

Abstract
Background/Purpose:
Wilms tumor (WT) is the most common childhood kidney cancer globally. Our prior unbiased proteomic screen of WT disparities revealed increased expression of Fragile X-Related 1 (FXR1) in Kenyan specimens where survival is dismal. FXR1 is an RNA-binding protein that associates with poor outcomes in multiple adult cancers. The aim of this study therefore was to validate and characterize the FXR1 expression domain in WT.
Methods:
Quantitative FXR1 gene expression was compared between WT, adjacent, adult, and fetal kidney specimens. The cellular and subcellular expression domain of FXR1 was characterized across these tissues using immunoperoxidase staining. RNA-sequencing of FXR1 was performed from WT and other pediatric malignancies to examine its broader target potential.
Results:
FXR1 was detected in all clinical WT specimens evaluated (n=82), and as a result appeared independent of demographic, histology, or adverse event. Specific cytosolic staining was strongest in blastema, intermediate and variable in epithelia, and weakest in stroma. When present, areas of skeletal muscle differentiation stained strongly for FXR1. qPCR revealed increased FXR1 expression in WT compared to adult and adjacent kidney (p<0.0002) but was similar to fetal kidney (p=0.648). RNA-sequencing revealed expression of FXR1 in multiple pediatric tumors, greatest in rhabdomyosarcoma and WT.
Conclusions:
FXR1 was expressed consistently across this broad sampling of WT and most robustly in the primitive blastema. Notably, FXR1 labeled a specific self-renewing progenitor population of the fetal kidney.
Keywords: Wilms tumor, Nephroblastoma, FXR1, Embryonal tumor, Pediatric cancer
1. Introduction
Wilms tumor (WT) is the most common kidney cancer of childhood, affecting approximately 1 in 10,000 children age 15 years or younger [1]. In developed countries, overall survival from WT currently exceeds 90% at 5 years. However, survival among certain subgroups, including those with specific genomic alterations (e.g., copy number gain at 1q and dual loss of heterozygosity at 1p and 16q), unfavorable histology, and relapsed disease, is much poorer [2]. Furthermore, WT poses a profound cancer health disparity to black children of sub-Saharan African ancestry. Survival from WT among black children living in sub-Saharan Africa is dismal and alarmingly less than 50% at two years [3–5]. In addition to socioeconomic inequalities and barriers to care, differences in the molecular profile of WT confer treatment resistance and also contribute to poorer outcomes from WT in sub-Saharan Africa [3, 4, 6]. Taken together, these survival disparities underscore the need to elucidate adverse WT biology with the ultimate goal to identify genetic markers that guide risk stratification and personalized therapies globally.
WT biology appears to recapitulate aberrantly that of the embryonic kidney, and specific gene expression profiles of WT and fetal kidney have been shown to overlap [7–9]. Histologically, WT demonstrates most commonly a triphasic pattern of cell types, comprised of blastema, primitive epithelia, and stroma that together resemble the developing kidney but show no functional architecture or organization. The blastema represents the least differentiated cellular component, is the malignant analogue of the nephron progenitor (i.e., metanephric mesenchyme) and the putative cancer stem cell of WT, and confers a more aggressive phenotype when predominant or persistent after neoadjuvant therapy [7, 10, 11]. The epithelial component consists of structures appearing as primitive tubules and glomeruli. The stroma is comprised of mostly undifferentiated mesenchymal cells but can also demonstrate heterologous muscle or cartilaginous differentiation [12]. The molecular pathways that fuel perpetuation of the blastema and block its terminal differentiation therefore are candidate targets for novel and future therapies.
To explore the biologic basis of the WT cancer health disparity globally, our laboratory previously identified through an unbiased proteomic screen a highly discriminating increase in expression of the Fragile X Related 1 protein (FXR1) among Kenyan relative to North American WT [4]. The FXR1 gene is an autosomal paralog of Fragile X Mental Retardation 1 (FMR1), and together with FXR2, comprise the Fragile X Gene Family. As a member of the RNA-induced silencing complex (i.e., RISC), the FXR1 protein binds RNA and therefore is implicated in translational editing and silencing [13]. FXR1 is critical to normal myogenesis and muscle development and is embryonic lethal when silenced, although its contribution to nephrogenesis has yet to be established [14]. FXR1 has been identified as a candidate driver at the 3q26-29 amplicon, which has been implicated as a critical region of genetic alteration in multiple human cancers [15, 16]. Interestingly, FXR1 expression has been associated recently with poor prognosis in several adult human cancers including breast, ovarian, head and neck, and non-small cell lung cancer, perhaps due to its regulation of two oncogenes, protein kinase C iota and epithelial cell transforming 2 [17]. Although FXR1 has not previously been analyzed in WT, allelic imbalances in the region of the FXR1 gene on 3q have been shown to occur at higher frequency in relapsed WT [18].
Applying the rationale that FXR1 emerged as a key discriminating protein between North American and Kenyan WT specimens (i.e., where survival is alarmingly low) and associates with poor prognosis in certain adult cancers, we hypothesized that FXR1 detection would align similarly with aggressive phenotypes of WT. Therefore, this seminal study was designed foremost to validate and characterize its expression domain across a broad sampling of tissues.
