

Abstract
Adequate reprogramming of cellular metabolism in response to stresses or suboptimal growth conditions involves a myriad of coordinated changes that serve to promote cell survival. As protein synthesis is an energetically expensive process, its regulation under stress is of critical importance. Reprogramming of mRNA translation involves well-understood stress-activated kinases that target components of translation initiation machinery resulting in the robust inhibition of general translation and promotion of the translation of stress-responsive proteins. Translational arrest of mRNAs also results in the accumulation of transcripts in cytoplasmic foci called stress granules. Recent studies point on the key roles of transfer RNA (tRNA) in the stress-induced translational reprogramming. These include stress-specific regulation of tRNA pools, codon-biased translation influenced by tRNA modifications, tRNA miscoding, and tRNA cleavage. In combination, signal transduction pathways and tRNA metabolism changes regulate translation during stress resulting in the adaptation and cell survival. This review will examine molecular mechanisms that regulate protein synthesis in response to stress.
Keywords: stress, translation, translation initiation, translational reprogramming, tRNA
Graphical Abstract
Translational control contributes to various aspects of cell homeostasis. As mRNA translation is energy-expensive, its regulation is critical. Reprogramming of mRNA translation during stress involves signal transduction pathways and tRNA metabolism changes that aim on cell survival. This review examines molecular mechanisms that regulate protein synthesis in response to stress.
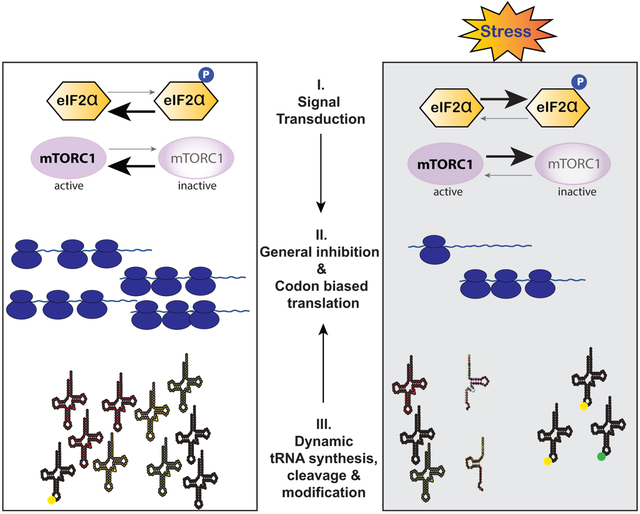
1. Introduction
Protein synthesis and its control are central to gene expression. Regulation of translation contributes to many aspects of cell physiology such as growth, differentiation and survival. Maintenance of protein homeostasis is an energy expensive process that needs coordinated regulation of transcription and translation to provide optimal levels of required proteins. In the same time, the survival of cells exposed to different adverse conditions (stresses) requires fast and efficient reprogramming of mRNA translation, the process aiming on the conservation of energy and repair of stress-induced damage. Such changes in stress-responsive protein synthesis target both global translation and the translation of specific mRNAs [1].
Although the changes in mRNA expression levels play an obvious role in the determination of the cellular level of a protein, number of studies show that there is no strict correlation between these two phenomena, with some abundant mRNAs translated poorly and vice versa [2][3][4]. Generally, the number of ribosomes translating a specific mRNA is the most critical parameter of the rate of the synthesis of a specific protein. Translation rates are not uniform among different mRNAs and are commonly determined by the proteins bound to them to form messenger ribonucleoprotein complexes (mRNPs). Specific protein factors within mRNPs dictate translational status (e.g., stimulation or inhibition of translation) and localization (e.g. sequestration into RNA granules) of mRNAs [5]. Recruitment of such factors into mRNPs is determined by the presence of specific sequence or structural motifs in each mRNA, which interact with specific RNA-binding proteins (RBPs). Another factor determining translational rates is the availability of tRNAs and codon composition of an mRNA [6]. In this case, a number of housekeeping genes have a bias towards codon usage profile correlated with the most abundant tRNAs, thus ensuring higher translational output to maintain vital metabolic processes [1]. Here, we examine the targets and mechanisms of translational control and discuss translation related pathways that contribute to cell survival under stress.
2. Translation initiation at the crossroads of protein synthesis regulation
Protein synthesis is a cyclic process that can be divided into four main stages: initiation, elongation, termination and ribosome recycling. Although modulation of translation can happen at any step of protein synthesis, most regulatory events occur at the initiation step. As a general rule, once translation enters into elongation phase, ribosomes are committed to complete synthesis of a nascent polypeptide to avoid formation of a potentially harmful incomplete protein. The initiation step requires coordinated action of multiple trans-acting factors, many of which are targets for tight regulation. In eukaryotes, mRNA translation starts with the assembly of a complex consisting of the small 40S ribosomal subunit with associated proteins known as eukaryotic initiation factors (eIFs). Association of 40S with eIF1, eIF1A, eIF5 factors and multisubunit eIF3 complex is followed by the binding of the heterotrimeric ternary complex consisting of initiator transfer RNA (methionyl-transfer RNA or Met-tRNAi), eIF2 and GTP. This culminates in the formation of a large 43S pre-initiation complex (PIC), which is capable of recognition of the correct initiation codon (typically AUG) at the start of an open reading frame (ORF) within mRNA (reviewed in [7]).
Recognition of the start codon by the PIC requires initial attachment of the 43S to an mRNA. Most eukaryotic mRNAs are marked by 7-methylguanosine cap at their 5’-ends (5`-cap) and polyadenylated tail (polyA tail) at their 3’-ends. Interaction of the cap-binding eIF4F complex (consisting of scaffolding protein eIF4G, cap-binding protein eIF4E and an RNA helicase eIF4A) with 5’-cap activates an mRNA for translation. The poly(A)-binding protein (PABP) binds to poly(A) tails and interacts with the cap-bound eIF4G, thus bringing mRNA 5’- and 3’-termini in the close proximity to form so called a “closed-loop” mRNA. eI4F-bound mRNA then recruits 43S PIC at the vicinity of 5’-cap, facilitating intermediate 43S:mRNA complex formation[8]. Upon binding, 43S PIC-bound eIF3 further interacts with eIF4G to stabilize mRNA:40S interactions and posits the 43S PIC for scanning through 5’-untranslated region (UTR) of an mRNA [9].
5’UTR scanning involves movement of the 43S PIC along the mRNA in the 5’−3’ direction searching for the AUG codon in the appropriate context (“Kozak sequence”) [10]. Such context promotes the recognition of the start codon by the base pairing between AUG codon and P site-bound Met-tRNAi anticodon. Efficient codon-anticodon interaction halts 43S PIC scanning, signals to the associated eIFs in the PIC and triggers conformational changes in the pre-initiation complex to form 48S PIC[11]. Such PIC rearrangements lead to the formation of a “closed” stable conformation that causes release of specific eIFs (such as eIF1, eIF5 and eIF2:GDP) and recruitment of the large 60S subunit to form an 80S ribosome that is ready to begin translation elongation (reviewed in [7]).
