

Abstract
Background
Chronic Lymphocytic Leukemia (CLL) is the most frequent lymphoproliferative disorder in western countries and is characterized by a remarkable clinical heterogeneity. During the last decade, multiple genomic studies have identified a myriad of somatic events driving CLL proliferation and aggressivity. Nevertheless, and despite the mounting evidence of inherited risk for CLL development, the existence of germline variants associated with clinical outcomes has not been addressed in depth.
Methods
Exome sequencing data from control leukocytes of CLL patients involved in the International Cancer Genome Consortium (ICGC) was used for genotyping. Cox regression was used to detect variants associated with clinical outcomes. Gene and pathways level associations were also calculated.
Results
Single nucleotide polymorphisms in PPP4R2 and MAP3K4 were associated with earlier treatment need. A gene-level analysis evidenced a significant association of RIPK3 with both treatment need and survival. Furthermore, germline variability in pathways such as apoptosis, cell-cycle, pentose phosphate, GNα13 and Nitric oxide was associated with overall survival.
Conclusion
Our results support the existence of inherited conditionants of CLL evolution and points towards genes and pathways that may results useful as biomarkers of disease outcome. More research is needed to validate these findings.
Electronic supplementary material
The online version of this article (10.1186/s12885-019-5628-y) contains supplementary material, which is available to authorized users.
Keywords: Chronic lymphocytic leukemia, Germline, Polymorphism, Association, Prognosis
Background
Chronic Lymphocytic Leukemia (CLL) is the most frequent lymphoproliferative disease in western countries, and it shows remarkable clinical heterogeneity [1]. Recently, some studies demonstrated a wealth of genomic and epigenomic differences that determine part of its clinical aggressivity [2], such as point mutations in NOTCH1, SF3B1, ATM, TP53 and POT1, and the absence of somatic hypermutation in the IGHV locus.
Inherited predisposition to the development of CLL has been addressed by various genome wide association studies (GWAS) during the last years. In this regard, dozens of common variants at genes such as BCL2, EOMES, CASP10 and POT1 have been associated with significant risk of CLL development [3–5]. Similarly, GWAS studies in other lymphoproliferative disorders such as follicular lymphoma and diffuse large B cell lymphoma have found evidence for the association of germline variants with overall survival (OS) and progression-free survival [6, 7]. Despite this evidence, analysis of CLL clinical evolution have been limited almost exclusively to acquired somatic events.
In this paper, we addressed for the first time to our knowledge the association of common genomic variants with time to treatment (TTT) and OS of CLL patients participating in the Spanish ICGC cohort. Our results suggest the existence of polymorphisms at some genes (e.g., PPP4R2 and MAP3K4) significantly associated with TTT. Moreover, we found significant associations with TTT and OS both at the gene and pathway-level, which could shed new light about CLL biology and its mechanisms of progression.
Methods
Data source
We applied for access to the International Cancer Genome Consortium (ICGC) CLL sequencing data [8] deposited in the European Genome-Phenome Database (EGA). The Data Access Committee approved access to this data under DACO-1040945. We downloaded exome-seq data from control non-tumoral samples from patients with CLL under the accession code EGAD00001001464.
Data preprocessing
Exome-seq data were previously aligned to the reference genome (GRCh37.75) using bwa [9] as described in Puente et al [10]. Briefly, 3 μg of genomic DNA were used for paired-end sequencing library construction, followed by enrichment in exomic sequences using the SureSelect Human All Exon 50 Mb v4 kit or the SureSelect Human All Exon 50 Mb + UTR kits (Agilent Technologies). Next, DNA was pulled down using magnetic beads with streptavidin, followed by 18 cycles of amplification. Sequencing was performed on an Illumina GAIIx or on a HiSeq2000 sequencer (2x76bp). Duplicate read removal, sorting and indexing was done using samtools [11]. Base quality score recalibration was made with BamUtil [12] using a logistic regression model.
Variant detection and filtering
Platypus2 [13] was run on genotyping mode. All dbSNP variants [14] were used as input for genotyping. We used the following specifications: “minVarFreq = 0.02”, “minReads = 2”, “maxReads = 8000”, “assemble = 1”, “minBaseQual = 20”, “trimSoftClipped = 1”, “minPosterior = 20”, “sbThreshold = 0.01”, “badReadsWindow = 15” and “badReadsThreshold = 15”, at least 10 reads covering a position and 2 reads covering a variant, a minimum genotype quality (GQ) of 20 Phred, genotype likelihood (GL) below − 3, maximum homopolymer run (HP) below 11, minimum variant quality adjusted per read depth (QD) above 2 and minimum median minimum base quality for bases around variant (MMLQ) above 10. Variants labeled by platypus as “HapScore”, “SC”, “strandBias” and “MQ” were discarded. Heterozygous loci with variant allele frequency (VAF) < 35% or > 70% were also discarded.
Sample filtering
We used principal component analysis (PCA) to detect outliers in our study cohort. Similarly, identity-by-descent (IBD) was used to discard all individuals with a degree of relatedness equivalent to third degree or higher. PCAs and IBD data were computed on a linkage disequilibrium (LD) pruned dataset (LD upper threshold of 0.2) using the Bioconductor [15] package SNPRelate [16]. Our final filtered dataset contained 426 cases. Among these, 253 were males and 173 were females. By IGHV status, there were 146 unmutated cases and 273 mutated cases; and by clinical staging, the data contained 47 monoclonal B-cell lymphocytosis (MBL), 332 Binet A, 37 Binet B and 8 Binet C cases. Information about clinical staging and IGHV mutation status was not available for 2 and 7 cases, respectively.
Regression analysis
Cox regression and assumption of proportional hazards was performed with the survival R package [17, 18]. Variables with p-value < 0.2 in a univariate model were selected as covariates for the GWAS. In the cases of TTT these were “donor sex”, “IGHV mutation status” and “Binet stage”; whilst in the case of overall survival we used “IGHV mutation status”, “Binet stage” and “donor age at diagnosis” as covariates. Three association models were computed: an additive model, a dominant model and a recessive model. P-values were adjusted using the Benjamini-Hochberg (BH) method.
Due to the heterogeneity of exome-seq coverage and quality metrics, many variables had incomplete data. We included in the analysis variables with at least 25% call rate, a minimum of 10 events (progression or death). A minimum allele frequency of 1% was selected as the lowest threshold. Furthermore, we only analyzed polymorphisms where Platypus called at least 10 minor alleles (additive model) or genotypes (dominant and recessive models).
Inflation values were estimated with the R package QQperm [19]. Briefly, a random distribution of p-values was created by randomly permuting phenotype variables. Then, the association p-values are compared with the null. This method doesn’t consider the null distribution to be distributed uniformly.
Gene-level analysis
VEGAS2 [20] was used to calculate LD-adjusted association p-values for TTT and OS. Briefly, VEGAS2 takes GWAS p-values, and then uses a simulation-based approach using information from population variant reference panels to adjust for LD effects. We used the 1000 Genomes phase 3 data from the iberian population in Spain as our reference population, since all patients of this cohort were of Spanish origin [21]. Only variants falling within the 5′ and 3′ coordinates of RefSeq genes were included. P-values were adjusted for multiple testing using the BH method.
Pathways and gene ontology (GO) analysis
GSEA4GWAS version 1.1 [22] was used for testing significant associations in pathways and biological process annotations. Our input pathways were “Canonical Pathways” and “Gene Ontology Biological Process”. Maximum distance was set to 20 Kb, and the major histocompatibility complex region was masked from the analysis. P-values were adjusted with the BH method.