2. Materials and Methods
2.1. qPCR analysis of WT, adjacent kidney, and total RNA of human fetal and adult kidney
Quantitative polymerase chain reaction (qPCR) was used to assess FXR1 expression among 20 primary WT and 12 adjacent kidneys randomly selected from our laboratory repository of embryonal tumors. RNA was isolated and purified from snap-frozen WT tissues using RNA-bee (Tel-Test, Friendswood, TX). Included in this qPCR analysis were two independent human fetal kidney total RNA samples (Catalog # - 636584, Clontech, Mountain View, CA; and Catalog # - 540169, Agilent, Santa Clara, CA) and two independent human adult kidney total RNA samples (Catalog # - 636529, Clontech, Mountain View, CA; and 540013, Agilent, Santa Clara, CA). Reverse transcription was performed using Superscript II (Life Technologies, Carlsabad, CA) for synthesis of cDNA. qPCR experiments were performed on the BioRad iCycler using iQ SYBR Green Super Mix (BioRad). Primers utilized were:
FXR1 Forward: 5’-TGG GCT AGT CAC AGT TGC AG-3’
FXR1 Reverse: 5’-TTC CCT TGG GAG GTT TCT TT-3’
Changes in level of mRNA expression were assessed against a standard curve generated using human embryonic kidney cells (HEK293) cDNA.
2.2. WT tissue samples
The FXR1 expression domain across WT cell types and subcellular compartments was characterized using immunoperoxidase staining of four tissue microarrays (TMA) comprised of a broad sampling of consecutive WT specimens archived in our laboratory repository [19]. Briefly, the four TMAs include 299 total clinical and experimental punches: 43 primary WT, 5 metastases, 7 adjacent kidneys, 9 human fetal kidneys, 12 heterotopic patient-derived xenografts, heterotransplanted human WiT49 cells, and 2 rodent models of WT [19]. To increase study power, additional WT specimens not included in the TMAs were immunostained for FXR1 (n=26 institutional, n=13 Kenyan). The Vanderbilt IRB approved all aspects of this study (#100734 and #020888).
2.3. Immunohistochemical analysis
Slides were placed on the Leica Bond Max IHC Stainer. All steps besides dehydration, clearing, and cover slipping were performed on the Bond Max. Slides were de-paraffinized. Heat induced antigen retrieval was performed on the Bond Max using their Epitope Retrieval 2 solution for 20 minutes. Slides were incubated with anti-FXRl (Catalog # - HPA018246, Sigma, St. Louis, MO) at a dilution of 1:250 for 60 minutes. The Bond Polymer Refine detection system was used for visualization. Slides were then dehydrated, cleared and cover slipped. Staining was scored as absent (0), weak (1), or strong (2) by a pediatric pathologist who was blinded to all other data.
2.4. St. Jude Children’s Research Hospital Pediatric Cancer (PeCan) Data Portal
The online, interactive St. Jude Children’s Research Hospital Pediatric Cancer (PeCan) Data Portal (https://pecan.stjude.cloud/home) was utilized to visualize somatic mutations in the FMR family of genes and the relative gene expression of FXR1 and FXR2 among 4,469 pediatric cancer specimens currently uploaded to the data portal. To gain insight into the potential for FXR1 as a broader target among multiple pediatric cancers, 1,976 specimens had available RNA-sequencing data and were analyzed according to tumor type.
2.5. Statistical analysis
qPCR normalized expression was summarized and plotted using the median and interquartile range (IQR). Immunohistochemical staining scores from multiple sections derived from a single tumor were averaged, and then overall scores for each cellular compartment (blastema, epithelia, stroma, and skeletal muscle differentiation) were summarized and plotted using the median and interquartile range (IQR). The Wilcoxon rank sums or matched-pairs test was applied to two-group continuous outcomes (Kruskal-Wallis for more than two groups). All analyses were performed using Stata 15 (StataCorp, LLC, College Station, TX). Statistical significance was set atp < 0.05.
3. Results
3.1. qPCR for quantification of FXR1
Quantitative PCR was employed as an initial means to document differences in FXR1 gene expression between snap frozen WT (n=20), adjacent kidney (i.e., from which a tumor arose, n=12), and normal adult (n=2 independent samples of total RNA) and fetal kidneys (n=2 independent samples of total RNA). When comparing FXR1 expression in WT with these various renal tissues, expression was significantly greater in WT and fetal kidney than in adjacent and normal adult kidney (p<0.0002, Figure 1A). Among paired samples (i.e., WT and adjacent kidney from the same patient; n=7), FXR1 also was expressed in significantly higher quantities in WT compared to adjacent kidney (p=0.018, Figure 1B).
Figure 1. FXR1 Expression in North American Wilms Tumor.