Although most of the cellular translation is believed to proceed as described above (canonical translation initiation), other alternative modes of translation initiation are also described. These mechanisms are not considered in this review since they are involved in translation of only a minor fraction of mRNAs and have a limited impact on global translational profile.
3. Reprogramming of protein synthesis under stress
Cells encounter a wide range of environmental and physiological stresses that activate an intrinsic network of adaptive changes (stress responses). Stress stimuli include both abiotic (temperature changes, ultraviolet (UV) irradiation, hypoxia, oxidative stress etc.) and biotic (viral infections, exposure to toxins, etc.) conditions, which are specifically recognized to trigger dynamic reprogramming of gene expression. As protein synthesis is energetically and metabolically expensive, control of cellular translation is a major checkpoint in the integrative stress response (ISR), a global program that diverts anabolic energy towards stress adaptation and restoration of cellular homeostasis.
The arrest of translation initiation is a major hallmark of stress-induced translational control. mRNA translation arrest results from the modification of components of translational machinery or their availability, as well as from the subcellular changes in the localization of mRNPs. Although targets of regulation at the initiation step are numerous, two translation initiation factors are main targets of translational control, namely eIF2 and eIF4E (Figure 1). As most eukaryotic mRNAs require the action of these factors for their translation, interference with eIF2, eIF4E or both affects most of translatome, and has rapid and robust outcome, which is an essential feature of ISR. Functions of these initiation factors is regulated by signal transduction pathways, which are highly sensitive to stresses and physiological stimuli. Importantly, stress-induced translation initiation is a reversible process, where upon removal of stress, translation is resumed to reach optimal rates.
Figure 1.

A. The translation initiation ternary complex (TC) consisting of eIF2-GTP-Met-tRNAi is a part of 43S pre-initiation complex (PIC) required for cap-dependent translation initiation. Decoding of AUG ‘start’ codon at P-site leads to GTP hydrolysis and releases of eIF2-GDP. Phosphorylation of eIF2 at Ser-51 of the α-subunit by stress induced kinases GCN2 (UV and amino acid starvation), HRI (heme deficiency, oxidative stress and heat shock), PERK (hypoxia, ER stress and proteostasis) and PKR (viral infection and dsRNA) prevents GDP to GTP exchange by nucleotide exchange factor eIF2B, the step required to regenerate the TC. This stress-mediated eIF2α phosphorylation results in global cap-dependent translational inhibition, stimulates stress granule (SG) assembly and promotes selective translation of stress-responsive mRNAs. B. mTORC1 is a positive regulator of cap-dependent translation. Increased activity of mTORC1 results in the phosphorylation of translational repressor eIF4E-binding proteins (4E-BPs) and ribosomal S6 kinases (S6K½). These phosphorylation events are proposed to regulate translation of TOP/TOP like mRNAs. Treatment with mTOR inhibitors (e.g., rapamycin or Torin) or starvation conditions decreases the activity of mTORC1, thus inhibiting global translation and promoting stress responses.
3.1. Stress induced eIF2α phosphorylation promotes translational reprogramming and assembly of stress granules
The binding of Met-tRNAi to the 40S ribosomal subunit mediated by the ternary complex is a critical step of translation initiation. Availability of the ternary complex for the assembly of the 43S PIC is regulated by the stress-induced phosphorylation of the α-subunit of eIF2 at serine 51. Phosphorylated eIF2α (p-eIF2α) inhibits GDP/GTP exchange mediated by factor eIF2B thus resulting in the depletion of translation-competent ternary complex available for the assembly of 43S PIC. Since eIF2α is in excess over eIF2B, even subtle changes in levels of p-eIF2α have significant impact on cellular translation (Figure 1A).
In mammals, phosphorylation of eIF2α is accomplished by four distinct protein kinases. They include heme-regulated inhibitor kinase (HRI) [12], protein kinase RNA (PKR) [13], general control non-repressible-2 (GCN2) [14] and PKR-like ER kinase (PERK) [15]. Each of these eIF2α kinases possess distinct regulatory domains that serve to sense specialized stress signals or ligands. HRI is activated in response to oxidative stress, low levels of heme, and heat shock. PKR plays essential role in innate immunity to viral infections and is activated by the presence of double-stranded RNAs. GCN2 is activated by amino acid starvation and accumulation of uncharged tRNAs in the cytosol as well as by UV irradiation. Finally, PERK is activated in response to ER stress, presence of unfolded proteins, calcium release, and changes in lipid composition.
Regardless of the nature of stress, activation of eIF2α kinases results in the elevation of p-eIF2α levels and consequent inhibition of global protein synthesis [16]. By lowering rate of global translation, cells reshape proteome to optimize gene expression and rewire signaling pathways to cope with stress and promote cell survival. While translation of most mRNAs (such as encoded by housekeeping genes) is potently inhibited, translation of specific mRNAs encoding stress-responsive and pro-survival proteins is paradoxically increased in response to elevated p-eIF2α levels. Many such proteins are key ISR regulatory factors that affect diverse cellular processes. One of the important ISR genes preferentially translated under stress is ATF4, a basic zipper transcription factor that regulates expression of many genes involved in stress responses [15]. ATF4 mRNA (like many other mRNAs of ISR genes) features upstream ORFs (uORFs [17]) in its 5’-UTR, which are typically inhibitory to translation of downstream ORFs including the primary coding sequence (CDS). Consequently, when levels of p-eIF2α are low and the ternary complex is abundant, ribosomes initiate at 5’-proximal uORFs and then terminate before reaching the CDS [15] [18][19]. Depending on mRNA and number of uORFs, this process can also include several events of termination/reinitiation. In contrast, when levels of p-eIF2α are high and the levels of the ternary complex are low, reinitiation at uORFs becomes less frequent, which allows the scanning 40S ribosomal subunit to reach a CDS (which typically has an AUG in strong Kozak context) and, eventually, initiate translation. However, it should be noted that the specific features of uORFs (such as their length, placement in 5’-UTRs, combinations with other ORFs) significantly influence efficiency of translation in response to p-eIF2α. Moreover, the presence of other secondary structures in mRNAs and binding of specific RBPs to 5’-UTRs further determine translation efficiency of a given mRNA (detailed discussion about stress-induced changes obtained from genome-wide translation studies can be found at [20])
An important aspect of general translation repression is an accumulation of untranslated mRNPs in the cytosol upon inhibition of translation initiation and disassembly of polysomes [21]. These untranslated mRNPs interact with specific factors (such as G3BP1, TIA1, TIAR etc), which possess specific regions that tend to aggregate. Some of these proteins are RBPs that directly bind mRNAs in a sequence- or secondary structure-dependent manner, others interact with translation machinery [22]. Consequently, translationally-arrested mRNPs via multiple RNA:RNA, protein:protein and RNA:proten interactions are condensed into non-membrane-enclosed subcellular compartments called stress granules (SGs) [23]. SGs are dynamic entities in the equilibrium with polysomes that function as sites of mRNA triage and sort mRNAs for storage, reinitiation or degradation [24]. Importantly, an increase in the pool of translationally arrested mRNPs (such as by elevating p-eIF2α levels) promotes SG formation, whereas reduction in translationally arrested mRNPs (as during stress relief) promotes SG disassembly [25] [26]. Further details on the functional interaction between SGs and translational control can be found in the recent reviews [27][28][29] and will not be covered here in depth.