Results
Genomic polymorphisms associated with treatment-free survival
We created three models to analyze variant association with time to first treatment: an additive, a dominant and a regressive model. PCA plots (Additional file 1: Figure S1) and lambda inflation values (lambda values of 1.06, 1.03 and 1.04, Fig. 1) revealed no significant inflation or population stratification. In the additive model we observed 6 polymorphisms associated with TTT (BH adjusted p-value < 0.05), and other 6 showed association with BH adjusted p-value in the range of 0.05–0.1 (Fig. 2, Table 1, Additional file 9 Table S1). These variants were located in MAP3K4, PEX26 (4 variants), PPP4R2 (2 variants), TTLL12/TSPO, TXNRD2, ZCCHC7, MKI67IP and MARCH10. Notably, rs537453728 at MAP3K4 broke the genome-wide association p-value (4.53 × 10− 8). Other 391 variants were suggestively associated with TTT (BH adjusted p-value 0.1–0.5).
Fig. 1.

QQplots for the TTT and OS additive, dominant and recessive models
Fig. 2.
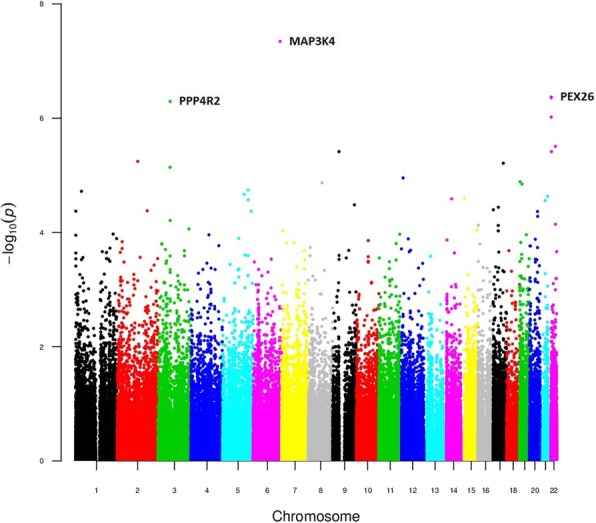
Manhattan plot of the additive TTT model results
Table 1.
Results of the additive, dominant and recessive TTT models. Polymorphisms with a BH-adjusted P-value < 0.1 are show
| rs ID | P-value | BH-adjusted P-value | Reference | Alternative | Gene Symbol | Hazard Ratio | MAF |
|---|---|---|---|---|---|---|---|
| ADDTIVE MODEL | |||||||
| rs537453728 | 4.53E-08 | 5.39 E-3 | CT | C | MAP3K4 | 7.08 | 0.01 |
| rs361807 | 4.31E-07 | 0.01 | T | C | PEX26 | 4.55 | 0.1 |
| rs361946 | 4.31E-07 | 0.01 | A | G | PEX26 | 4.55 | 0.1 |
| rs1043278 | 4.31E-07 | 0.01 | A | G | PEX26 | 4.55 | 0.1 |
| rs7620924 | 5.08E-07 | 0.01 | A | G | PPP4R2 | 0.24 | 0.49 |
| rs5992169 | 9.55E-07 | 0.02 | G | A | PEX26 | 4.45 | 0.11 |
| rs9463 | 3.11E-06 | 0.05 | G | A | TTLL12 | 3.18 | 0.42 |
| rs55715863 | 3.84E-06 | 0.05 | C | A | TXNRD2 | 5.24 | 0.01 |
| rs3780333 | 3.84E-06 | 0.05 | A | G | ZCCHC7 | 4.8 | 0.27 |
| rs17016977 | 5.69E-06 | 0.07 | A | G | MKI67IP | 5.53 | 0.01 |
| rs72842201 | 6.13E-06 | 0.07 | A | G | MARCH.10 | 2.99 | 0.05 |
| rs3172278 | 7.23E-06 | 0.07 | T | C | PPP4R2 | 4.04 | 0.49 |
| DOMINANT MODEL | |||||||
| rs3172278 | 2.40E-08 | 1.23E-03 | T | C | PPP4R2 | 0.09 | 0.49 |
| rs2247870 | 1.60E-07 | 4.10E-03 | G | A | GPR98 | 0.4 | 0.47 |
| rs9463 | 3.07E-07 | 5.26E-03 | G | A | TTLL12 | 0.17 | 0.42 |
| rs28656102 | 1.22E-06 | 0.02 | T | C | PPP4R2 | 0.18 | 0.42 |
| rs1045960 | 6.22E-06 | 0.05 | C | T | PPP4R2 | 0.21 | 0.42 |
| rs62039297 | 6.29E-06 | 0.05 | G | A | SRL | 0.2 | 0.26 |
| rs9873229 | 9.72E-06 | 0.07 | C | T | PPP4R2 | 0.22 | 0.44 |
| RECESSIVE MODEL | |||||||
| rs7620924 | 8.07E-11 | 1.08E-05 | A | G | PPP4R2 | 11.77 | 0.49 |
| rs537453728 | 4.53E-08 | 3.04E-03 | CT | C | MAP3K4 | 0.14 | 0.01 |
| rs9310254 | 7.49E-07 | 0.03 | T | C | PPP4R2 | 5.56 | 0.49 |
The dominant and the recessive models evidenced very significant enrichment of variants at PPP4R2 (lowest p-value at rs7620924: 8.07 × 10− 11). Variants at GPR98, MAP3K4 and TTLL12 were also significantly associated with TTT (BH adjusted p-value < 0.05) (Figs. 3 and 4, Table 1, Additional file 9: Tables S2 and S3).
Fig. 3.
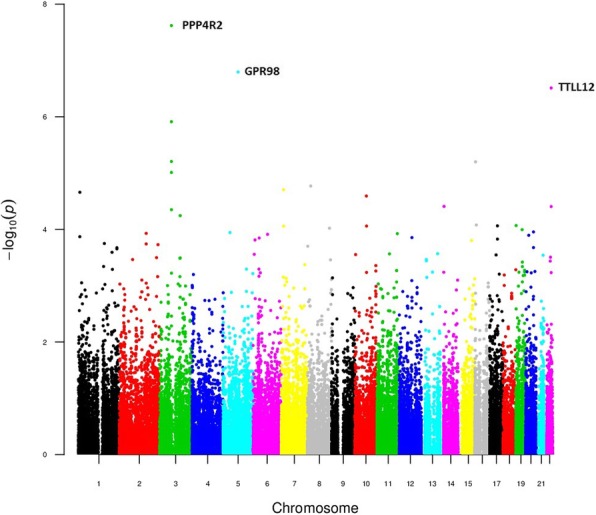
Manhattan plot of the dominant TTT model results
Fig. 4.
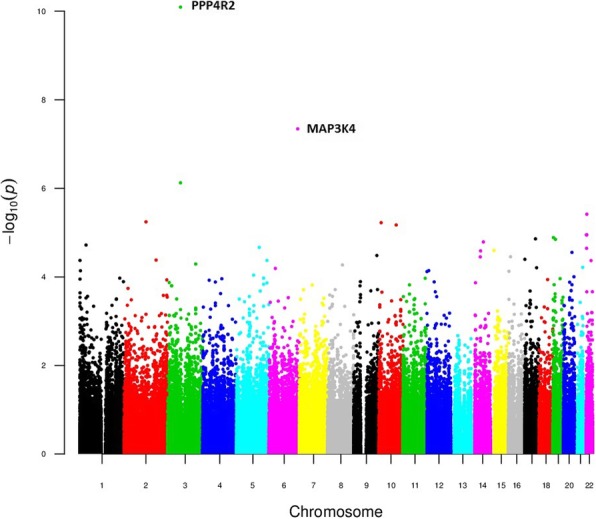
Manhattan plot of the recessive TTT model result
Some of this polymorphisms are associated with functional changes in their corresponding genes. According to dbSNP [14], rs2247870 induces a missense change in the GPR98 gene. In a similar fashion, a search in the HaploReg database [23] points toward functional implications of some of these variants. For example, rs7620924 is located in a lymphocyte-specific enhancer region that is strongly associated with PPP4R2 expression in whole blood (p-value 2.18 × 10− 34) and in lymphoblastoid cells (p-value 2.36 × 10− 8). Similarly, the polymorphisms rs361807, rs361946, rs5992169 and rs1043278 in PEX36 are associated with PEX36 expression in lymphoblastoid cells (minimum p-value 6.66 × 10− 10). rs9463 in TTLL12 strongly correlates with the expression of the adjacent gene TSPO (p-value 9.81 × 10− 198), and to a lower extent, with TTLL12 (p-value 9.57 × 10− 4) in blood cells. Other polymorphisms suggestively associated with TTT such as those in TXNRD2 and ZCCHC7 are also significantly associated with the expression of their respective genes in blood cells. On the contrary, no functional information exists about the intronic variant rs537453728 within the MAP3K4 gene.