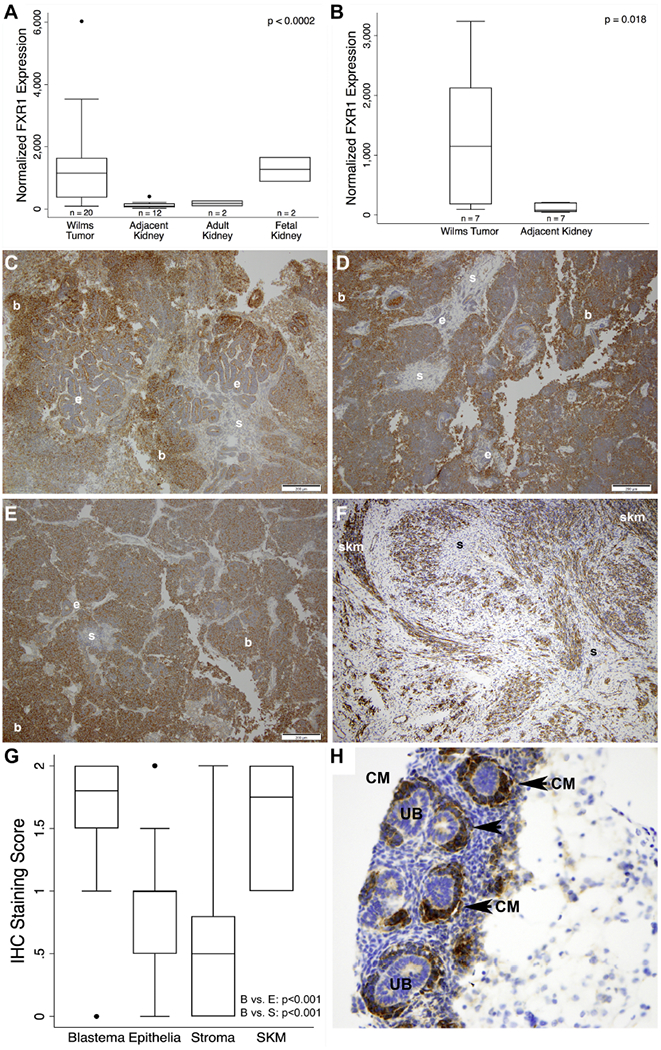
(A) Quantitative PCR of FXR1 in primary WT (n=20), adjacent kidney (n=12), adult kidney total RNA (n=2), and fetal kidney total RNA (n=2). FXR1 in WT was significantly increased (~10-fold) in WT relative to adjacent and adult kidney but was similar to fetal kidney (p<0.0002). (B) When controlling for matched WT and adjacent kidney specimens (n=7), a similar increase in FXR1 expression was observed (p=0.018). (C-F) Representative expression pattern of FXR1 in 4 different WT specimens (10× magnification; bars = 200 um). Expression showed a gradient of intensity from consistently intense in blastema, intermediate and variable in epithelia, and weak to absent in stroma. Heterologous skeletal muscle showed intense FXR1 staining (F). (G) Scoring (absent = 0, weak = 1, or strong = 2) with a pediatric pathologist confirmed these observations (n=64 patients, p<0.001). Boxes represent interquartile range (IQR) with whiskers spanning to 1.5*IQR. Dots represent values outside of 1.5*IQR. (H) Human fetal kidney showed highly specific detection of FXR1 in the cap mesenchyme (arrowhead and CM, cap mesenchyme; UB, ureteric bud; b, blastema; e, epithelia; s, stroma; skm, skeletal muscle).
3.2. Immunohistochemistry for localization of FXR1
Peroxidase-based immunohistochemical (IHC) staining of the FXR1 protein was performed across a broad spectrum of WT and related tissue samples to define the cell type, subcellular compartment, and clinical phenotype of its expression domain (Figure 1C-F). Among 64 unique North American primary WT specimens, 44 included a blastemal component, 39 an epithelial component, 46 a stromal component, and 9 showed heterologous skeletal muscle differentiation. Descriptively, staining of the epithelial component was intermediate and variable, but was mostly absent in more mature, tubular structures and was weakly positive in immature epithelial structures. Absent to weak FXR1 staining was observed in the stroma except in cases demonstrating skeletal muscle differentiation, where staining was strong (Figure 1F). After strict scoring, differences in intensity of FXR1 detection across these three classic WT cell types were highly significant (p<0.001 for blastema vs. epithelia and blastema vs. stroma; Figure 1G). Subcellular expression appeared restricted to the cytosol in all three cellular elements of WT. Interestingly, human fetal kidney showed specific and intense staining of FXR1 restricted to the cap mesenchyme (i.e., the nephron progenitor and putative embryonic ancestor of malignant blastema; Figure 1H).
Metastatic deposits in lymph nodes, lung and liver (n=5 patients, 13 punches) showed a similar staining pattern of FXR1 to that of primary tumors, with consistently strong detection again in blastema and weak to absent staining in the epithelia and stroma (Figure 2A-C, 2E-F). These observations were uniform between North American and Kenyan WT (n=13). FXR1 expression using IHC appeared similar between favorable and unfavorable histologies (Figure 2D).
Figure 2. Adverse behavior and high-risk WT.
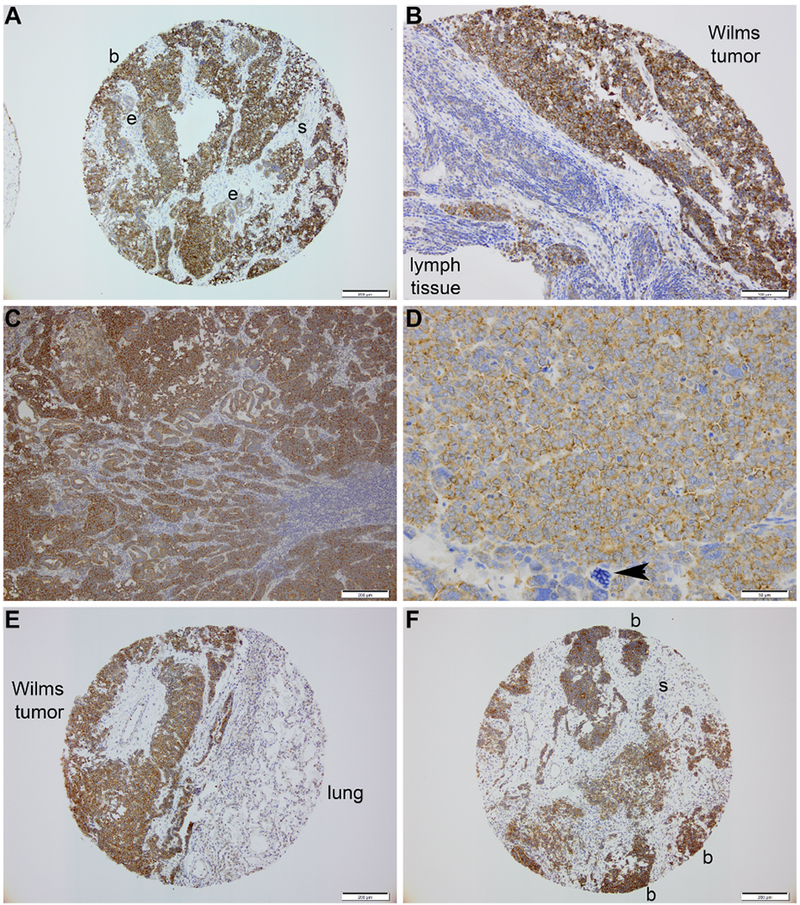