3.2. mTOR steers translation during stress adaptation
The second major pathway contributing to translational control under stress is dependent on the activity of a protein kinase called mammalian target of rapamycin (mTOR) (Figure 1B). It is a member of the phosphatidylinositol kinase-related kinase family that forms two different multisubunit complexes, mTORC1 and mTORC2 [30]. While mTORC2 regulates cytoskeleton and cellular proliferation by sensing different growth factors, mTORC1 senses metabolic stress by assessing cellular nutrient (e.g., intracellular amino acid levels) and energetic (AMP:ATP ratio) status. Main targets of mTORC1 are proteins directly involved in the regulation of translation or protein kinases that regulate activity of translational machinery.
Two classes of mTORC1 targets are especially relevant for translation regulation under stress. First includes the family of small phosphoproteins termed eIF4E-binding proteins (4EBPs), which directly bind to eIF4E [31]. Under optimal conditions, mTORC1 constitutively phosphorylates 4EBPs and their phosphorylated variants (p-4EBPs) cannot bind eIF4E. In response to stress, mTORC1 is inactivated and p-4EBPs become dephosphorylated. Dephosphorylated 4EBPs bind to cap-associated eIF4E. Since binding sites of eIF4G and 4EBPs on eIF4E are overlapping [32], 4EBPs interfere with eIF4F complex assembly by sequestering the cap-binding protein, resulting in the inhibition of translation. The second class consists of S6 kinases (S6Ks) [33], which target and phosphorylate ribosomal protein S6 (RPS6) [33], a component of the 40S subunit, and eIF4B [34], a translation initiation factor that promotes the helicase activity of eIF4A. Phosphorylation of these S6K targets is believed to promote translation, although molecular details of S6K-mediated translation stimulations are unclear. In the same time, inactivation of mTORC1 negatively influences S6Ks and is proposed to have inhibitory effect on translation (reviewed in [35]). Interestingly, mTORC1 also directly binds eIF3 and stimulates interactions between eIF4G and eIF3 on 40S subunit [36]. Such interaction enhances recruitment of 40S ribosomal subunits to the cap-bound eIF4F and promotes assembly of PICs.
The effect of mTORC1 inhibition on global mRNA translation has been examined using various mTORC1 inhibitors. Although mTORC1 inhibition generally results on global repression of protein synthesis, polysome and ribosome profiling approaches identified subsets of mRNAs that are strongly impacted under mTORC1 inhibition [37][38][39]. The most prominent class of mRNAs associated with mTORC1 signaling represents transcripts containing 5’-terminal oligopyrimidine tracts (5’TOPs). 5’TOPs are defined by a cytosine immediately after mRNA cap, followed by uninterrupted 4–15 nucleotide pyrimidine-rich element in the context of a relatively short and unstructured 5’UTR (reviewed in [35][40][41]). This motif is commonly found in transcripts encoding components of translational machinery such as ribosomal proteins, translation initiation and elongation factors. Such coordination of 5’TOP transcripts translation by nutrient- and mitogen-activated mTORC1 provides an efficient and robust mechanism of ribosome biogenesis regulation. In addition to 5’TOP mRNAs, other subsets of mRNAs were found to be mTORC1-sensitive. They include transcripts bearing TOP-like motifs (consisting of a stretch of at least 5 pyrimidines within 4 nucleotides from the cap structure) [40], those with long and complex 5’UTRs and, in contrast, with mRNAs very short and unstructured 5’UTRs [41].
In summary, although, the molecular mechanisms of translation regulation by eIF2α phosphorylation and mTOR signaling are well-understood, details to explain preferential inhibition or induction of translation of some specific mRNA targets are lacking. It may suggest that under stress other alternative or additive pathways exist that regulate mRNA translation. One of such emerging themes in stress-induced protein synthesis reprogramming is a modulation of tRNA metabolism.
3.3. tRNA metabolism changes impact translatome under stress
Transfer RNA represents 10–15% of the total RNA in cell with >500 human tRNA genes known [42][43]. Being the second most abundant non-coding RNA (ncRNA) and energetically expensive to make, its expression is tightly regulated depending on the cell type and its physiological state [44][45]. Cellular abundance of tRNAs at any given instance may dictate codon usage, and hence the speed and accuracy of protein synthesis [46]. Therefore, regulating abundance of specific tRNA pools presents a dynamic mechanism of modulating translation.
Transcription of tRNA genes by RNA polymerase III (Pol III) is tightly regulated, and Pol III activity demonstrates dynamic and heterogeneous adaptation to various growth and stress stimuli (Figure 2). One such mechanism of dynamic regulation of tRNA transcription is mediated through MAF1, constitutive repressor of Pol III transcription that is regulated by mTOR [47][48][49]. Starvation deactivates mTOR inducing dephosphorylation of MAF1, which translocates into the nucleus and inhibits recruitment of Pol III on tRNA genes [50]. Such MAF1-dependent Pol III repression results in a different transcriptional outcome with two classes of tRNA genes revealing distinct responsiveness to stress. The first class of tRNA genes do not change their expression under both favorable and stress conditions (“stable” tRNA genes), whereas second class (“unstable” tRNA genes) dynamically adapt to stress. This results in the increased stoichiometry of tRNAs transcribed from stable “housekeeping” tRNA genes, which is proposed to modulate translation of mRNAs enriched in cognate codons and encoding stress-responsive proteins [51][52]. This ensures metabolic efficacy of synthesis and turnover of Pol III transcripts essential to maintain cellular homeostasis. The heterogenous transcription of tRNA genes under stress is further supported in a recent report using quantification of stress-specific tRNA abundances as a function of translational efficiency (through high-resolution transcriptomics and ribosome profiling) in yeast [53]. The analysis suggests a stress-dependent regulation of selective tRNA isodecoders that facilitate codon-biased translation of selective transcripts to maintain protein homeostasis. For example, an increased abundance of tRNALeu(CAA) under multiple stresses may facilitate increased translation of UUG-enriched transcripts [53].