Genomic polymorphisms associated with overall survival
We created an additive, a dominant and a recessive model to investigate variant association with OS. No significant inflation was observed (lambda values of 1.02, 1.01 and 1.01 for additive, dominant and recessive models, respectively). No variant achieved BH-adjusted p-values below 0.05 in any model (Additional file 2: Figure S2, Additional file 3: Figure S3 Additional file 4: Figure S4, Table 2, Additional file 9: Tables S4-S6). Nevertheless, 20 variants were below 0.06 in the additive models, with the top variants falling in TTC32, WDR35 and CLIP1. Notably, rs2304588 at TTC32 achieved the lowest p-value (7.6 × 10− 7, Bonferroni-adjusted p-value 0.067, BH-adjusted p-value 0.056). Overall, the number of variants with BH adjusted p-values below 0.1 was 25 in the additive model, 4 in the dominant model and 18 in the recessive model.
Table 2.
Results of the additive, dominant and recessive OS models. Polymorphisms with a BH-adjusted P-value < 0.1 are show
| rs ID | P-value | BH-adjusted P-value | Reference | Alternative | Symbol | Hazard Ratio | MAF |
|---|---|---|---|---|---|---|---|
| ADDITIVE | |||||||
| rs2304588 | 7.36E-07 | 0.06 | A | G | TTC32 | 6.59 | 0.08 |
| rs2293671 | 1.43E-06 | 0.06 | T | C | WDR35 | 4.97 | 0.08 |
| rs139689179 | 2.17E-06 | 0.06 | G | C | CLIP1 | 33.51 | 0.02 |
| rs149301745 | 3.45E-06 | 0.06 | ACTT | A | SPATA5L1 | 6.04 | 0.04 |
| rs56127964 | 3.88E-06 | 0.06 | A | G | ACO1 | 7.51 | 0.02 |
| rs1800502 | 4.65E-06 | 0.06 | A | G | CFTR | 6.23 | 0.024 |
| rs1060742 | 4.88E-06 | 0.06 | T | C | WDR35 | 4.05 | 0.08 |
| rs72860063 | 4.98E-06 | 0.06 | A | G | KIF6 | 11.19 | 0.02 |
| rs7563410 | 6.46E-06 | 0.06 | C | A | TTC32 | 3.97 | 0.08 |
| rs28502265 | 6.48E-06 | 0.06 | T | G | WDR35 | 3.98 | 0.08 |
| rs769479742 | 7.23E-06 | 0.06 | T | G | PDE1C | 7.39 | 0.03 |
| rs76774695 | 7.96E-06 | 0.06 | C | T | PARP6 | 10.54 | 0.01 |
| rs148915854 | 8.41E-06 | 0.06 | T | TA | FAM184B | 5.19 | 0.07 |
| rs114314465 | 9.74E-06 | 0.06 | A | C | SLC4A5 | 10.69 | 0.02 |
| rs3732783 | 9.77E-06 | 0.06 | T | C | DRD3 | 13.34 | 0.09 |
| rs117149407 | 9.79E-06 | 0.06 | G | A | EYA1 | 14.24 | 0.03 |
| rs12194408 | 1.14E-05 | 0.06 | C | G | PPIL1 | 7.15 | 0.02 |
| rs1790557 | 1.16E-05 | 0.06 | C | T | TENM4 | 7.98 | 0.25 |
| rs35034822 | 1.27E-05 | 0.06 | T | G | PROM2 | 14.77 | 0.02 |
| rs147203128 | 1.29E-05 | 0.06 | G | A | CYTH2 | 12.94 | 0.02 |
| chr2:95944632 | 1.42E-05 | 0.06 | T | G | PROM2 | 14.47 | 0.02 |
| rs200619224 | 1.49E-05 | 0.06 | A | C | CDC23 | 6.15 | 0.02 |
| rs75827446 | 1.57E-05 | 0.06 | T | C | TNL2 | 43.14 | 0.02 |
| rs1053361 | 1.68E-05 | 0.06 | G | C | RHCE | 11.75 | 0.02 |
| rs7270676 | 2.44E-05 | 0.09 | T | C | SEMG1 | 6.84 | 0.04 |
| DOMINANT | |||||||
| rs7512589 | 3.71E-06 | 0.07 | G | A | CELA2A | 0.21 | 0.29 |
| rs3737697 | 3.93E-06 | 0.07 | C | T | CELA2A | 0.21 | 0.29 |
| rs62039297 | 9.09E-06 | 0.1 | G | A | SRL | 0.06 | 0.26 |
| rs10783474 | 1.08E-05 | 0.1 | G | A | SCN8A | 0.11 | 0.16 |
| RECESSIVE | |||||||
| rs2304588 | 7.36E-07 | 0.05 | A | G | TTC32 | 0.15 | 0.08 |
| rs2293671 | 1.12E-06 | 0.05 | T | C | WDR35 | 0.18 | 0.08 |
| rs139689179 | 2.17E-06 | 0.06 | G | C | CLIP1 | 0.03 | 0.02 |
| rs149301745 | 3.45E-06 | 0.06 | ACTT | A | SPATA5L1 | 0.17 | 0.04 |
| rs1060742 | 4.18E-06 | 0.06 | T | C | WDR35 | 0.23 | 0.08 |
| rs72860063 | 4.98E-06 | 0.06 | A | G | KIF6 | 0.09 | 0.02 |
| rs7563410 | 5.41E-06 | 0.06 | C | A | TTC32 | 0.24 | 0.08 |
| rs28502265 | 5.59E-06 | 0.06 | T | G | WDR35 | 0.24 | 0.08 |
| rs769479742 | 7.23E-06 | 0.07 | T | G | PDE1C | 0.14 | 0.03 |
| rs76774695 | 7.96E-06 | 0.07 | C | T | PARP6 | 0.09 | 0.01 |
| rs114314465 | 9.74E-06 | 0.08 | A | C | SLC4A5 | 0.09 | 0.02 |
| rs117149407 | 9.79E-06 | 0.08 | G | A | EYA1 | 0.07 | 0.03 |
| rs12194408 | 1.14E-05 | 0.08 | C | G | PPIL1 | 0.14 | 0.02 |
| rs35034822 | 1.27E-05 | 0.08 | T | G | PROM2 | 0.07 | 0.02 |
| chr2:95944632 | 1.42E-05 | 0.08 | T | G | PROM2 | 0.07 | 0.02 |
| rs200619224 | 1.49E-05 | 0.08 | A | C | CDC23 | 0.16 | 0.02 |
| rs75827446 | 1.57E-05 | 0.08 | T | C | TLN2 | 0.02 | 0.02 |
| rs1053361 | 1.68E-05 | 0.09 | G | C | RHCE | 0.09 | 0.02 |
Gene-based association with TTT and OS
Gene-level integration of p-values for the TTT model using VEGAS2 evidenced the association of 5 genes with BH adjusted p-values < 0.05 and 10 genes with BH adjusted p-values in the range of 0.05–0.1 (Table 3, Additional file 9: Table S7). The most significant genes were AURKAIP1, NIFK, RIPK3, SIK1 and ZCCHC7.
