

Abstract
Aberrant activation of epithelial to mesenchymal transition (EMT) associated factors were highly correlated with increased mortality in cancer patients. SNAIL family of transcriptional repressors comprised of three members, each of which were essentially associated with gastrulation and neural crest formation. Among which, SNAI1 and SNAI2 were efficiently induced during EMT and their expressions were correlated with poor clinical outcome in patients with breast, colon and ovarian carcinoma. In an ovarian cancer cell lines panel, we identified that SNAI1 and SNAI2 expressions were mutually exclusive, where SNAI1 predominantly represses SNAI2 expression. Detailed analysis of SNAI2 promoter region revealed that SNAI1 binds to two E-box sequences that mediated transcriptional repression. Through epigenetic inhibitor treatments, we identified that inhibition of histone deacetylase (HDAC) activity in SNAI1 overexpressing cells partially rescued SNAI2 expression. Importantly, we demonstrated a significant deacetylation of histone H3 and significant enrichments of HDAC1 and HDAC2 corepressors in both E-box regions of SNAI2 promoter. Our results suggested that SNAI1 repression on SNAI2 expression was predominantly mediated through the recruitment of the histone deacetylation machinery. Utilization of HDAC inhibitors would require additional profiling of SNAI1 activity and combined targeting of SNAI1 and HDACs might render efficient cancer treatment.
Subject terms: Gene silencing, Ovarian cancer
Introduction
Epithelial-Mesenchymal Transition (EMT) is a reversible process, where epithelial cells lose apico-basal polarity and intercellular junctions to form invasive and motile mesenchymal cells or cells with the mesenchymal phenotype, express mesenchymal markers and acquire the front-rear polarity1. EMT process is conserved throughout evolution as it contributes to embryogenesis and organ development. In addition, EMT has been implicated during carcinoma progression such as mediating therapeutic resistance2,3. Several transcription factors essential during gastrulation have been shown to play central roles in orchestrating the EMT process.
The SNAIL family is the best studied EMT transcription factor4. SNAI1 was first discovered as snail in Drosophila in 1984. Grau Y et al., found that mutants at the snail locus are zygotically embryonic lethal, though affecting dorsoventral patterning indicating that snail plays essential roles during embryogenesis5. SNAI2 was first described by Nieto et al., ten years later than the discovery of SNAI1. In this study, SNAI2 was named as slug and found to be critical in chick embryo mesoderm formation and neural crest emigration during gastrulation, evidenced by the inhibition of slug specifically impeded the normal change in cell behaviour6. Belonging to the zinc finger transcription factor family, SNAI1 and SNAI2 proteins are small in size while maintaining the ability of binding or recruiting several co-regulators. SNAI1 and SNAI2 share a conserved organization, given that both of them are composed of a group of 4 (SNAI1) to 5 (SNAI2) C2H2 type zinc fingers at C-terminal and a SNAG domain at N-terminal7. SNAI2 is characterized by a centrally located slug domain which is absent in SNAI1. The SNAG domain of SNAI1, has been shown to bind corepressor, such as HDAC1/2 for histone deacetylation8, the protein arginine methyltransferase 5 (PRMT5) for its nuclear translocation9, the coREST for the formation of SNAI1-LSD1-CoREST repressive complex10, and polycomb repressive complex 2 (PRC2) for gene repression11. In the case of SNAI2, co-repressors NCoR and CtBP1 interact with SNAG and SLUG domain of SNAI2, respectively12.
Being the prototype of the family, the regulation of SNAI1 transcription has been extensively studied and reviewed13,14. The regulation of SNAI2 transcription on the contrary is less characterized. The expression of SNAI2 is known to be regulated by several transcription factors. KLF4 and FOXA1 were reported to form reinforcing regulatory loops with SNAI2 in prostate cancer cell lines15. Another reciprocal transcriptional repression was reported between SOX3 and SNAI2. This antagonistic relationship is involved in the regulation of subdivision of the early embryo into ectodermal and mesendodermal lineages16. A short splice variant of the Per-Arnt-Sim transcription factor Singleminded-2 (SIM2), has been shown to repress SNAI2 in a dose-dependent manner in breast cancer cell lines17. ELF5, an ETS (E-twenty-six)-domain transcription factor which suppressed SNAI2 during normal mammary gland development18.