Figure 2.

MAF1 is a negative regulator of Pol III-mediated transcription of tRNA genes. Under normal growth conditions, mTORC1 mediated phosphorylation of MAF1 prevents its binding to with Pol III. This ensures optimal transcription of tRNA genes to meet cellular metabolic needs. Decreased activity of mTORC1 under conditions of stress leads to decreased phosphorylation of MAF1 thereby promoting its association with Pol III. This results in differential transcription of tRNA genes that alters the cellular tRNA pool to favor translation of stress-responsive proteins.
Although differential transcription of tRNA genes under optimal versus stress conditions is not well understood, it could be mediated through multiple mechanisms of regulation such as polymerase subunits recruitment and/ or trans-acting factors that affect chromatin organization. It has been shown that MAF1 mediated adjustment of tRNA synthesis may also impact tRNA maturation and processing, further shaping a pool of tRNAs available for translation [54][55]. In addition, tRNA availability for protein synthesis is also governed by the rates of tRNA aminoacylation. In yeast, accumulation of uncharged tRNAs upon conditions of nutrient deprivation and osmotic stress is sensed through direct interactions with Gcn2p (yeast orthologue of GCN2 kinase), which then mediates phosphorylation of eIF2α[56][57]. Besides tRNA availability regulation, post-transcriptional structural and sequence dependent tRNA processing programs exist that expand the scope of stress dependent translational modulation, as described below.
3.3.1. Dynamic tRNA modifications fine-tune translation under stress.
An average eukaryotic tRNA undergoes 11–13 modifications with a high variance in amount and location of modification [58]. These modifications greatly increase the number of cellular tRNA isotypes [59][60]. While there are several conserved locations for tRNA modifications within 70–90 nucleotides long tRNAs, specific modification at the anticodon loop are vital to reprogram translation under stress [61]. According to their importance, perturbations in tRNA modifications are broadly associated with multiple human pathologies (discussed in[62][63][64]).
The modification at the wobble base greatly alters the kinetics of the codon:anti-codon interactions, thereby altering the codon dependent stability and expression of specific transcripts [61][65]. The most common modification of tRNA at the wobble position is methylation of nucleotide bases or ribose sugar catalyzed by tRNA methyltransferases (Trms) [66]. As methylation of wobble base in the anticodon loop plays an important role in maintaining translational fidelity [67][68], cells modulate methylation and demethylation of tRNA bases to tune translational accuracy in response to stress (Figure 3). For example, Trm9 mediated 5-methyl-U (m5U) modifications at positions U34 of tRNAGlu(UUC) and tRNAArg(UCU) in Saccharomyces cerevisiae are important for maintaining translational fidelity in response to DNA damage caused by alkylating agents [69][70]. Another modification at wobble base, 5-methyl-C (m5C) at position C34 of tRNALeu(CAA) by methyltransferase Trm4 contributes to codon biased translation of stress response proteins [71]. Similarly, stress-responsive expression of selenoproteins in mammals is enhanced by increased methylation at positions U34 of selenocystyl-tRNA[Ser]Sec [72][73] where m5U modification greatly increases selenocysteine incorporation at the UGA stop-codon.
Figure 3.

Stress alters tRNA metabolism to dynamically reprogram translation to achieve pro-survival equilibrium until optimal conditions are reached. Selective methylation of nucleotide bases in the anti-codon loop by Transfer RNA methyltransferases (Trms) under conditions of oxidative and xenobiotic stress helps maintain translational fidelity and promote codon biased translation of specific transcripts called Modification Tunable Transcripts (MoTTs). Nutrient starvation and ROS conditions promote misacylation of tRNAs that increase decoding of near-cognate tRNAs leading to increased synthesis of stress responsive proteins. Further, stress-mediated cleavage of tRNAs by RNases like angiogenin (ANG) generates tRNA halves (5` and 3` tiRNAs) that facilitate cell viability. Mechanistically, tiRNA interact with translation machinery components and inhibit mRNA translation in eIF2α-independent manner. tRNA fragments smaller that tiRNAs called tRFs, which mimic miRNAs in size, are speculated to regulate protein synthesis in RNAi like/sequence specific manner.
It is important to note that Trm4- and Trm9-mediated methylation activities are stress specific, and they increase the abundance of specific tRNA species. Such increase stabilizes transcripts enriched in degenerate codons, having distinct codon usage profile than an average transcript, and coding for stress response proteins [74] [75]. Thus, the modulation of the level of methylation in response to specific stress increases the proteomic output from these transcripts in a codon biased manner. Such transcripts, whose expression is fine-tuned as a consequence of modification levels of tRNAs are termed as Modification Tunable Transcripts (MoTTs) [76] [77].
Methylation of the nucleotides outside the anticodon loop that ensure structural stability also play an important role in regulating protein synthesis. TRM6 mediated methylation of A58 (m1A58) increases the stability of Met-tRNAi necessary for optimal translational output [78]. Interestingly, during starvation antagonistic activity of ALKBH1, a demethylase, greatly reduces m1A58 thereby significantly decreasing Met-tRNAi abundance to facilitate translational repression [79]. All the above instances emphasize the importance of sequence-specific methylation in modulating translation, which is also aided by the finding that loss of function of one or more of these methyltransferases increases sensitivity to specific stress [80].
Components of URM1 pathway in Saccharomyces cerevisiae catalyze thiolation of uridine bases (5-methoxycarbonylmethyl-2-thiouridine) at wobble position 34 of tRNALys(UUU), tRNAGlu(UUC) and tRNAGln(UUG). These modifications promote efficient translation of transcripts enriched in AAA, GAA and CAA codons[81]. Most URM1-sensitive transcripts include proteins involved in ribosome biogenesis and translation regulation[81]. Temperature dependent hypothiolation during heat shock response results in downregulation of translation and upregulation of catabolic process as an adaptive response to maintain proteomic homeostasis[81][82].
3.3.2. tRNAs promote miscoding to favor adaptive translation under stress
Accuracy of protein synthesis is dependent on: 1) accuracy of aminoacylation by aminoacyl tRNA synthetase (aaRS), and 2) accuracy of decoding of cognate aminoacyl tRNA by the ribosome [83]. Interestingly, increased translational fidelity decreases proteomic output and inhibits cell growth, highlighting the tradeoff between accuracy and speed [84][85]. Therefore, cells must balance translational yield and accuracy by mechanisms that are not canonical. Translational miscoding is one such mechanism that refers to non-canonical decoding of aminoacyl tRNA to the mRNA codon at the ribosomal A-site [86]. Previously considered as aberrant and unfavorable, translational miscoding has been recently appreciated as an adaptive mechanism to maintain metabolic equilibrium under different physiological states. Miscoding increases translational capacity and optimizes output by selecting ‘suitable’ substrate from the pool of active aminoacyl tRNAs [87].
