.1101/cshperspect.a020610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Borovski T, Vermeulen L, Sprick MR, Medema JP. One renegade cancer stem cell? Cell Cycle. 2009;8(6):803–8. doi: 10.4161/cc.8.6.7935. https://doi.org/10.4161/cc.8.6.7935. [DOI] [PubMed] [Google Scholar]
- 39.Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444(7120):756–60. doi: 10.1038/nature05236. https://doi.org/10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 40.Liu G, Yuan X, Zeng Z, Tunici P, Ng H, Abdulkadir IR, et al. Analysis of gene expression and chemoresistance of CD133+cancer stem cells in glioblastoma. Mol Cancer. 2006;5:67. doi: 10.1186/1476-4598-5-67. https://doi.org/10.1186/1476-4598-5-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, et al. Identification of human brain tumour initiating cells. Nature. 2004;432(7015):396–401. doi: 10.1038/nature03128. https://doi.org/10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 42.Ailles LE, Weissman IL. Cancer stem cells in solid tumors. Curr Opin Biotechnol. 2007;18(5):460–6. doi: 10.1016/j.copbio.2007.10.007. https://doi.org/10.1016/j.copbio.2007.10.007. [DOI] [PubMed] [Google Scholar]
- 43.Welte Y, Adjaye J, Lehrach HR, Regenbrecht CR. Cancer stem cells in solid tumors:Elusive or illusive? Cell Commun Signal. 2010;8(1):6. doi: 10.1186/1478-811X-8-6. https://doi.org/10.1186/1478-811X-8-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Azzarelli R, Simons BD, Philpott A. The developmental origin of brain tumours:A cellular and molecular framework. Development. 2018;145(10):dev162693. doi: 10.1242/dev.162693. https://doi.org/10.1242/dev.162693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim KH, Seol HJ, Kim EH, Rheey J, Jin HJ, Lee Y, et al. Wnt/beta-catenin signaling is a key downstream mediator of MET signaling in glioblastoma stem cells. Neuro Oncol. 2013;15(2):161–71. doi: 10.1093/neuonc/nos299. https://doi.org/10.1093/neuonc/nos299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yamanaka R. Cell-and peptide-based immunotherapeutic approaches for glioma. Trends Mol Med. 2008;14(5):228–35. doi: 10.1016/j.molmed.2008.03.003. https://doi.org/10.1016/j.molmed.2008.03.003. [DOI] [PubMed] [Google Scholar]
- 47.Zacher A, Kaulich K, Stepanow S, Wolter M, Köhrer K, Felsberg J, et al. Molecular diagnostics of gliomas using next generation sequencing of a glioma-tailored gene panel. Brain Pathol. 2017;27(2):146–59. doi: 10.1111/bpa.12367. https://doi.org/10.1111/bpa.12367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Aldape K, Zadeh G, Mansouri S, Reifenberger G, von Deimling A. Glioblastoma:Pathology, molecular mechanisms and markers. Acta Neuropathol. 2015;129(6):829–48. doi: 10.1007/s00401-015-1432-1. https://doi.org/10.1007/s00401-015-1432-1. [DOI] [PubMed] [Google Scholar]
- 49.Omuro A, De Angelis LM. Glioblastoma and other malignant gliomas:A clinical review. JAMA. 2013;310(17):1842–50. doi: 10.1001/jama.2013.280319. https://doi.org/10.1001/jama.2013.280319. [DOI] [PubMed] [Google Scholar]
- 50.Ho VK, Reijneveld JC, Enting RH, Bienfait HP, Robe P, Baumert BG, et al. Changing incidence and improved survival of gliomas. Eur J Cancer. 2014;50(13):2309–18. doi: 10.1016/j.ejca.2014.05.019. https://doi.org/10.1016/j.ejca.2014.05.019. [DOI] [PubMed] [Google Scholar]
- 51.Czapski B, Baluszek S, Herold-Mende C, Kaminska B. Clinical and immunological correlates of long term survival in glioblastoma. Contemp Oncol (Pozn) 2018;22(1A):81–5. doi: 10.5114/wo.2018.73893. https://doi.org/10.5114/wo.2018.73893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Monticelli M, Zeppa P, Zenga F, Altieri R, Mammi M, Bertero L, et al. The post-surgical era of GBM:How molecular biology has impacted on our clinical management. A review. Clin Neurol Neurosurg. 2018;170:120–6. doi: 10.1016/j.clineuro.2018.05.015. https://doi.org/10.1016/j.clineuro.2018.05.015. [DOI] [PubMed] [Google Scholar]