Table 3.
VEGAS2 gene-level analysis of the additive TTT and OS models. Genes with a BH-adjusted P-value < 0.1 are shown
| Gene | nSNPs | nSims | Test | P value | TopSNP | TopSNP.p value | BH-ajdusted p-value |
|---|---|---|---|---|---|---|---|
| VEGAS2 Analysis of TTT | |||||||
| AURKAIP1 | 2 | 1.00E+06 | 26.72 | 1.00E-06 | rs2765035 | 2.28E-04 | 6.90E-03 |
| NIFK | 4 | 1.00E+06 | 36.22 | 1.00E-06 | rs17016977 | 5.69E-06 | 6.90E-03 |
| RIPK3 | 7 | 1.00E+06 | 22.32 | 2.00E-06 | rs28379107 | 0.02 | 9.20E-03 |
| SIK1 | 3 | 1.00E+06 | 31.07 | 9.00E-06 | rs3746951 | 2.32E-05 | 0.03 |
| ZCCHC7 | 2 | 1.00E+06 | 23.2 | 1.10E-05 | rs3780333 | 3.84E-06 | 0.03 |
| ABCA7 | 18 | 1.00E+06 | 88.04 | 2.30E-05 | rs3752230 | 3.22E-04 | 0.05 |
| PEX26 | 7 | 1.00E+06 | 83.49 | 2.70E-05 | rs361807 | 4.31E-07 | 0.05 |
| AARS | 3 | 1.00E+06 | 17.21 | 4.10E-05 | rs2070203 | 2.34E-03 | 0.06 |
| NIFK-AS1 | 3 | 1.00E+06 | 24.72 | 4.50E-05 | rs17016977 | 5.69E-06 | 0.06 |
| TSPO | 2 | 1.00E+06 | 24.36 | 4.60E-05 | rs6971 | 7.18E-05 | 0.06 |
| MARCH.10 | 2 | 1.00E+06 | 20.45 | 4.80E-05 | rs72842201 | 6.13E-06 | 0.06 |
| LOC574538 | 3 | 1.00E+06 | 24.15 | 7.20E-05 | rs56204927 | 1.94E-04 | 0.08 |
| SEC24D | 10 | 1.00E+06 | 44.69 | 7.60E-05 | rs115446044 | 1.09E-03 | 0.08 |
| TTLL12 | 9 | 1.00E+06 | 54.06 | 9.90E-05 | rs9463 | 3.11E-06 | 0.1 |
| GALNT11 | 4 | 1.00E+06 | 15.18 | 1.07E-04 | rs146169444 | 0.01 | 0.1 |
| VEGAS2 Analysis of OS | |||||||
| CLUAP1 | 7 | 1.00E+06 | 57.76 | 1.00E-06 | rs78851263 | 3.27E-04 | 6.17E-03 |
| TTC32 | 5 | 1.00E+06 | 66.51 | 1.00E-06 | rs2304588 | 7.36E-07 | 6.17E-03 |
| DRD3 | 2 | 1.00E+06 | 21.54 | 4.00E-06 | rs3732783 | 9.77E-06 | 0.02 |
| RAD51AP2 | 7 | 1.00E+06 | 22.31 | 6.00E-06 | rs62130401 | 0.02 | 0.02 |
| RIPK3 | 8 | 1.00E+06 | 18.64 | 1.30E-05 | rs3212251 | 0.01 | 0.03 |
| EPYC | 2 | 1.00E+06 | 17.78 | 2.00E-05 | rs76171854 | 3.25E-04 | 0.03 |
| TENM4 | 13 | 1.00E+06 | 75.05 | 2.10E-05 | rs1790557 | 1.16E-05 | 0.03 |
| GTPBP1 | 6 | 1.00E+06 | 17.81 | 2.30E-05 | rs16999297 | 6.42E-03 | 0.03 |
| GAMT | 2 | 1.00E+06 | 17.56 | 2.80E-05 | rs266809 | 8.65E-05 | 0.03 |
| DCP1B | 16 | 1.00E+06 | 61.9 | 3.00E-05 | rs150660202 | 7.13E-03 | 0.03 |
| MIR548D1 | 3 | 1.00E+06 | 17.35 | 3.30E-05 | rs12141159 | 4.48E-04 | 0.03 |
| MIR548AA1 | 3 | 1.00E+06 | 17.35 | 3.40E-05 | rs12141159 | 4.48E-04 | 0.03 |
| WDR35 | 14 | 1.00E+06 | 89.29 | 5.90E-05 | rs2293671 | 1.43E-06 | 0.06 |
| BNIPL | 6 | 1.00E+06 | 23.11 | 7.40E-05 | rs955955 | 2.91E-03 | 0.06 |
| KARS | 3 | 1.00E+06 | 15.43 | 1.05E-04 | rs148298278 | 1.28E-04 | 0.09 |
| ACO1 | 13 | 1.00E+06 | 57.84 | 1.26E-04 | rs56127964 | 3.88E-06 | 0.1 |
| PARP6 | 3 | 1.00E+06 | 21.16 | 1.33E-04 | rs76774695 | 7.96E-06 | 0.1 |
Similarly, we observed 12 genes associated with OS at a BH-adjusted p-value < 0.05 (Table 2, Additional file 9: Table S8), namely CLUAP1, TTC32, DRD3, RAD51AP2, RIPK3, EPYC, TENM4, GTPBP1, GAMT, DCP1B, MIR548D1 and MIR548AA1. Other 5 genes were associated with OS with BH-adjusted p-values in the range of 0.05–0.1. These genes were WDR35, BNIPL, KARS, ACO1 and PARP6.
Pathway-level variation significantly associated with TTT and OS
We used GSEA4GWAS to analyze associations with TTT and OS at the pathway level. No significant enrichment neither in “Canonical pathways” nor in “Biological Process” terms was observed for the TTT variable. Nevertheless, a different scenario was found for the OS variable. Significant “Canonical pathways” annotations (BH-adjusted p-values < 0.05) were “Pentose Phosphate pathway”, “Gluconeogenesis”, “Glycolysis”, “GNα13 pathway”, “Nitric Oxide pathway”, “Apoptosis”, “Glycolysis and Gluconeogenesis”, “Tumor Necrosis Factor pathway”, “Ovarian Infertility genes”, “Bile Acid Biosynthesis”, “Keratinocyte pathway” and “Glycine Serine and Threonine pathway” (Table 4, Additional file 5: Figure S5 and Additional file 6: Figure S6). Other pathways had evidence of suggestive association (BH-adjusted p-value < 0.25), such as “Notch Signalling pathway”, “EGF pathway”, “JNK MAPK pathway” and “PDGF pathway” among others. Among “GO Biological Processes” terms, the following were associated with OS with a BH-adjusted p-value < 0.05: “Induction of Apoptosis by Extracellular Signals”, “Mitosis”, “M phase”, “M phase of mitotic cell cycle” and “Negative regulation of Developmental Process”. Terms with BH-adjusted p-value < 0.25 were “Anti Apoptosis”, “Chromosome Segregation”, “Vasculature Development” and “Negative Regulation of Apoptosis”, among others (Table 4, Additional file 7: Figure S7 and Additional file 8: Figure S8).
Table 4.