There are several examples of tRNA-mediated translational miscoding in response to physiological stress in prokaryotes and lower eukaryotes [88]. The degeneracy of the genetic code that allows for decoding of aminoacyl tRNA with non-complementary wobble base also tolerates selection of near-cognate tRNAs that incorporates a different amino acid (Figure 3). Depletion of the amino acid Asparagine results in decreased abundance of tRNAAsn(GUU) and tRNAAsn(AUU) in E.coli. This results in increased decoding of near-cognate tRNALys(UUU) at AAC and AAU codons as a pro-survival response [89]. Oxidative stress-induced mischarging of tRNA in yeast results in mutant protein phenotype leading to increased fitness in response to reactive oxygen species (ROS). Similarly, ROS also induces methionine misacylation in HeLa cells, where approximately 1% of the cellular tRNAs were misacylated with methionine. This results in increased incorporation of methionine in protein to promote cell viability [90]. Such adaptive mistranslation mechanism is stimulated by phosphorylation of methionyl-tRNA synthetase (MetRS) by kinases ERK½, which causes promiscuous misacylation of non-cognate tRNAs with methionine [91]. The stress-induced misacylation with methionine is conserved in archaea and bacteria suggesting a vital role of increased methionine incorporation for translational reprograming during stress [92][93].
3.3.3. Stress-induced tRNA cleavage
A relatively underappreciated phenomenon of stress-induced translational reprograming is cleavage of tRNAs. For a long time these tRNA fragments were considered as degradation products but recent work highlights their role in modulating protein synthesis during cellular stress, proliferation and differentiation [94][95]. One such stress dependent cleavage of tRNAs is catalyzed by the ribonuclease Angiogenin (ANG), which target the anticodon loop to produce tRNA halves called tRNA-derived stress-induced RNAs (tiRNAs). The 5` and 3` halves are called 5`-tiRNA (30–35 nucleotides) and 3`-tiRNA (40–50 nucleotides), respectively (Figure 3) [96][97].
ANG is a stress responsive ribonuclease of the RNase A superfamily, which is induced under conditions of starvation, oxidative stress, hypoxia and heat shock [98]. Stress-activated ANG cleaves approximately 1–3% of cytoplasmic tRNA to generate tiRNAs [96]. Certain 5`tiRNAs are biologically active and perturb global translation [99]. Specifically, 5’-tiRNAs derived from tRNAAla and tRNACys (5`tiRNAAla and 5`tiRNACys) potently inhibit cap-dependent translation. They uniquely possess 5` terminal oligo guanidine (5`-TOG) motifs that form G-quadruplexes (G4s), non-canonical RNA structures rich in guanines[100]. Mechanistically, G4-tiRNAs inhibit translation by displacing the eIF4F complex from the cap structures of mRNA. The dissociation of cap-binding complex from mRNAs stimulate formation of SGs in eIF2α-independent manner [101][102]. An RNA/DNA-binding protein YB-1, an RNA chaperon, is capable of binding to 5`TOG motifs to promote SG formation downstream of 5`tiRNA-dependent displacement of eIF4F complex [103][102] [104]. Interestingly, ANG-mediated modulation of protein synthesis through tiRNAs plays a neuroprotective role under various stresses and loss-of-function mutations within ANG coding sequences are associated with neurodegenerative diseases [105] [106].
Stress specific cleavage of both mature and pre- tRNAs by ribonucleases like angiogenin and Dicer can results in tRNA fragments smaller than tiRNAs called tRNA-derived fragments (tRFs) that are 15–26 nucleotide in length [107]. Recent work identifies the role of tRFs in translational inhibition [108][109]. tRFs are similar in size as miRNAs and have been shown to associate with argonaute proteins, they are proposed to inhibit translation in a manner similar to miRNAs (Figure 3) [110][111]. Other data suggests that these tRNA fragments may be involved in translational regulation of certain transcripts by targeting their translation in a sequence-independent manner, although details are largely unknown [112] [113]. Interestingly, in humans fragment derived from tRNALeu stimulates expression of RPS28 in sequence dependent manner [114], in contrast to many other examples where tRFs work as inhibitors of translation.
4. Emerging topics and future perspectives
Translational control is an integral part of normal cell physiology to ensure the cell’s survival under stress. Several distinct translation related regulatory stress responses are known, and the list is growing. Although recent years have seen substantial progress to characterize new players in translational control, further examples remain to be identified. Development of novel technologies such as ribosome profiling or tRNA-sequencing to monitor and quantify different aspects of translation and related processes has dramatically advanced this field.
One emerging theme in translational control is the interplay between mRNAs (codon profile) and tRNAs (availability, modification and aminoacylation profile) that can efficiently modulate both global translation and translation of specific mRNA subgroups. This allows cells to maintain translatome homeostasis under optimal growth conditions and efficiently reshape protein synthesis under suboptimal conditions of stress. It is also important to underscore the emerging roles of the messenger RNA modifications in translation regulation. One of the most studied modifications is N6-methyladenosine (m6A) that plays diverse roles in RNA metabolism. m6A is the most abundant modification of mRNAs in eukaryotes conserved from yeast to humans with over 25% of human transcripts containing methylated sites [115][116][117]. The m6A are abundant near termination codons and in 3ÙTRs [118]. Dynamic m6A modification is regulated by the ‘writers’ (methyltransferases), ‘readers’ (trans-acting protein co-factors) and ‘erasers’ (demethylases) that alter stability and proteomic output of specific transcripts [119]. Surprisingly, m6A residues within 5’UTR can behave as an m6A-induced ribosome engagement site (MIRES), which promote cap-independent translation initiation that does not require cap structure and eIF4E. Importantly, m6A modification in 5ÙTR is dynamic and stress-responsive; for example, it is induced by heat shock or oxidative stress. One of such MIRESes is found in 5’UTR of Hsp70 mRNA where it promotes Hsp70 translation under heat shock. Moreover, m6A modifications in the 5’UTR of ATF4 mRNA (containing uORFs) regulate ribosome scanning and selection of start site under conditions of amino acid starvation. Additionally, dose dependent response to sodium arsenite increases m6A signals at a 5ÙTR and 5’-vicinity of CDS of mRNAs, which promotes their targeting to SGs via interaction with YTHDF3 protein [120]. All this suggest that global and alternative translation is modulated by m6A modifications within mRNAs. We agree here that m6A (and likely other RNA modifications) broadly contribute to stress response by targeting ‘housekeeping’ transcripts to SGs and stimulating alternative translation of pro-survival transcripts; we also speculate that it provides a dynamic synergy between tRNA pools and modified/unmodified mRNAs to rapidly alter proteome for translational response to stress.