(A-B) Blastema within a primary WT (A) and lymph node metastasis (B) from same patient stain intensely for FXR1 and in an identical pattern. (C) Nodal metastasis of a Kenyan WT shows intense blastemal staining for FXR1. (D) Unfavorable histology Kenyan WT – arrowhead denotes enlarged, hyperchromatic and multipolar mitotic figure consistent with anaplasia. (E) Interface of a pulmonary metastasis and normal lung parenchyma; note intensity of FXR1 detection in the blastemal elements to the left and its absence in the lung parenchyma. (F) FXR1 detection in a hepatic metastasis of WT shows identical immunostaining pattern of FXR1 as primary specimens. (b, blastema; e, epithelia; s, stroma)
To explore experimental WT models that would be useful for future mechanistic studies, we characterized FXR1 expression in numerous heterotopic patient-derived xenografts (n=12), heterotransplanted human WiT49 cells (n=4 separate subcutaneous tumors), and 2 rodent models of WT. These experimental samples demonstrated similar patterns of FXR1 expression using peroxidase-based IHC (Figure 3A-F) [8, 20–22]. Notably, FXR1 expression was retained through multiple propagations of WT (Figure 3A-D), both favorable and unfavorable histologies, in immunodeficient mice, in a de novo model of murine nephroblastoma, and after heterotransplantation of the human WT cell line, WiT49 (Figure 3E, F).
Figure 3. FXR1 expression in experimental models of WT.
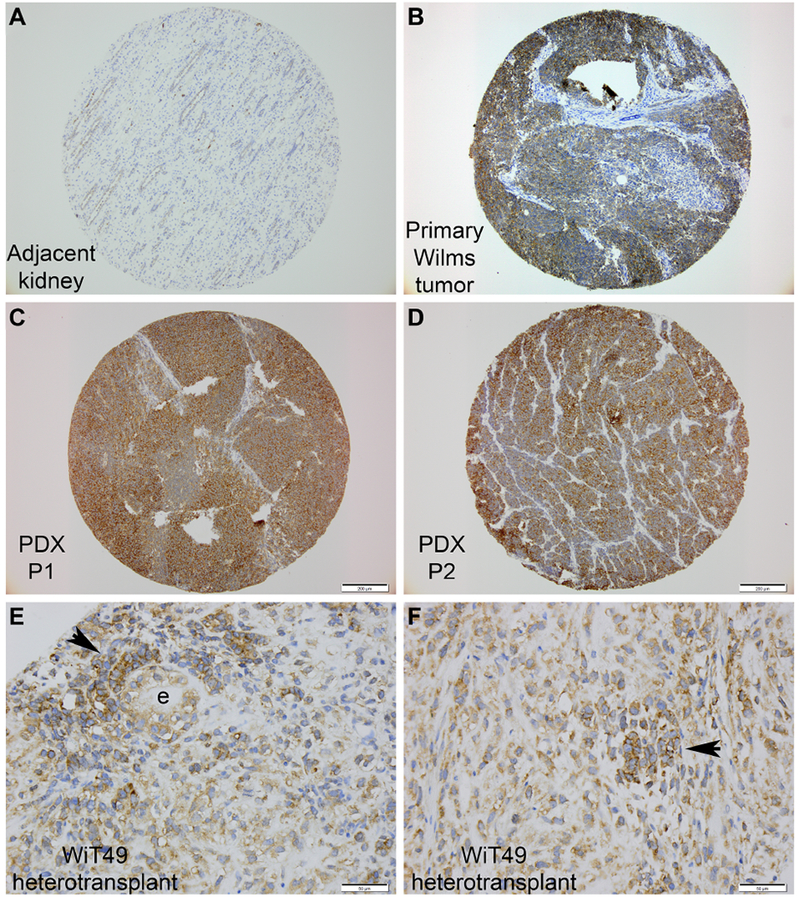
(A-D) Specimens originating from the same patient and then after propagation into immunodeficient mice. (A) Adjacent kidney, (B) primary WT, (C) after one propagation in mice, and (D) after second propagation in mice. (PDX, patient-derived xenograft; P1, 2 = propagation number). (E, F) Two different WiT49 tumors after injection of this cell line into immunodeficient mice. (Arrowhead denotes blastemal-like clusters; e, epithelia.)
3.3. FXR1 expression and patient outcomes
The specificity and consistency of FXR1 staining patterns across this diverse sampling of WT specimens precluded association with any evaluated demographic, histology or outcome in this study. Similarly, among the subset of patients included in the qPCR analysis, normalized expression of FXR1 did not associate with vital status (100% survival at 5 years) or relapse (p=0.91).
3.4. FXR1/2 mutations and gene expression in WT
We queried the St. Jude Children’s Research Hospital Pediatric Cancer (PeCan) Data Portal (https://pecan.stjude.cloud/home) for somatic mutations in the FMR family of genes and the relative expression of FXR1 and FXR2 among 4,469 pediatric cancer specimens currently uploaded to the data portal [23]. For WT, PeCan portal data are included from 3 genomic studies, totaling 201 WT specimens [24–26]. No somatic mutations were detected in FXR1 for any pediatric cancer type examined. For FXR2, 6 mutations in 4 cancer subtypes (neuroblastoma, osteosarcoma, WT, and B-cell acute lymphoblastic leukemia [ALL]) were visualized (Figure 4A). Interestingly, 4 of 6 unfavorable histology WT specimens contained a TP53-FXR2 fusion transcript (TP53 chr17:7576854 reverse → FXR2 chr17:7499314 reverse). The functional significance of this transcript is unknown. No additional mutations in the FMR family of genes were detected in WT. A total of 1,976 pediatric solid tumor specimens, including 196 WT, had available RNA-sequencing data. FXR1 gene expression in the context of WT and several other pediatric tumors is depicted in Figure 4B. Interestingly, while FXR1 gene expression was significantly higher in WT relative to osteosarcoma, neuroblastoma and adrenocortical tumors, rhabdomyosarcoma revealed the highest overall expression, which fits with its necessary function in myogenesis and its detection in skeletal muscle elements of WT.