- 53.Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352(10):997–1003. doi: 10.1056/NEJMoa043331. https://doi.org/10.1056/NEJMoa043331. [DOI] [PubMed] [Google Scholar]
- 54.Thomas L, Di Stefano AL, Ducray F. Predictive biomarkers in adult gliomas:The present and the future. Curr Opin Oncol. 2013;25(6):689–94. doi: 10.1097/CCO.0000000000000002. https://doi.org/10.1097/CCO.0000000000000002. [DOI] [PubMed] [Google Scholar]
- 55.Dunn GP, Rinne ML, Wykosky J, Genovese G, Quayle SN, Dunn IF, et al. Emerging insights into the molecular and cellular basis of glioblastoma. Genes Dev. 2012;26(8):756–84. doi: 10.1101/gad.187922.112. https://doi.org/10.1101/gad.187922.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kitambi SS, Toledo EM, Usoskin D, Wee S, Harisankar A, Svensson R, et al. RETRACTED:Vulnerability of glioblastoma cells to catastrophic vacuolization and death induced by a small molecule. Cell. 2014;157(2):313–28. doi: 10.1016/j.cell.2014.02.021. https://doi.org/10.1016/j.cell.2014.02.021. [DOI] [PubMed] [Google Scholar]
- 57.Yuan GQ, Wei NL, Mu LY, Wang XQ, Zhang YN, Zhou WN, et al. A 4-miRNAs signature predicts survival in glioblastoma multiforme patients. Cancer Biomark. 2017;20(4):443–52. doi: 10.3233/CBM-170205. https://doi.org/10.3233/CBM-170205. [DOI] [PubMed] [Google Scholar]
- 58.Gately L, McLachlan SA, Philip J, Ruben J, Dowling A. Long-term survivors of glioblastoma:A closer look. J Neurooncol. 2018;136(1):155–62. doi: 10.1007/s11060-017-2635-1. https://doi.org/10.1007/s11060-017-2635-1. [DOI] [PubMed] [Google Scholar]
- 59.Tykocki T, Eltayeb M. Ten-year survival in glioblastoma. A systematic review. J Clin Neurosci. 2018;54:7–13. doi: 10.1016/j.jocn.2018.05.002. https://doi.org/10.1016/j.jocn.2018.05.002. [DOI] [PubMed] [Google Scholar]
- 60.Fan X, Li Y, Shan X, You G, Wu Z, Li Z, et al. Seizures at presentation are correlated with better survival outcomes in adult diffuse glioma:A systematic review and meta-analysis. Seizure. 2018;59:16–23. doi: 10.1016/j.seizure.2018.04.018. https://doi.org/10.1016/j.seizure.2018.04.018. [DOI] [PubMed] [Google Scholar]
- 61.Berendsen S, Varkila M, Kroonen J, Seute T, Snijders TJ, Kauw F, et al. Prognostic relevance of epilepsy at presentation in glioblastoma patients. Neuro Oncol. 2016;18(5):700–6. doi: 10.1093/neuonc/nov238. https://doi.org/10.1093/neuonc/nov238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dobran M, Nasi D, Chiriatti S, Gladi M, Somma LD, Iacoangeli M, et al. Prognostic factors in glioblastoma:Is there a role for epilepsy? Neurol Med Chir (Tokyo) 2018;58(3):110–5. doi: 10.2176/nmc.oa.2017-0167. https://doi.org/10.2176/nmc.oa.2017-0167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Flanigan PM, Jahangiri A, Kuang R, Truong A, Choi S, Chou A, et al. Improved survival with decreased wait time to surgery in glioblastoma patients presenting with seizure. Neurosurgery. 2017;81(5):824–33. doi: 10.1093/neuros/nyx084. https://doi.org/10.1093/neuros/nyx084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Reifenberger G, Weber RG, Riehmer V, Kaulich K, Willscher E, Wirth H, et al. Molecular characterization of long-term survivors of glioblastoma using genome- and transcriptome-wide profiling. Int J Cancer. 2014;135(8):1822–31. doi: 10.1002/ijc.28836. https://doi.org/10.1002/ijc.28836. [DOI] [PubMed] [Google Scholar]
- 65.Michaelsen SR, Urup T, Olsen LR, Broholm H, Lassen U, Poulsen HS, et al. Molecular profiling of short-term and long-term surviving patients identifies CD34 mRNA level as prognostic for glioblastoma survival. J Neurooncol. 2018;137(3):533–42. doi: 10.1007/s11060-017-2739-7. https://doi.org/10.1007/s11060-017-2739-7. [DOI] [PubMed] [Google Scholar]