GSEA4GWAS analysis of the additive OS model. Gene Ontology Biological Process and Canonical Pathways terms with a BH-adjusted P-value < 0.25 are shown
| Pathway/Gene set name | P-value | FDR | Significant genes/Selected genes/All genes |
|---|---|---|---|
| GSEA4GWAS Biological Process results for the Additive OS model | |||
| INDUCTION OF APOPTOSIS BY EXTRACELLULAR SIGNALS | < 0.001 | 0.01 | 9/21/27 |
| MITOSIS | < 0.001 | 0.02 | 23/68/82 |
| M PHASE | < 0.001 | 0.02 | 31/96/114 |
| M PHASE OF MITOTIC CELL CYCLE | < 0.001 | 0.04 | 23/70/85 |
| NEGATIVE REGULATION OF DEVELOPMENTAL PROCESS | 0.001 | 0.05 | 36/148/197 |
| ANTI APOPTOSIS | < 0.001 | 0.05 | 20/83/118 |
| CHROMOSOME SEGREGATION | 0.003 | 0.1 | 10/29/32 |
| CELL CYCLE PHASE | 0.001 | 0.13 | 38/144/170 |
| CELL CYCLE PROCESS | < 0.001 | 0.19 | 42/160/193 |
| REGULATION OF MITOSIS | 0.004 | 0.19 | 11/36/41 |
| VASCULATURE DEVELOPMENT | 0.007 | 0.24 | 14/43/55 |
| SKELETAL DEVELOPMENT | 0.011 | 0.25 | 23/80/103 |
| GAMETE GENERATION | 0.011 | 0.25 | 18/76/114 |
| NEGATIVE REGULATION OF APOPTOSIS | 0.008 | 0.25 | 21/112/150 |
| NEGATIVE REGULATION OF PROGRAMMED CELL DEATH | 0.008 | 0.25 | 21/112/151 |
| GSEA4GWAS Cannonical Pathways results for the Additive OS model | |||
| PENTOSE PHOSPHATE PATHWAY | < 0.001 | 1.67E-03 | 8/19/25 |
| GLUCONEOGENESIS | < 0.001 | 2.00E-03 | 18/47/53 |
| GLYCOLYSIS | < 0.001 | 2.00E-03 | 18/47/53 |
| HSA00030 PENTOSE PHOSPHATE PATHWAY | < 0.001 | 0.01 | 8/21/26 |
| ST GA13 PATHWAY | < 0.001 | 0.02 | 11/29/37 |
| NO1PATHWAY | < 0.001 | 0.03 | 11/26/31 |
| HSA04210 APOPTOSIS | 0.001 | 0.03 | 19/68/84 |
| HSA00010 GLYCOLYSIS AND GLUCONEOGENESIS | 0.001 | 0.03 | 17/58/64 |
| ST TUMOR NECROSIS FACTOR PATHWAY | < 0.001 | 0.04 | 7/20/29 |
| OVARIAN INFERTILITY GENES | 0.001 | 0.04 | 8/21/25 |
| BILE ACID BIOSYNTHESIS | 0.002 | 0.04 | 9/23/27 |
| KERATINOCYTEPATHWAY | 0.001 | 0.04 | 12/37/46 |
| GLYCINE SERINE AND THREONINE METABOLISM | 0.006 | 0.05 | 9/28/37 |
| BREAST CANCER ESTROGEN SIGNALING | 0.001 | 0.06 | 21/78/101 |
| GLYCEROLIPID METABOLISM | 0.008 | 0.09 | 13/36/45 |
| NOS1PATHWAY | 0.01 | 0.1 | 7/20/22 |
| HSA00120 BILE ACID BIOSYNTHESIS | 0.003 | 0.11 | 11/36/38 |
| HSA04330 NOTCH SIGNALING PATHWAY | 0.011 | 0.11 | 10/35/47 |
| FRUCTOSE AND MANNOSE METABOLISM | 0.01 | 0.11 | 7/21/25 |
| EGFPATHWAY | 0.009 | 0.11 | 8/24/27 |
| HSA05218 MELANOMA | 0.01 | 0.11 | 13/58/71 |
| ST JNK MAPK PATHWAY | 0.011 | 0.12 | 10/33/40 |
| PDGFPATHWAY | 0.011 | 0.12 | 8/24/27 |
| TYROSINE METABOLISM | 0.015 | 0.12 | 8/24/32 |
| HSA05223 NON SMALL CELL LUNG CANCER | 0.009 | 0.12 | 13/47/54 |
| HSA00760 NICOTINATE AND NICOTINAMIDE METABOLISM | 0.019 | 0.14 | 7/20/24 |
| HSA00051 FRUCTOSE AND MANNOSE METABOLISM | 0.013 | 0.14 | 10/35/42 |
| BUTANOATE METABOLISM | 0.017 | 0.14 | 7/26/29 |
| HSA00260 GLYCINE SERINE AND THREONINE METABOLISM | 0.018 | 0.15 | 8/36/45 |
| HSA00561 GLYCEROLIPID METABOLISM | 0.022 | 0.19 | 15/49/58 |
| STRIATED MUSCLE CONTRACTION | 0.023 | 0.2 | 11/31/39 |
| PPARAPATHWAY | 0.023 | 0.2 | 11/43/57 |
| HSA04510 FOCAL ADHESION | 0.015 | 0.2 | 53/171/200 |
| G1PATHWAY | 0.04 | 0.21 | 4/22/28 |
| HSA05222 SMALL CELL LUNG CANCER | 0.024 | 0.21 | 22/78/87 |
| HSA05010 ALZHEIMERS DISEASE | 0.029 | 0.21 | 7/23/28 |
| HIVNEFPATHWAY | 0.029 | 0.22 | 13/46/58 |
| HSA04662 B CELL RECEPTOR SIGNALING PATHWAY | 0.041 | 0.23 | 15/57/64 |
| HSA05214 GLIOMA | 0.035 | 0.24 | 14/56/64 |
| NFATPATHWAY | 0.039 | 0.24 | 11/46/53 |
Discussion
In this work we present evidence that suggest the existence of germline variation modulating CLL’s clinical aggressivity. The most remarkable finding was the strong and recessive association of rs7620924 near PPP4R2 with short time to treatment. The implication of PPP4R2 in the regulation of cell survival and DNA repair in hematopoietic and leukemia cells has been recently reported [24]. Indeed, different studies have identified PPP4R2 as a modulator of protein phosphatase 4 (PPP4), which regulates DNA repair through non-homologous end joining [25]. Concordantly, the ablation of PPP4 activity in mice increases genomic instability and aborgates class switch recombination in B cells, leading to an abnormal immune response [26]; and its function also seems to be essential in V(D) J recombination during normal B cell maturation [27]. Other polymorphisms associated with time to first treatment were located in MAP3K4, PEX26 and TTLL12. MAP3K4 participates in the TRAIL/MAP3K4/p38/HSP27/Akt pathway, thereby modulating processes such as autophagy and cell migration. Indeed, MAP3K4 is affected by recurrent loss-of-function mutations in different types of cancers [28–32]. Conversely, less is known about the peroxisome-related gene PEX26 [33]; the G-protein GPR98; and TTLL12, which participates in chromosome stability and mitosis-related processes [34]. On the contrary, although we did not find any variant significantly associated with overall survival, we devised some variants with suggestive associations. The two most significant ones were in the TTC32/WDR35 and CLIP1 loci, the last of which is overexpressed in Reed-Sternberg cells of Hodgkin lymphoma [35, 36].