Figure 4. FMR Family Mutations and gene expression in WT.
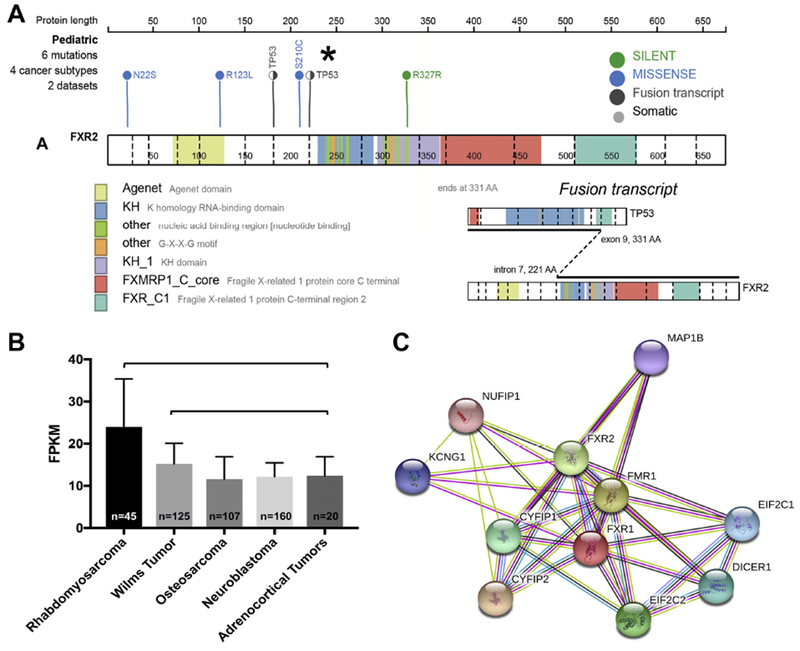
No deleterious mutations were found in FXR1 for any pediatric cancer subtype. (A) 6 mutations in 4 pediatric cancer subtypes were found in FXR2: (N22S missense in B-cell ALL, R123L missense in neuroblastoma, TP53-FXR2 fusion transcript in WT, S210C missense in neuroblastoma, and R327R silent mutation in neuroblastoma). A TP53-FXR2 fusion transcript was detected in 4 of 6 unfavorable histology WT specimens from 201 analyzed. (B) FXR1 expression levels in 5 pediatric cancer types. The vertical axis represents the range of FPKM (fragments per kilobase of transcript per million mapped reads) values from RNA-sequencing analysis. Higher FPKM values indicate greater gene expression of FXR1. Rhabdomyosarcoma showed the greatest FXR1 expression relative to the other pediatric tumor types tested (p<0.0001). WT also showed significantly greater expression of FXR1 relative to osteosarcoma and neuroblastoma (p<0.0001) and to adrenocortical tumors (p=0.0147). (D) STRING analysis shows interactions of FXR1 protein with other members of the RISC, including DICER1. STRING analysis summarizes predicted interactions for a protein of interest. In the case of FXR1 and Dicer, the existing evidence for a meaningful, specific association includes co-expression evidence (black line), experimental evidence of interaction between putative homologs in other species (lavender line), and textmining evidence (co-mentioned in PubMed abstracts, yellow line; http://version-10-5.string-db.org/cgi/network.pl?networkId=CzbW5arHZE0x).
4. Discussion
Although recently identified as a marker of poor prognosis in several adult cancers, the FXR1 protein, which emerged in our prior unbiased proteomic analysis of Kenyan and North American WT as a candidate discriminating molecule, has not been previously characterized in WT. The purpose of the current study then was to describe broadly the FXR1 expression domain in WT. Foremost from these current studies, FXR1 appears to be a ubiquitous marker of predominantly undifferentiated and self-renewing cell types in both WT and embryonic kidney. Specifically, qPCR demonstrated greater FXR1 expression in WT and human fetal kidney when compared to adjacent and normal adult human kidney. Immunohistochemical analysis demonstrated consistent expression of FXR1 across a broad sampling of WT, with strongest detection in blastema, intermediate and variable detection in the epithelia, and absent to weak detection in the stroma. Interestingly, areas of heterologous skeletal muscle differentiation within WT stained intensely for FXR1, in keeping with the protein’s previously described role in myogenesis [14]. FXR1 was also detected using IHC in the cap mesenchyme (i.e., the nephron progenitor pool and presumed embryonic ancestor of WT blastema) of normally developing human fetal kidney at 18-weeks gestation. Unlike adult cancers, however, no correlation between FXR1 expression and oncologic outcome was observed in this cohort due to its broad yet highly specific detection across all WT types analyzed herein.
Having established that WT and human fetal kidney express FXR1 at ~ 10-fold higher levels than adjacent and normal adult kidney and having localized the strongest expression of FXR1 to the blastemal component (i.e., the putative cancer stem cell) and areas of skeletal muscle differentiation within WT, the question arises whether FXR1 drives tumor initiation and/or perpetuates disease progression. The FXR1 protein has been previously identified as a marker of pluripotent human embryonic stem cells, which supports our observation that its expression was significantly more intense among less differentiated cellular compartments of WT [27, 28]. Persistent activation of FXR1 beyond embryonic development and in the malignant context may represent an unchecked cell survival pathway.