- 66.Gerber NK, Goenka A, Turcan S, Reyngold M, Makarov V, Kannan K, et al. Transcriptional diversity of long-term glioblastoma survivors. Neuro Oncol. 2014;16(9):1186–95. doi: 10.1093/neuonc/nou043. https://doi.org/10.1093/neuonc/nou043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hartmann C, Hentschel B, Simon M, Westphal M, Schackert G, Tonn JC, et al. Long-term survival in primary glioblastoma with versus without isocitrate dehydrogenase mutations. Clin Cancer Res. 2013;19(18):5146–57. doi: 10.1158/1078-0432.CCR-13-0017. https://doi.org/10.1158/1078-0432.CCR-13-0017. [DOI] [PubMed] [Google Scholar]
- 68.Li F, Li Y, Zhang K, Li Y, He P, Liu Y, et al. FBLN4 as candidate gene associated with long-term and short-term survival with primary glioblastoma. Onco Targets Ther. 2017;10:387–95. doi: 10.2147/OTT.S117165. https://doi.org/10.2147/OTT.S117165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sandmann T, Bourgon R, Garcia J, Li C, Cloughesy T, Chinot OL, et al. Patients with proneural glioblastoma may derive overall survival benefit from the addition of bevacizumab to first-line radiotherapy and temozolomide:Retrospective analysis of the AVAglio trial. J Clin Oncol. 2015;33(25):2735–44. doi: 10.1200/JCO.2015.61.5005. https://doi.org/10.1200/JCO.2015.61.5005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cantero D, Rodríguez de Lope Á, Moreno de la Presa R, Sepúlveda JM, Borrás JM, Castresana JS, et al. Molecular study of long-term survivors of glioblastoma by gene-targeted next-generation sequencing. J Neuropathol Exp Neurol. 2018;77(8):710–6. doi: 10.1093/jnen/nly048. https://doi.org/10.1093/jnen/nly048. [DOI] [PubMed] [Google Scholar]
- 71.Niyazi M, Zehentmayr F, Niemöller OM, Eigenbrod S, Kretzschmar H, Schulze-Osthoff K, et al. MiRNA expression patterns predict survival in glioblastoma. Radiat Oncol. 2011;6:153. doi: 10.1186/1748-717X-6-153. https://doi.org/10.1186/1748-717X-6-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Srinivasan S, Patric IR, Somasundaram K. A ten-microRNA expression signature predicts survival in glioblastoma. PLoS One. 2011;6(3):e17438. doi: 10.1371/journal.pone.0017438. https://doi.org/10.1371/journal.pone.0017438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhang W, Zhang J, Yan W, You G, Bao Z, Li S, et al. Whole-genome microRNA expression profiling identifies a 5-microRNA signature as a prognostic biomarker in Chinese patients with primary glioblastoma multiforme. Cancer. 2013;119(4):814–24. doi: 10.1002/cncr.27826. https://doi.org/10.1002/cncr.27826. [DOI] [PubMed] [Google Scholar]
- 74.Hermansen SK, Sørensen MD, Hansen A, Knudsen S, Alvarado AG, Lathia JD, et al. A 4-miRNA signature to predict survival in glioblastomas. PLoS One. 2017;12(11):e0188090. doi: 10.1371/journal.pone.0188090. https://doi.org/10.1371/journal.pone.0188090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sperduto CM, Chakravarti A, Aldape K, Burger P, Papermaster GB, Sperduto P, et al. Twenty-year survival in glioblastoma:A case report and molecular profile. Int J Radiat Oncol Biol Phys. 2009;75(4):1162–5. doi: 10.1016/j.ijrobp.2008.12.054. https://doi.org/10.1016/j.ijrobp.2008.12.054. [DOI] [PubMed] [Google Scholar]
- 76.Smoll NR, Schaller K, Gautschi OP. The cure fraction of glioblastoma multiforme. Neuroepidemiology. 2012;39(6):63–9. doi: 10.1159/000339319. https://doi.org/10.1159/000339319. [DOI] [PubMed] [Google Scholar]
- 77.Walid MS. Prognostic factors for long-term survival after glioblastoma. Perm J. 2008;12(4):45–8. doi: 10.7812/tpp/08-027. [DOI] [PMC free article] [PubMed] [Google Scholar]