Another theme is how tRNA modifications affect tRNA cleavage and biological activities of tRNA-derived molecules. As it was recently shown by Guzzi et. al. a presence of specific modification in a tRF can greatly modulate its biological activity. This work suggests the role of pseudouridylation in selective regulation of translation via generation of tRFs to impact stem cell differentiation during early embryogenesis. The stem cell abundant pseudouridine synthetase PUS7 promotes synthesis and pseudouridylation (ψ) of TOG-containing tRNAs that in turn are sources for 5`tRFs referred as mini TOGs (mTOGs). Ψ-containing 5`tRFs inhibit association of PABP with the eIF4F cap-binding complex thereby inhibiting translation [121]. In contrast, Ψ-lacking mTOG-tRFs fail to inhibit translation. These recently identified phenomena suggest existence of additional mechanisms of dynamic alteration of RNA modifications in translation machinery (tRNAs and mRNAs) to fine tune responses to various physiological conditions including stress.
Because aberrant translation control-related stress responses are tightly linked to many human pathologies, a better understanding of the underlining molecular mechanisms is required. Such understanding will enable to interfere with these pathological processes and open new avenues for the development of targeted therapeutics and have a great potential for drug discovery.
Acknowledgements
The authors would like to thank Dr. Shawn Lyons and Dr. Prakash Kharel for reading and providing comments on this manuscript. Research in the laboratory of P.I. was supported by the NIH (GM126150) and Brigham and Women’s Hospital (Fund to Sustain Research Excellence).
Abbreviations:
- CDS
coding sequence
- eIF
eukaryotic initiation factor
- ISR
integrative stress response
- mRNP
messenger ribonucleoprotein complex
- ORF
open reading frame
- PIC
pre-initiation complex
- RBP
RNA-binding protein
- SG
stress granule
- UTR
untranslated region
Footnotes
Conflict of Interest
The authors declare no conflict of interest
References
- [1].Hershey JWB, Sonenberg N, Mathews MB, Cold Spring Harb. Perspect. Biol. 2018, a032607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Greenbaum D, Colangelo C, Williams K, Gerstein M, Genome Biol. 2003, 4, 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Beyer A, Hollunder J, Nasheuer H-P, Wilhelm T, Mol. Cell. Proteomics 2004, 3, 1083. [DOI] [PubMed] [Google Scholar]
- [4].Maier T, Güell M, Serrano L, FEBS Lett. 2009, 583, 3966. [DOI] [PubMed] [Google Scholar]
- [5].Mitchell SF, Parker R, Mol. Cell 2014, 54, 547. [DOI] [PubMed] [Google Scholar]
- [6].Gingold H, Pilpel Y, Mol. Syst. Biol. 2014, 7, 481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Jackson RJ, Hellen CUT, V Pestova T, Nat. Rev. Mol. Cell Biol. 2010, 11, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kapp LD, Lorsch JR, Annu. Rev. Biochem. 2004, 73, 657. [DOI] [PubMed] [Google Scholar]
- [9].V Pestova T, Kolupaeva VG, Genes Dev. 2002, 16, 2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kozak M, Cell 1978, 15, 1109. [DOI] [PubMed] [Google Scholar]
- [11].Hinnebusch AG, Annu. Rev. Biochem. 2014, 83, 779. [DOI] [PubMed] [Google Scholar]
- [12].Lu L, Han AP, Chen JJ, Mol. Cell. Biol. 2001, 21, 7971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Srivastava SP, Kumar KU, Kaufman RJ, J. Biol. Chem. 1998, 273, 2416. [DOI] [PubMed] [Google Scholar]
- [14].Wek SA, Zhu S, Wek RC, Mol. Cell. Biol. 1995, 15, 4497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D, Mol. Cell 2000, 5, 897. [DOI] [PubMed] [Google Scholar]
- [16].Wek RC, Cold Spring Harb. Perspect. Biol. 2018, 10, a032870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Iacono M, Mignone F, Pesole G, Gene 2005, 349, 97. [DOI] [PubMed] [Google Scholar]
- [18].Lu PD, Harding HP, Ron D, J. Cell Biol. 2004, 167, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Vattem KM, Wek RC, Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 11269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Andreev DE, O’Connor PBF, Loughran G, Dmitriev SE, Baranov PV, Shatsky IN, Nucleic Acids Res. 2017, 45, 513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kedersha NL, Gupta M, Li W, Miller I, Anderson P, J. Cell Biol. 1999, 147, 1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kedersha N, Panas MD, Achorn CA, Lyons S, Tisdale S, Hickman T, Thomas M, Lieberman J, McInerney GM, Ivanov P, Anderson P, J. Cell Biol. 2016, 212, 845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Anderson P, Kedersha N, Ivanov P, Biochim. Biophys. Acta 2015, 1849, 861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kedersha N, Ivanov P, Anderson P, Trends Biochem. Sci. 2013, 38, 494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kedersha N, Cho MR, Li W, Yacono PW, Chen S, Gilks N, Golan DE, Anderson P, J. Cell Biol. 2000, 151, 1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kedersha N, Stoecklin G, Ayodele M, Yacono P, Lykke-Andersen J, Fritzler MJ, Scheuner D, Kaufman RJ, Golan DE, Anderson P, J. Cell Biol. 2005, 169, 871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ivanov P, Kedersha N, Anderson P, Cold Spring Harb. Perspect. Biol. 2018, a032813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Panas MD, Ivanov P, Anderson P, J. Cell Biol. 2016, 215, 313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Protter DSW, Parker R, Trends Cell Biol. 2016, 26, 668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Saxton RA, Sabatini DM, Cell 2017, 168, 960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Merrick WC, Pavitt GD, Cold Spring Harb. Perspect. Biol. 2018, 10, a033092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Mader S, Lee H, Pause A, Sonenberg N, Mol. Cell. Biol. 1995, 15, 4990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Pende M, Um SH, Mieulet V, Sticker M, Goss VL, Mestan J, Mueller M, Fumagalli S, Kozma SC, Thomas G, Mol. Cell. Biol. 2004, 24, 3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Shahbazian D, Roux PP, Mieulet V, Cohen MS, Raught B, Taunton J, Hershey JWB, Blenis J, Pende M, Sonenberg N, EMBO J. 2006, 25, 2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Meyuhas O, Kahan T, Biochim. Biophys. Acta - Gene Regul. Mech. 2015, 1849, 801. [DOI] [PubMed] [Google Scholar]