FXR1 functions as an RNA-binding protein within the RNA-induced silencing complex (RISC) through which microRNA (miRNA) and small interfering RNA (siRNA) guide degradation of complementary mRNA [29]. RNA-binding proteins regulate gene expression through mRNA silencing in embryonic development and more recently have been implicated in cancer biology, particularly disease progression. [30–32]. The specific role of FXR1 to bind and edit mRNA in the malignant context, including WT, is largely unknown, but initial experiments point to interactions with the Dicer protein as a key mediator (Figure 4C). Dicer facilitates mRNA interference through its role in maturation of small non-coding RNAs (e.g., miRNA) and loading of the derived small RNAs onto RISC [33]. Somatic DICER1 mutations have been identified in a variety of human cancers, including WT, and impair expression of key tumor suppressor miRNAs, specifically the let-7 family [26]. This latter family of miRNAs has been shown to regulate activity of several WT oncogenes promoting either adverse behavior (e.g., MYCN) or blastemal maintenance (e.g., LIN28) [26]. Interestingly, DICER1 mRNA has been identified as an FXR1 target in human embryonic kidney (HEK293) cells [34]. Taken together, FXR1 may exert its deleterious effects in initiation and progression of human cancers, and perhaps WT, through interaction with DICER1 (Figure 4C).
Another area of emerging interest is the relationship between FXR1 and the p53 tumor suppressor. Homozygous deletion of TP53 on 17p occurs in a variety of cancers, and importantly anaplastic WT, and has been associated with co-deletion of nearby FXR2 [35–37]. In the presence of these alterations, FXR1 is upregulated and considered potentially as a compensatory mechanism to promote perpetual cellular proliferation. Indeed, FXR1 knockdown inhibits cell growth in FXR2/TP53 co-deletion cell lines, raising the question of whether inhibition of FXR1 might serve as a therapeutic target for tumors that harbor TP53 homozygous deletions [37]. While diffuse anaplasia is rare in WT (~ 5% of cases), characterization of FXR1 function in the context of FXR2/TP53 co-deletion is warranted, and our analysis indeed detected this dual mutant in 4 of 6 unfavorable histology specimens.
Certain limitations temper interpretation of the current study results. First, our laboratory contains a limited number of snap-frozen adjacent kidney specimens to quantify and compare more robustly FXR1 expression with WT. With only seven matched pairs, it was difficult to draw definitive conclusions regarding the generalizability of our observation that FXR1 is more abundantly expressed in WT as compared to adjacent kidney. Nonetheless, the striking differences in FXR1 expression using unpaired WT and adjacent kidney specimens (~10-fold), as well as total RNA from normal adult kidneys, strongly suggests a true correlation. Similarly, using IHC, we detected far greater FXR1 expression in WT than adjacent kidneys, albeit more qualitative.
While these data provide compelling evidence for increased expression of FXR1 in WT that is localized predominantly to the blastema, the mechanism for its overexpression and functional significance in any oncogenic state remains unproven. Interestingly, given the essentially ubiquitous yet highly specific detection across both favorable and unfavorable histologies and among those experiencing or not experiencing an adverse event, FXR1 expression levels did not correlate with any specific outcome variable tested. This study was not powered directly to detect an impact of FXR1 expression on outcome and was conducted principally to validate FXR1 retention in WT and to characterize its expression domain broadly. To evaluate FXR1 as a prognostic feature of adverse WT behavior, future considerations will require a tightly controlled expression analysis of relapsed versus not relapsed specimens, anaplastic versus favorable histology, and higher risk versus very low risk tumors. Sufficient study power will necessitate collaboration through the COG renal tumor biospecimen archive, which our otherwise robust repository cannot satisfy. However, we do feel confident from the breadth and depth of this analysis that FXR1 labels undifferentiated and self-renewing small round blue cells which confer an embryonic phenotype to WT.
From these current studies as well, FXR1 was detected robustly in all Kenyan and North American WT specimens analyzed. While its expression appeared almost 2-fold greater among Kenyan WT in our prior unbiased proteomic analysis, the current, less sensitive studies were not designed to address any additional quantitative comparison with North American samples. We are confident that the previous quantitative difference at the protein level was indeed real but may have resulted from analyzing blastema that persisted after neoadjuvant therapy in the Kenyan population compared to chemotherapy-naïve specimens in the North American specimens. Properly controlling for these variables also will be necessary to identify any impact on outcomes.
In summary, FXR1 expression appears to be a marker of undifferentiated blastema and could represent a potential mechanism for maintenance of this cancer stem cell population and other poorly differentiated elements of WT. Interestingly, PeCan data demonstrated the highest level of FXR1 expression in rhabdomyosarcoma (reminiscent of its critical role in skeletal muscle development) and second highest in WT, suggesting that this embryonic gene may play a role in multiple embryonal tumors of mesodermal origin. If FXR1 indeed represents a targetable molecular pathway of stemness and perpetuity, it is reasonable to speculate that its inhibition could induce terminal differentiation as a novel therapeutic approach to WT. The interaction of FXR1 with DICER1 and analysis of FXR1 expression in the context of co-deleted FXR2/TP53 are the subject of future functional and mechanistic studies.
