
Extended Data Fig. 10. The workflow of Native MS mixing experiment.
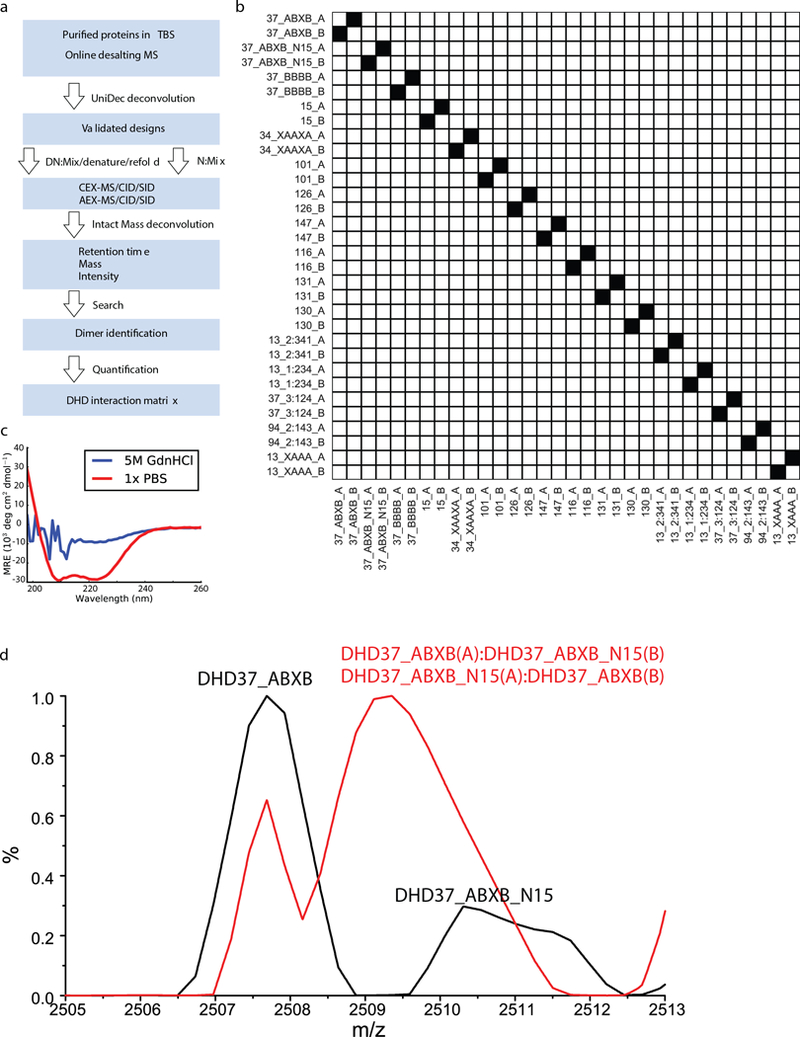
a, Protein samples were characterized using online-desalting coupled to native MS and deconvoluted using UniDec software. Proteins shown expected masses were mixed in equimolar ratio, and the final mix was divided into two parts: in the experimental group (DN), proteins were denatured by 5M GdnHCl at 75°C and refoled into 150mM AmAc; in the control mixing experiment (N), denaturation and refolding steps were omitted. Sample mixtures in each group was further equally divided into 3 parts and was individually injected on LC-MS with WCX and WAX respectively. LC-MS analysis was performed for mixtures in full MS mode and MSMS mode with HCD and SID, respectively. Data were deconvoluted using Intact Mass™. The deconvoluted mass lists from Intact Mass™ were searched against a theoretical mass list of all possible monomer, dimer, trimer and tetramer combinations. Dimers were identified based on the full MS runs and MSMS runs with both subunits being detected at the same retention time. b, In the control mixing experiment (N), after mixing all 16 proteins in solution without the denaturation and renaturation steps, no exchange among proteins were observed. c, CD data for a mixture of purified DHDs in PBS (red) or 5M GdnHCl and 75°C (blue). Protein mixture was fully denatured under the latter condition. d, A mixing experiment of DHD_37_ABXB and 15N labeled DHD_37_ABXB with (red) or without (black) the denaturation and refolding steps. MS peaks merged after subunit exchange due to the similarity in the masses of 15N labeled and unlabeled subunits. Two biologically independent experiments (b-d) were performed.