- [36].Harris TE, Chi A, Shabanowitz J, Hunt DF, Rhoads RE, Lawrence JC, EMBO J. 2006, 25, 1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Larsson O, Morita M, Topisirovic I, Alain T, Blouin M-J, Pollak M, Sonenberg N, Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 8977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Thoreen CC, Chantranupong L, Keys HR, Wang T, Gray NS, Sabatini DM, Nature 2012, 485, 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Hsieh AC, Liu Y, Edlind MP, Ingolia NT, Janes MR, Sher A, Shi EY, Stumpf CR, Christensen C, Bonham MJ, Wang S, Ren P, Martin M, Jessen K, Feldman ME, Weissman JS, Shokat KM, Rommel C, Ruggero D, Nature 2012, 485, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Ivanov P, Kedersha N, Anderson P, Genes Dev. 2011, 25, 2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Gandin V, Masvidal L, Hulea L, Gravel S-P, Cargnello M, McLaughlan S, Cai Y, Balanathan P, Morita M, Rajakumar A, Furic L, Pollak M, Porco JA, St-Pierre J, Pelletier J, Larsson O, Topisirovic I, Genome Res. 2016, 26, 636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Chan PP, Lowe TM, Nucleic Acids Res. 2016, 44, D184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Parisien M, Wang X, Pan T, RNA Biol. 2013, 10, 1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Dittmar KA, Goodenbour JM, Pan T, PLoS Genet. 2006, 2, e221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Mahlab S, Tuller T, Linial M, RNA 2012, 18, 640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Novoa EM, Ribas de Pouplana L, Trends Genet. 2012, 28, 574. [DOI] [PubMed] [Google Scholar]
- [47].Shor B, Wu J, Shakey Q, Toral-Barza L, Shi C, Follettie M, Yu K, J. Biol. Chem. 2010, 285, 15380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Michels AA, Robitaille AM, Buczynski-Ruchonnet D, Hodroj W, Reina JH, Hall MN, Hernandez N, Mol. Cell. Biol. 2010, 30, 3749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Boguta M, Graczyk D, Trends Biochem. Sci. 2011, 36, 451. [DOI] [PubMed] [Google Scholar]
- [50].Desai N, Lee J, Upadhya R, Chu Y, Moir RD, Willis IM, J. Biol. Chem. 2005, 280, 6455. [DOI] [PubMed] [Google Scholar]
- [51].Graczyk D, Cieśla M, Boguta M, Biochim. Biophys. Acta - Gene Regul. Mech. 2018, 1861, 320. [DOI] [PubMed] [Google Scholar]
- [52].Orioli A, Bioessays 2017, 39, 1600158. [DOI] [PubMed] [Google Scholar]
- [53].Torrent M, Chalancon G, de Groot NS, Wuster A, Madan Babu M, Sci. Signal. 2018, 11, eaat6409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Arimbasseri AG, Maraia RJ, Trends Biochem. Sci. 2016, 41, 546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Karkusiewicz I, Turowski TW, Graczyk D, Towpik J, Dhungel N, Hopper AK, Boguta M, J. Biol. Chem. 2011, 286, 39478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Zaborske JM, Narasimhan J, Jiang L, Wek SA, Dittmar KA, Freimoser F, Pan T, Wek RC, J. Biol. Chem. 2009, 284, 25254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Pan T, Cell Res. 2018, 28, 395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Czerwoniec A, Dunin-Horkawicz S, Purta E, Kaminska KH, Kasprzak JM, Bujnicki JM, Grosjean H, Rother K, Nucleic Acids Res. 2009, 37, D118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Saikia M, Fu Y, Pavon-Eternod M, He C, Pan T, RNA 2010, 16, 1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Clark WC, Evans ME, Dominissini D, Zheng G, Pan T, RNA 2016, 22, 1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].El Yacoubi B, Bailly M, de Crécy-Lagard V, Annu. Rev. Genet. 2012, 46, 69. [DOI] [PubMed] [Google Scholar]
- [62].Zhang X, Cozen AE, Liu Y, Chen Q, Lowe TM, Trends Mol. Med. 2016, 22, 1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Pereira M, Francisco S, Varanda AS, Santos M, Santos MAS, Soares AR, Int. J. Mol. Sci. 2018, 19, 3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Bednářová A, Hanna M, Durham I, VanCleave T, England A, Chaudhuri A, Krishnan N, Front. Mol. Neurosci. 2017, 10, 135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Agris PF, Nucleic Acids Res. 2004, 32, 223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Towns WL, Begley TJ, DNA Cell Biol. 2012, 31, 434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Capra JD, Peterkofsky A, J. Mol. Biol. 1968, 33, 591. [DOI] [PubMed] [Google Scholar]
- [68].Karlsborn T, Tükenmez H, Mahmud AKMF, Xu F, Xu H, Byström AS, RNA Biol. 2014, 11, 1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Bjork GR, Ericson JU, Gustafsson CED, Hagervall TG, Jonsson YH, Wikstrom PM, Annu. Rev. Biochem. 1987, 56, 263. [DOI] [PubMed] [Google Scholar]
- [70].Johansson MJO, Esberg A, Huang B, Björk GR, Byström AS, Mol. Cell. Biol. 2008, 28, 3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Chan CTY, Pang YLJ, Deng W, Babu IR, Dyavaiah M, Begley TJ, Dedon PC, Nat. Commun. 2012, 3, 937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Carlson BA, Xu X-M, Gladyshev VN, Hatfield DL, Um34 in Selenocysteine tRNA is Required for the Expression of Stress-Related Selenoproteins in Mammals, Fine-Tuning of RNA Functions by Modification and Editing. Springer, Berlin, Heidelberg, 2005, pp. 431–438. [Google Scholar]
- [73].Xia L, Nordman T, Olsson JM, Damdimopoulos A, Björkhem-Bergman L, Nalvarte I, Eriksson LC, Arnér ESJ, Spyrou G, Björnstedt M, J. Biol. Chem. 2003, 278, 2141. [DOI] [PubMed] [Google Scholar]
- [74].Begley U, Dyavaiah M, Patil A, Rooney JP, DiRenzo D, Young CM, Conklin DS, Zitomer RS, Begley TJ, Mol. Cell 2007, 28, 860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Maraia RJ, Blewett NH, Bayfield MA, Nat. Chem. Biol. 2008, 4, 162. [DOI] [PubMed] [Google Scholar]