5. Conclusions
We demonstrated here that FXR1 expression is elevated significantly in WT when compared to adjacent and normal adult kidney. Moreover, normalized FXR1 expression in WT closely resembles that of human fetal kidney versus adjacent or normal adult kidney in which FXR1 is expressed less abundantly. Immunostaining for FXR1 principally labels blastema in WT and the nephron progenitor population in human fetal kidney, in keeping with a previously described role of FXR1 in stem cell maintenance. This initial evidence for FXR1 overexpression in WT in conjunction with emerging evidence of its association with poor outcomes from adult cancers, suggests that its dysregulation may also contribute to WT pathogenesis. Indeed, further investigation is warranted to establish a mechanism by which overexpression of FXR1 may drive WT maintenance.
DR. MARTIN: This paper is now open for discussion. Dr. Phelps, I have one question, were any of your Wilms tumor specimens anaplastic histology?
DR. PHELPS: Yes, there was at least one anaplastic, well, there were at least two anaplastic histology Wilms tumor within the analysis, yes.
DR. MARTIN: And your hypothesis was that this might be associated to a worse outcome, but your data didn’t really support that, is that correct?
DR. PHELPS: Right, yes.
DR. MARTIN: Any thoughts on that?
DR. PHELPS: One of our potential mechanisms, just as we’ve looked in the literature for correlations with FXR1, is that FXR2 is actually associated with the P53 mutation just because of their proximity on the chromosome 17. So those are often co-deleted. And it’s been shown that FXR1 expression is increased when FXR2 and P53 are co-deleted. So it's thought that FXR1–actually, potentially the driver could be this P53 mutation rather than abnormality in FXR1 itself.
[Applause]
MALE VOICE 1: So sort of a related question, do you have a sense of whether this is really an impactful driver mutation or abnormality, or is it just sort of a passenger consequence of arrested development?
DR. PHELPS: Yes, thank you for that question. We did not identify any actual sequence mutations in FXR1 itself when we looked at the St. Jude data, so there doesn’t appear to be a driver mutation.
MALE VOICE 1: A driver effect I mean. Is this contributing to Wilms tumor pathogenesis?
DR. PHELPS: I think that it could be, yes.
Acknowledgements:
The authors would like to acknowledge Sherry Smith, Kelly Boyd, Cindy Lowe, and the Vanderbilt Translational Pathology Shared Resource for technical assistance in performing the immunohistochemistry experiments.
Funding: This work was supported in part by the National Center for Advancing Translational Sciences [CTSA grant number UL1 TR002243 (HNL)] and by the National Cancer Institute [grant CA 161252 (PPM)]. Its contents are solely the responsibility of the authors and do not necessarily represent official views of the National Center for Advancing Translational Sciences or the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Conflicts: None
References
- [1].Breslow N, Olshan A, Beckwith JB, Green DM. Epidemiology of Wilms tumor. Med Pediatr Oncol 1993;21(3):172–81. [DOI] [PubMed] [Google Scholar]
- [2].Dome JS, Graf N, Geller JI, Fernandez CV, Mullen EA, Spreafico F, et al. Advances in Wilms Tumor Treatment and Biology: Progress Through International Collaboration. J Clin Oncol 2015;33(27):2999–3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Lovvorn HN 3rd, Pierce J, Libes J, Li B, Wei Q, Correa H, et al. Genetic and chromosomal alterations in Kenyan Wilms Tumor. Genes Chromosomes Cancer 2015;54(11):702–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Libes JM, Seeley EH, Li M, Axt JR, Pierce J, Correa H, et al. Race disparities in peptide profiles of North American and Kenyan Wilms tumor specimens. J Am Coll Surg 2014;218(4):707–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Axt J, Abdallah F, Axt M, Githanga J, Hansen E, Lessan J, et al. Wilms tumor survival in Kenya. J Pediatr Surg 2013;48(6):1254–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Murphy AJ, Axt JR, de Caestecker C, Pierce J, Correa H, Seeley EH, et al. Molecular characterization of Wilms’ tumor from a resource-constrained region of sub-Saharan Africa. Int J Cancer 2012;131(6):E983–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Li CM, Guo M, Borczuk A, Powell CA, Wei M, Thaker HM, et al. Gene expression in Wilms’ tumor mimics the earliest committed stage in the metanephric mesenchymal-epithelial transition. Am J Pathol 2002;160(6):2181–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Lovvorn HN, Westrup J, Opperman S, Boyle S, Shi G, Anderson J, et al. CITED1 expression in Wilms’ tumor and embryonic kidney. Neoplasia 2007;9(7):589–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Murphy AJ, Pierce J, de Caestecker C, Taylor C, Anderson JR, Perantoni AO, et al. SIX2 and CITED1, markers of nephronic progenitor self-renewal, remain active in primitive elements of Wilms’ tumor. J Pediatr Surg 2012;47(6):1239–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Vujanic GM, Sandstedt B. The pathology of Wilms’ tumour (nephroblastoma): the International Society of Paediatric Oncology approach. J Clin Pathol 2010;63(2):102–9. [DOI] [PubMed] [Google Scholar]
- [11].Shukrun R, Pode-Shakked N, Pleniceanu O, Omer D, Vax E, Peer E, et al. Wilms’ tumor blastemal stem cells dedifferentiate to propagate the tumor bulk. Stem Cell Reports 2014;3(1):24–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Al-Hussain T, Ali A, Akhtar M. Wilms tumor: an update. Adv Anat Pathol 2014;21(3):166–73. [DOI] [PubMed] [Google Scholar]