In a similar fashion, we detected variation on different genes associated with CLL evolution. The most relevant was the association of RIP3K with both time to treatment and overall survival. RIP3K encodes a protein that regulates necroptosis, a form of regulated cell death characterized by cell membrane permeabilization [37]. Other relevant genes associated with rapid progression were the pro-proliferative gene NIFK [38], the tumor suppressor SIK1 [39, 40] and ZCCHC7, which is encoded near the B-cell specific PAX5 super-enhancer locus [41, 42]. On the other hand, various genes were associated with overall survival, such as CLUAP1, which participates in tumor growth and cytoskeleton regulation [43–45]; and the enzyme GAMT, which converts S-adenosylmethionine to creatine in order to foster high energy demands [46]. Moreover, the BCL-2 interacting gene BNIPL [47, 48] was suggestively associated with survival and deserves further characterization. In the same direction, the most remarkable pathway-level association with survival was that of the pentose phosphate metabolic pathway, which fuels cells with metabolites for nucleotide and lipid biosynthesis, and provides reducing power to promote cell survival under stressful conditions [49]. Other pathways such as GNα13 and Nitric Oxide were also significant. Concordantly, recurrent inactivating mutations in the G-protein superfamily gene GNA13 have been described in B cell lymphomas [50–53], and the contribution of nitric oxide to apoptosis resistance in CLL cells has been addressed by various studies [54, 55].
The main limitation of this study is the lack of an independent cohort for validation of these findings. Furthermore, although inflation values were low, we assume that treatment heterogeneity could have an impact on overall survival associations. Nevertheless, the global results are not only statistical significant but also biologically plausible. Thus, we believe that this report will motivate further studies in order to confirm the effect of these variants and to determine their mechanisms of action in lymphoproliferative disorders.
Conclusions
Our results point towards the existence of germline variability as a determinant of CLL clinical aggressivity. Future studies to validate and characterize the activity of these variants in CLL are needed.
Additional files
{kind=link}
Figure S1. Principal component plots for the subjects included in the final analysis. (JPG 154 kb)
{kind=link}
Figure S2. Manhattan plot of the additive OS model results. (JPG 199 kb)
{kind=link}
Figure S3. Manhattan plot of the dominant OS model results. (JPG 222 kb)
{kind=link}
Figure S4. Manhattan plot of the recessive TTT model results. (JPG 193 kb)
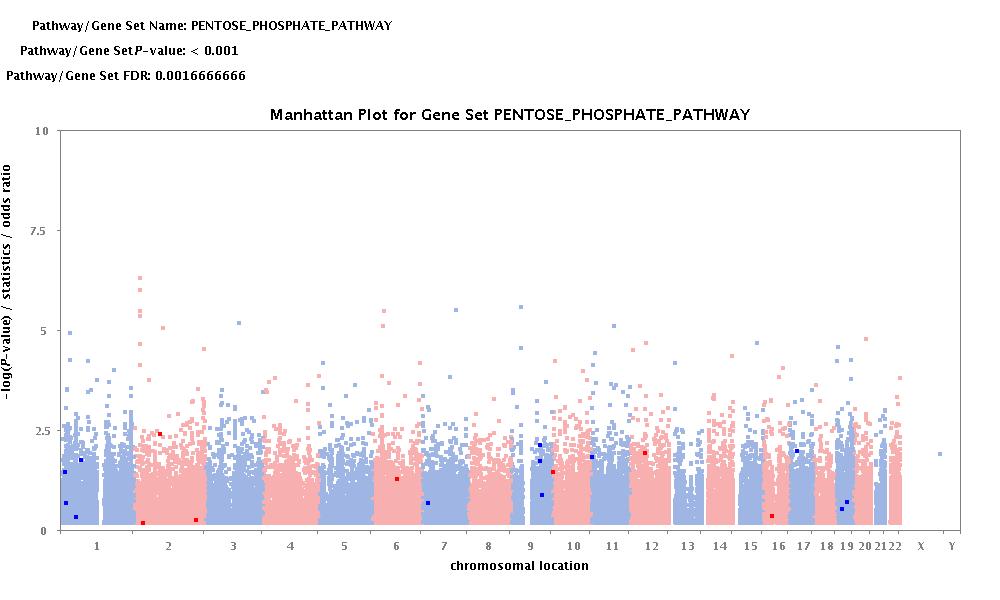{kind=link}
Figure S5. Manhattan plot for the Pentose Phosphate pathway. (JPG 56 kb)
{kind=link}
Figure S6. Manhattan plot for the GNα13 pathway. (JPG 54 kb)
{kind=link}
Figure S7. Manhattan plot for the “Induction of apoptosis by extracellular signal” biological process. (JPG 59 kb)
{kind=link}
Figure S8. Manhattan plot for the “Mitosis” biological process. (JPG 54 kb)
Table S1. GWAS results of the additive Cox regression model for TTT. Polymorphisms with a BH-adjusted P-value < 0.5 are shown. Table S2. GWAS results of the dominant Cox regression model for TTT. Polymorphisms with a BH-adjusted P-value < 0.5 are shown. Table S3. GWAS results of the recessive Cox regression model for TTT. Polymorphisms with a BH-adjusted P-value < 0.5 are shown. Table S4. GWAS results of the additive Cox regression model for OS. Polymorphisms with a BH-adjusted P-value < 0.5 are shown. Table S5. GWAS results of the dominant Cox regression model for OS. Polymorphisms with a BH-adjusted P-value < 0.5 are shown. Table S6. GWAS results of the recessive Cox regression model for OS. Polymorphisms with a BH-adjusted P-value < 0.5 are shown. Table S7. VEGAS2 gene-level results for association with TTT. Table S8. VEGAS2 gene-level results for association with OS. (XLSX 3081 kb)
Acknowledgements
We would like to thank the Supercomputing Center of Galicia (CESGA) for providing all the informatics resources needed for performing this research. We would also like to thank the International Cancer Genome Consortium and the European Genome Archive for providing and facilitating data access. The content of this paper is part of the doctoral thesis of Adrián Mosquera Orgueira to obtain a PhD at the Department of Medicine, University of Santiago de Compostela.
Funding
This research had no funding support.
Availability of data and materials
The data that support the findings of this study are available from the ICGC but restrictions apply to the availability of these data, which were used under license for the current study, and so are not publicly available. Data are however available from the authors upon reasonable request and with permission of the ICGC.
Abbreviations
- BH
Benjamini-Hochberg
- CLL
Chronic Lymphocytic Leukemia:
- EGA
European Genome-Phenome Database
- IBD
Identity By Descent
- ICGC
International Cancer Genome Consortium
- LD
Linkage Disequilibrium
- MBL
Monoclonal B cell lymphocytosis
- OS
Overall Survival:
- PCA
Principal Component Analysis
- TTT
Time to Treatment
- VAF
Variant allele frequency
Authors’ contributions
AMO, BAR and JLBL designed the study and wrote the manuscript; AMO performed the analysis; AMO, BAR, CAT, EMGS, ACA interpreted the results; JADA, NAV, NDV and MMPE were involved in manuscript drafting and in critical revision for important intellectual content. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Adrián Mosquera Orgueira, Email: adrian.mosquera@live.com.
Beatriz Antelo Rodríguez, Email: Beatriz.Antelo.Rodriguez@sergas.es.
Natalia Alonso Vence, Email: Natalia.Alonso.Vence@sergas.es.
José Ángel Díaz Arias, Email: Jose.Angel.Diaz.Arias@sergas.es.
Nicolás Díaz Varela, Email: Nicolas.Diaz.Varela@sergas.es.
Manuel Mateo Pérez Encinas, Email: Manuel.Mateo.Perez.Encinas@sergas.es.
Catarina Allegue Toscano, Email: catarina.allegue@usc.es.
Elena María Goiricelaya Seco, Email: elena.goiricelaya@rai.usc.es.
Ángel Carracedo Álvarez, Email: Angel.Carracedo.Alvarez@sergas.es.
José Luis Bello López, Email: Jose.Luis.Bello.Lopez@sergas.es.