- [76].Endres L, Dedon PC, Begley TJ, RNA Biol. 2015, 12, 603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Gu C, Begley TJ, Dedon PC, FEBS Lett. 2014, 588, 4287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Ozanick S, Krecic A, Andersland J, Anderson JT, RNA 2005, 11, 1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Liu F, Clark W, Luo G, Wang X, Fu Y, Wei J, Wang X, Hao Z, Dai Q, Zheng G, Ma H, Han D, Evans M, Klungland A, Pan T, He C, Cell 2016, 167, 816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Chan CTY, Dyavaiah M, DeMott MS, Taghizadeh K, Dedon PC, Begley TJ, PLoS Genet. 2010, 6, e1001247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Tyagi K, Pedrioli PGA, Nucleic Acids Res. 2015, 43, 4701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Damon JR, Pincus D, Ploegh HL, Mol. Biol. Cell 2015, 26, 270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Ibba M, Söll D, Science 1999, 286, 1893. [DOI] [PubMed] [Google Scholar]
- [84].Ruusala T, Andersson D, Ehrenberg M, Kurland CG, EMBO J. 1984, 3, 2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Wohlgemuth I, Pohl C, Mittelstaet J, Konevega AL, Rodnina MV, Philos. Trans. R. Soc. B Biol. Sci. 2011, 366, 2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Wohlgemuth I, Pohl C, V Rodnina M, EMBO J. 2010, 29, 3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Rodnina MV, in Adv. Protein Chem. Struct. Biol., 2012, pp. 95–128. [DOI] [PubMed] [Google Scholar]
- [88].Pan T, Annu. Rev. Genet. 2013, 47, 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Parker J, Johnston TC, Borgia PT, Mol. Gen. Genet. 1980, 180, 275. [DOI] [PubMed] [Google Scholar]
- [90].Netzer N, Goodenbour JM, David A, Dittmar KA, Jones RB, Schneider JR, Boone D, Eves EM, Rosner MR, Gibbs JS, Embry A, Dolan B, Das S, Hickman HD, Berglund P, Bennink JR, Yewdell JW, Pan T, Nature 2009, 462, 522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Lee JY, Kim DG, Kim B-G, Yang WS, Hong J, Kang T, Oh YS, Kim KR, Han BW, Hwang BJ, Kang BS, Kang M-S, Kim M-H, Kwon NH, Kim S, J. Cell Sci. 2014, 127, 4234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].SAKO Y, NOMURA N, UCHIDA A, ISHIDA Y, MORII H, KOGA Y, HOAKI T, MARUYAMA T, Int. J. Syst. Bacteriol. 1996, 46, 1070. [DOI] [PubMed] [Google Scholar]
- [93].Schwartz MH, Waldbauer JR, Zhang L, Pan T, Nucleic Acids Res. 2016, 44, gkw856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Lee YS, Shibata Y, Malhotra A, Dutta A, Genes Dev. 2009, 23, 2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Sun C, Fu Z, Wang S, Li J, Li Y, Zhang Y, Yang F, Chu J, Wu H, Huang X, Li W, Yin Y, Cancer Lett. 2018, 414, 16. [DOI] [PubMed] [Google Scholar]
- [96].Yamasaki S, Ivanov P, Hu G-F, Anderson P, J. Cell Biol. 2009, 185, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Thompson DM, Parker R, Cell 2009, 138, 215. [DOI] [PubMed] [Google Scholar]
- [98].Lyons SM, Fay MM, Akiyama Y, Anderson PJ, Ivanov P, RNA Biol. 2017, 14, 171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Emara MM, Ivanov P, Hickman T, Dawra N, Tisdale S, Kedersha N, Hu G-F, Anderson P, J. Biol. Chem. 2010, 285, 10959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Fay MM, Lyons SM, Ivanov P, J. Mol. Biol. 2017, 429, 2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Ivanov P, Emara MM, Villen J, Gygi SP, Anderson P, Mol. Cell 2011, 43, 613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Ivanov P, O’Day E, Emara MM, Wagner G, Lieberman J, Anderson P, Proc. Natl. Acad. Sci. 2014, 111, 18201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Lyons SM, Achorn C, Kedersha NL, Anderson PJ, Ivanov P, Nucleic Acids Res. 2016, 44, 6949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Lyabin DN, Eliseeva IA, Ovchinnikov LP, Wiley Interdiscip. Rev. RNA 2014, 5, 95. [DOI] [PubMed] [Google Scholar]
- [105].Kieran D, Sebastia J, Greenway MJ, King MA, Connaughton D, Concannon CG, Fenner B, Hardiman O, Prehn JHM, J. Neurosci. 2008, 28, 14056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Greenway MJ, Andersen PM, Russ C, Ennis S, Cashman S, Donaghy C, Patterson V, Swingler R, Kieran D, Prehn J, Morrison KE, Green A, Acharya KR, Brown RH, Hardiman O, Nat. Genet. 2006, 38, 411. [DOI] [PubMed] [Google Scholar]
- [107].Sobala A, Hutvagner G, Wiley Interdiscip. Rev. RNA 2011, 2, 853. [DOI] [PubMed] [Google Scholar]
- [108].Kuscu C, Kumar P, Kiran M, Su Z, Malik A, Dutta A, RNA 2018, 24, 1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Huang B, Yang H, Cheng X, Wang D, Fu S, Shen W, Zhang Q, Zhang L, Xue Z, Li Y, Da Y, Yang Q, Li Z, Liu L, Qiao L, Kong Y, Yao Z, Zhao P, Li M, Zhang R, Cancer Res. 2017, 77, 3194. [DOI] [PubMed] [Google Scholar]
- [110].Kumar P, Anaya J, Mudunuri SB, Dutta A, BMC Biol. 2014, 12, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Haussecker D, Huang Y, Lau A, Parameswaran P, Fire AZ, Kay MA, RNA 2010, 16, 673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Sobala A, Hutvagner G, RNA Biol. 2013, 10, 553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Luo S, He F, Luo J, Dou S, Wang Y, Guo A, Lu J, Nucleic Acids Res. 2018, 46, 5250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Kim HK, Fuchs G, Wang S, Wei W, Zhang Y, Park H, Roy-Chaudhuri B, Li P, Xu J, Chu K, Zhang F, Chua M-S, So S, Zhang QC, Sarnow P, Kay MA, Nature 2017, 552, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Bodi Z, Button JD, Grierson D, Fray RG, Nucleic Acids Res. 2010, 38, 5327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Hongay CF, Orr-Weaver TL, Proc. Natl. Acad. Sci. 2011, 108, 14855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, Cesarkas K, Jacob-Hirsch J, Amariglio N, Kupiec M, Sorek R, Rechavi G, Nature 2012, 485, 201. [DOI] [PubMed] [Google Scholar]
- [118].Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR, Cell 2012, 149, 1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Meyer KD, Jaffrey SR, Annu. Rev. Cell Dev. Biol. 2017, 33, 319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Anders M, Chelysheva I, Goebel I, Trenkner T, Zhou J, Mao Y, Verzini S, Qian S-B, Ignatova Z, Life Sci. alliance 2018, 1, e201800113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Guzzi N, Cieśla M, Ngoc PCT, Lang S, Arora S, Dimitriou M, Pimková K, Sommarin MNE, Munita R, Lubas M, Lim Y, Okuyama K, Soneji S, Karlsson G, Hansson J, Jönsson G, Lund AH, Sigvardsson M, Hellström-Lindberg E, Hsieh AC, Bellodi C, Cell 2018, 173, 1204. [DOI] [PubMed] [Google Scholar]