- [13].Siomi MC, Siomi H, Sauer WH, Srinivasan S, Nussbaum RL, Dreyfuss G. FXR1, an autosomal homolog of the fragile X mental retardation gene. EMBO J 1995;14(11):2401–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Mientjes EJ, Willemsen R, Kirkpatrick LL, Nieuwenhuizen IM, Hoogeveen-Westerveld M, Verweij M, et al. Fxr1 knockout mice show a striated muscle phenotype: implications for Fxr1p function in vivo. Hum Mol Genet 2004;13(13):1291–302. [DOI] [PubMed] [Google Scholar]
- [15].Wang J, Qian J, Hoeksema MD, Zou Y, Espinosa AV, Rahman SM, et al. Integrative genomics analysis identifies candidate drivers at 3q26-29 amplicon in squamous cell carcinoma of the lung. Clin Cancer Res 2013;19(20):5580–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Fields AP, Justilien V, Murray NR. The chromosome 3q26 OncCassette: A multigenic driver of human cancer. Adv Biol Regul 2016;60:47–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Qian J, Hassanein M, Hoeksema MD, Harris BK, Zou Y, Chen H, et al. The RNA binding protein FXR1 is a new driver in the 3q26-29 amplicon and predicts poor prognosis in human cancers. Proc Natl Acad Sci U S A 2015;112(11):3469–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Perotti D, Spreafico F, Torri F, Gamba B, D’Adamo P, Pizzamiglio S, et al. Genomic profiling by whole-genome single nucleotide polymorphism arrays in Wilms tumor and association with relapse. Genes Chromosomes Cancer 2012;51(7):644–53. [DOI] [PubMed] [Google Scholar]
- [19].Pierce J, Murphy AJ, Panzer A, de Caestecker C, Ayers GD, Neblett D, et al. SIX2 Effects on Wilms Tumor Biology. Transl Oncol 2014;7(6):800–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Clark PE, Polosukhina D, Love H, Correa H, Coffin C, Perlman EJ, et al. beta-Catenin and K-RAS synergize to form primitive renal epithelial tumors with features of epithelial Wilms’ tumors. Am J Pathol 2011;179(6):3045–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Yi Y, Polosukhina D, Love HD, Hembd A, Pickup M, Moses HL, et al. A Murine Model of K-RAS and beta-Catenin Induced Renal Tumors Expresses High Levels of E2F1 and Resembles Human Wilms Tumor. J Urol 2015;194(6):1762–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Murphy AJ, Pierce J, de Caestecker C, Ayers GD, Zhao A, Krebs JR, et al. CITED1 confers stemness to Wilms tumor and enhances tumorigenic responses when enriched in the nucleus. Oncotarget 2014;5(2):386–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Zhou X, Edmonson MN, Wilkinson MR, Patel A, Wu G, Liu Y, et al. Exploring genomic alteration in pediatric cancer using ProteinPaint. Nat Genet 2016;48(1):4–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Gadd S, Huff V, Walz AL, Ooms A, Armstrong AE, Gerhard DS, et al. A Children’s Oncology Group and TARGET initiative exploring the genetic landscape of Wilms tumor. Nat Genet 2017;49(10):1487–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Wegert J, Ishaque N, Vardapour R, Georg C, Gu Z, Bieg M, et al. Mutations in the SIX1/2 pathway and the DROSHA/DGCR8 miRNA microprocessor complex underlie high-risk blastemal type Wilms tumors. Cancer Cell 2015;27(2):298–311. [DOI] [PubMed] [Google Scholar]
- [26].Rakheja D, Chen KS, Liu Y, Shukla AA, Schmid V, Chang TC, et al. Somatic mutations in DROSHA and DICER1 impair microRNA biogenesis through distinct mechanisms in Wilms tumours. Nat Commun 2014;2:4802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Skottman H, Mikkola M, Lundin K, Olsson C, Stromberg AM, Tuuri T, et al. Gene expression signatures of seven individual human embryonic stem cell lines. Stem Cells 2005;23(9):1343–56. [DOI] [PubMed] [Google Scholar]
- [28].Li Y, Zhao X. Concise review: Fragile X proteins in stem cell maintenance and differentiation. Stem Cells 2014;32(7):1724–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Caudy AA, Myers M, Hannon GJ, Hammond SM. Fragile X-related protein and VIG associate with the RNA interference machinery. Genes Dev 2002;16(19):2491–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Stefani G, Slack FJ. Small non-coding RNAs in animal development. Nat Rev Mol Cell Biol 2008;9(3):219–30. [DOI] [PubMed] [Google Scholar]
- [31].Hayes J, Peruzzi PP, Lawler S. MicroRNAs in cancer: biomarkers, functions and therapy. Trends Mol Med 2014;20(8):460–9. [DOI] [PubMed] [Google Scholar]
- [32].Yao S MicroRNA biogenesis and their functions in regulating stem cell potency and differentiation. Biol Proced Online 2016;18:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Foulkes WD, Priest JR, Duchaine TF. DICER1: mutations, microRNAs and mechanisms. Nat Rev Cancer 2014;14(10):662–72. [DOI] [PubMed] [Google Scholar]
- [34].Ascano M, Mukherjee N, Bandaru P, Miller JB, Nusbaum J, Corcoran DL, et al. FMR1 targets distinct mRNA sequence elements to regulate protein expression. Nature 2012;492(7429):382–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Ooms AH, Gadd S, Gerhard DS, Smith MA, Guidry Auvil JM, Meerzaman D, et al. Significance of TP53 Mutation in Wilms Tumors with Diffuse Anaplasia: A Report from the Children’s Oncology Group. Clin Cancer Res 2016;22(22):5582–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Wegert J, Vokuhl C, Ziegler B, Ernestus K, Leuschner I, Furtwangler R, et al. TP53 alterations in Wilms tumour represent progression events with strong intratumour heterogeneity that are closely linked but not limited to anaplasia. J Pathol Clin Res 2017;3(4):234–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Fan Y, Yue J, Xiao M, Han-Zhang H, Wang YV, Ma C, et al. FXR1 regulates transcription and is required for growth of human cancer cells with TP53/FXR2 homozygous deletion. Elife 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]