References
- 1.Döhner H, et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med. 2000;343(26):1910–1916. doi: 10.1056/NEJM200012283432602. [DOI] [PubMed] [Google Scholar]
- 2.Nadeu F, et al. Clinical impact of the subclonal architecture and mutational complexity in chronic lymphocytic leukemia. Leukemia. 2018;32(3):645–653. doi: 10.1038/leu.2017.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Speedy HE, Di Bernardo MC, Sava GP, et al. A genome-wide association study identifies multiple susceptibility loci for chronic lymphocytic leukemia. Nat Genet. 2014;46(1):56–60. doi: 10.1038/ng.2843. [DOI] [PubMed] [Google Scholar]
- 4.Berndt SI, Skibola CF, Joseph V, et al. Genome-wide association study identifies multiple risk loci for chronic lymphocytic leukemia. Nat Genet. 2013;45(8):868–876. doi: 10.1038/ng.2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berndt SI, Camp NJ, Skibola CF, et al. Meta-analysis of genome-wide association studies discovers multiple loci for chronic lymphocytic leukemia. Meta-analysis of genome-wide association studies discovers multiple loci for chronic lymphocytic leukemia. Nat Commun. 2016;7:10933. doi: 10.1038/ncomms10933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baecklund F, Foo JN, Bracci P, et al. A comprehensive evaluation of the role of genetic variation in follicular lymphoma survival. BMC Med Genet. 2014;15:113. doi: 10.1186/s12881-014-0113-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ghesquieres H, Slager SL, Jardin F, et al. Genome-wide association study of event-free survival in diffuse large B-cell lymphoma treated with Immunochemotherapy. J Clin Oncol. 2015;33(33):3930–3937. doi: 10.1200/JCO.2014.60.2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ramsay AJ, et al. Next-generation sequencing reveals the secrets of the chronic lymphocytic leukemia genome. Clin Transl Oncol. 2013;15(1):3–8. doi: 10.1007/s12094-012-0922-z. [DOI] [PubMed] [Google Scholar]
- 9.Li H, et al. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Puente XS, et al. Non-coding recurrent mutations in chronic lymphocytic leukaemia. Nature. 2015;526(7574):519–524. doi: 10.1038/nature14666. [DOI] [PubMed] [Google Scholar]
- 11.Li H, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Breese MR, Liu Y. NGSUtils: a software suite for analyzing and manipulating next-generation sequencing datasets. Bioinformatics. 2013;29(4):494–496. doi: 10.1093/bioinformatics/bts731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rimmer A, et al. Integrating mapping-, assembly- and haplotype-based approaches for calling variants in clinical sequencing applications. Nat Genet. 2014;46(8):912–918. doi: 10.1038/ng.3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Database of single nucleotide polymorphisms (dbSNP). Bethesda (MD): National Center for biotechnology information, National Library of medicine. (dbSNP build ID: 150). Available from: http://www.ncbi.nlm.nih.gov/SNP/
- 15.Huber W, Carey VJ, Gentleman R, et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nat Methods. 2015;12(2):115–121. doi: 10.1038/nmeth.3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zheng X, Levine D, Shen J, Gogarten S, Laurie C, Weir B. A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics. 2012;28(24):3326–3328. doi: 10.1093/bioinformatics/bts606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Therneau T. A package for survival analysis in S. version 2.38. 2015. [Google Scholar]
- 18.Therneau TM, Grambsch PM. Modeling survival data: extending the cox model. New York: Springer; 2000. [Google Scholar]
- 19.Slave Petrovski, Quanli Wang. QQperm: permutation based QQ plot and inflation factor estimation. 2016. R package ersion 1.0.1. https://cran.r-project.org/web/packages/QQperm/index.html.
- 20.Mishra A, Macgregor S. VEGAS2: software for more flexible gene-based testing. Twin Res Hum Genet. 2015;18(1):86–91. doi: 10.1017/thg.2014.79. [DOI] [PubMed] [Google Scholar]
- 21.Castaño-Vinyals G, Aragonés N, Pérez-Gómez B, et al. Population-based multicase-control study in common tumors in Spain (MCC-Spain): rationale and study design. Gac Sanit. 2015;29(4):308–315. doi: 10.1016/j.gaceta.2014.12.003. [DOI] [PubMed] [Google Scholar]
- 22.Zhang K, Cui S, Chang S, Zhang L, Wang J. i-GSEA4GWAS: a web server for identification of pathways/gene sets associated with traits by applying an improved gene set enrichment analysis to genome-wide association study. Nucleic Acids Res. 2010;38(Web Server issue):W90–W95. doi: 10.1093/nar/gkq324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ward LD, Kellis M. HaploReg v4: systematic mining of putative causal variants, cell types, regulators and target genes for human complex traits and disease. Nucleic Acids Res. 2016;44(D1):D877–D881. doi: 10.1093/nar/gkv1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Herzig JK, Bullinger L, Tasdogan A, et al. Protein phosphatase 4 regulatory subunit 2 (PPP4R2) is recurrently deleted in acute myeloid leukemia and required for efficient DNA double strand break repair. Oncotarget. 2017;8(56):95038–95053. doi: 10.18632/oncotarget21119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu J, Xu L, Zhong J, et al. Protein phosphatase PP4 is involved in NHEJ-mediated repair of DNA double-strand breaks. Cell Cycle. 2012;11(14):2643–2649. doi: 10.4161/cc.20957. [DOI] [PubMed] [Google Scholar]
- 26.Chen Ming-Yu, Chen Ya-Ping, Wu Ming-Sian, Yu Guanni-Yi, Lin Wen-Jye, Tan Tse-Hua, Su Yu-Wen. PP4 Is Essential for Germinal Center Formation and Class Switch Recombination in Mice. PLoS ONE. 2014;9(9):e107505. doi: 10.1371/journal.pone.0107505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Su Yu-wen, Chen Ya-ping, Chen Ming-yu, Reth Michael, Tan Tse-Hua. The Serine/Threonine Phosphatase PP4 Is Required for Pro-B Cell Development through Its Promotion of Immunoglobulin VDJ Recombination. PLoS ONE. 2013;8(7):e68804. doi: 10.1371/journal.pone.0068804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kanchi KL, Johnson KJ, Lu C, et al. Integrated analysis of germline and somatic variants in ovarian cancer. Nat Commun. 2014;5:3156. 10.1038/ncomms4156. [DOI] [PMC free article] [PubMed]
- 29.Yang LX, Gao Q, Shi JY, et al. Mitogen-activated protein kinase kinase kinase 4 deficiency in intrahepatic cholangiocarcinoma leads to invasive growth and epithelial-mesenchymal transition. Hepatology. 2015;62(6):1804–1816. doi: 10.1002/hep.28149. [DOI] [PubMed] [Google Scholar]
- 30.Kim J, Kang D, Sun BK, et al. TRAIL/MEKK4/p38/HSP27/Akt survival network is biphasically modulated by the Src/CIN85/c-Cbl complex. Cell Signal. 2013;25(1):372–379. doi: 10.1016/j.cellsig.2012.10.010. [DOI] [PubMed] [Google Scholar]
- 31.Sollome JJ, Thavathiru E, Camenisch TD, Vaillancourt RR. HER2/HER3 regulates extracellular acidification and cell migration through MTK1 (MEKK4) Cell Signal. 2014;26(1):70–82. doi: 10.1016/j.cellsig.2013.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Keil E, Höcker R, Schuster M, et al. Phosphorylation of Atg5 by the Gadd45β-MEKK4-p38 pathway inhibits autophagy. Cell Death Differ. 2013;20(2):321–332. doi: 10.1038/cdd.2012.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weller S, Cajigas I, Morrell J, et al. Alternative splicing suggests extended function of PEX26 in peroxisome biogenesis. Am J Hum Genet. 2005;76(6):987–1007. doi: 10.1086/430637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brants J, Semenchenko K, Wasylyk C, et al. Tubulin tyrosine ligase like 12, a TTLL family member with SET- and TTL-like domains and roles in histone and tubulin modifications and mitosis. PLoS One. 2012;7(12):e51258. doi: 10.1371/journal.pone.0051258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Perez F, Diamantopoulos GS, Stalder R, Kreis TE. CLIP-170 highlights growing microtubule ends in vivo. Cell. 1999;96(4):517–527. doi: 10.1016/S0092-8674(00)80656-X. [DOI] [PubMed] [Google Scholar]
- 36.Sahin U, Neumann F, Tureci O, et al. Hodgkin and reed-Sternberg cell-associated autoantigen CLIP-170/restin is a marker for dendritic cells and is involved in the trafficking of macropinosomes to the cytoskeleton, supporting a function-based concept of Hodgkin and reed-Sternberg cells. Blood. 2002;100(12):4139–4145. doi: 10.1182/blood.V100.12.4139. [DOI] [PubMed] [Google Scholar]
- 37.Krysko O, Aaes TL, Kagan VE, et al. Necroptotic cell death in anti-cancer therapy. Immunol Rev. 2017;280(1):207–219. doi: 10.1111/imr.12583. [DOI] [PubMed] [Google Scholar]
- 38.Lin TC, Su CY, Wu PY, et al. The nucleolar protein NIFK promotes cancer progression via CK1α/β-catenin in metastasis and Ki-67-dependent cell proliferation. Elife. 2016;17:5. doi: 10.7554/eLife.11288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Selvik LK, Rao S, Steigedal TS, et al. Salt-inducible kinase 1 (SIK1) is induced by gastrin and inhibits migration of gastric adenocarcinoma cells. PLoS One. 2014;9(11):e112485. doi: 10.1371/journal.pone.0112485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hong B, Zhang J, Yang W. Activation of the LKB1-SIK1 signaling pathway inhibits the TGF-β-mediated epithelial-mesenchymal transition and apoptosis resistance of ovarian carcinoma cells. Mol Med Rep. 2018;17(2):2837–2844. doi: 10.3892/mmr.2017.8229. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 41.Núñez-Enríquez JC, Bárcenas-López DA, Hidalgo-Miranda A, et al. Gene expression profiling of acute lymphoblastic leukemia in children with very early relapse. Arch Med Res. 2016;47(8):644–655. doi: 10.1016/j.arcmed.2016.12.005. [DOI] [PubMed] [Google Scholar]
- 42.Bertrand P, Bastard C, Maingonnat C, et al. Mapping of MYC breakpoints in 8q24 rearrangements involving non-immunoglobulin partners in B-cell lymphomas. Leukemia. 2007;21(3):515–523. doi: 10.1038/sj.leu.2404529. [DOI] [PubMed] [Google Scholar]
- 43.Beyer T, Bolz S, Junger K, et al. CRISPR/Cas9-mediated genomic editing of Cluap1/IFT38 reveals a new role in actin arrangement. Mol Cell Proteomics. 2018;17(7):1285–1294. doi: 10.1074/mcp.RA117.000487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ishikura H, Ikeda H, Abe H, et al. Identification of CLUAP1 as a human osteosarcoma tumor-associated antigen recognized by the humoral immune system. Int J Oncol. 2007;30(2):461–467. [PubMed] [Google Scholar]
- 45.Takahashi M, Lin YM, Nakamura Y. Isolation and characterization of a novel gene CLUAP1 whose expression is frequently upregulated in colon cancer. Oncogene. 2004;23(57):9289–9294. doi: 10.1038/sj.onc.1208100. [DOI] [PubMed] [Google Scholar]
- 46.Yan YB. Creatine kinase in cell cycle regulation and cancer. Amino Acids. 2016;48(8):1775–1784. doi: 10.1007/s00726-016-2217-0. [DOI] [PubMed] [Google Scholar]
- 47.Qin W, Hu J, Guo M, et al. BNIPL-2, a novel homologue of BNIP-2, interacts with Bcl-2 and Cdc42GAP in apoptosis. Biochem Biophys Res Commun. 2003;308(2):379–385. doi: 10.1016/S0006-291X(03)01387-1. [DOI] [PubMed] [Google Scholar]
- 48.Xie L, Qin W, Li J, et al. BNIPL-2 promotes the invasion and metastasis of human hepatocellular carcinoma cells. Oncol Rep. 2007;17(3):605–610. [PubMed] [Google Scholar]
- 49.Patra KC, Hay N. The pentose phosphate pathway and cancer. Trends Biochem Sci. 2014;39(8):347–354. doi: 10.1016/j.tibs.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Love C, Sun Z, Jima D, et al. The genetic landscape of mutations in Burkitt lymphoma. Nat Genet. 2012;44(12):1321–1325. doi: 10.1038/ng.2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Muppidi JR, Schmitz R, Green JA, et al. Loss of signalling via Gα13 in germinal Centre B-cell-derived lymphoma. Nature. 2014;516(7530):254–258. doi: 10.1038/nature13765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Healy JA, Nugent A, Rempel RE, et al. GNA13 loss in germinal center B cells leads to impaired apoptosis and promotes lymphoma in vivo. Blood. 2016;127(22):2723–2731. doi: 10.1182/blood-2015-07-659938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Morin RD, Mungall K, Pleasance E, et al. Mutational and structural analysis of diffuse large B-cell lymphoma using whole-genome sequencing. Blood. 2013;122(7):1256–1265. doi: 10.1182/blood-2013-02-483727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kolb JP, Roman V, Mentz F, et al. Contribution of nitric oxide to the apoptotic process in human B cell chronic lymphocytic leukaemia. Leuk Lymphoma. 2001;40(3–4):243–257. doi: 10.3109/10428190109057923. [DOI] [PubMed] [Google Scholar]
- 55.Zhao H, Dugas N, Mathiot C, et al. B-cell chronic lymphocytic leukemia cells express a functional inducible nitric oxide synthase displaying anti-apoptotic activity. Blood. 1998;92(3):1031–1043. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Principal component plots for the subjects included in the final analysis. (JPG 154 kb)
Figure S2. Manhattan plot of the additive OS model results. (JPG 199 kb)
Figure S3. Manhattan plot of the dominant OS model results. (JPG 222 kb)
Figure S4. Manhattan plot of the recessive TTT model results. (JPG 193 kb)
Figure S5. Manhattan plot for the Pentose Phosphate pathway. (JPG 56 kb)
Figure S6. Manhattan plot for the GNα13 pathway. (JPG 54 kb)
Figure S7. Manhattan plot for the “Induction of apoptosis by extracellular signal” biological process. (JPG 59 kb)
Figure S8. Manhattan plot for the “Mitosis” biological process. (JPG 54 kb)
Table S1. GWAS results of the additive Cox regression model for TTT. Polymorphisms with a BH-adjusted P-value < 0.5 are shown. Table S2. GWAS results of the dominant Cox regression model for TTT. Polymorphisms with a BH-adjusted P-value < 0.5 are shown. Table S3. GWAS results of the recessive Cox regression model for TTT. Polymorphisms with a BH-adjusted P-value < 0.5 are shown. Table S4. GWAS results of the additive Cox regression model for OS. Polymorphisms with a BH-adjusted P-value < 0.5 are shown. Table S5. GWAS results of the dominant Cox regression model for OS. Polymorphisms with a BH-adjusted P-value < 0.5 are shown. Table S6. GWAS results of the recessive Cox regression model for OS. Polymorphisms with a BH-adjusted P-value < 0.5 are shown. Table S7. VEGAS2 gene-level results for association with TTT. Table S8. VEGAS2 gene-level results for association with OS. (XLSX 3081 kb)
Data Availability Statement
The data that support the findings of this study are available from the ICGC but restrictions apply to the availability of these data, which were used under license for the current study, and so are not publicly available. Data are however available from the authors upon reasonable request and with permission of the ICGC.