Though being highly similar in terms of the structure, SNAI1 and SNAI2 have been shown to display context-dependent functional roles. Recent data have suggested that SNAI1 and SNAI2 are differentially expressed in normal mammary glands and in mammary tumours that distinctly induces EMT program. SNAI1 occupies far more promoters than SNAI2 does, suggesting a more exclusive role of SNAI219. There is scattered information regarding how SNAI1 and SNAI2 regulate each other and how SNAI1 and SNAI2 is chosen under different contexts to execute exclusive functions.
In this study, we have reported the negative regulation of SNAI1 on SNAI2 expression in ovarian cancer via the recruitment of the histone deacetylase (HDAC) corepressor to the proximal E-box binding sites.
Results
SNAI1 expression shows negative correlation with SNAI2
By using an in-house panel of ovarian cancer cell lines, SGOCL, we firstly investigated the SNAI1 and SNAI2 mRNA expression and protein abundance. At the transcript level, there was a negative correlation (Rho = −0.3436; p = 0.0229) between SNAI1 and SNAI2 mRNA expressions across the SGOCL panel (Fig. 1a). The SGOCL panel was characterized into four phenotypes constituting the EMT spectrum: Epithelial (E), Intermediate Epithelial (IE), Intermediate Mesenchymal (IM) and Mesenchymal (M) and the delineation for each cell line was determined based on morphological examination and immunofluorescence staining of E-cadherin, Pan-cytokeratin and Vimentin20. The protein abundance of SNAI1 and SNAI2 in the SGOCL panel further showed a mutually exclusive pattern (Fig. 1b). With the exception of two mesenchymal cell lines, SNAI1 was predominantly expressed in cell lines with an epithelial-like phenotype (E and IE). In contrast, expression of SNAI2 was undetectable in epithelial-like (E), high expression in intermediate cell lines (IE and IM) and moderate expression in all cell lines with mesenchymal-like phenotype (M). To validate the presence of differential expression between SNAI1 and SNAI2 in other cancers, we subjected the lung adenocarcinoma cell line, A549 to TGFβ treatment. The results showed a gradual downregulation of epithelial marker, E-cadherin expression and simultaneous upregulation of mesenchymal genes VIM, SNAI1 and SNAI2 in all analysed time points (Supplementary Fig. 1). Notably, a trend of reciprocal expression of SNAI1 and SNAI2 was observed during the treatment.
Figure 1.
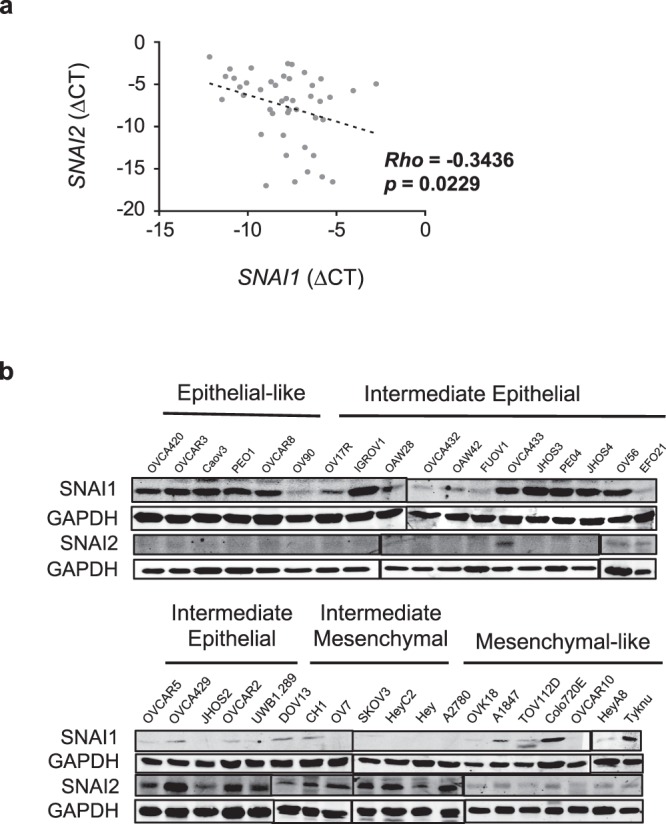
SNAI1 negatively correlates with SNAI2. (a) mRNA expression correlation between SNAI1 and SNAI2, quantified through qRT-PCR in the SGOCL collection of ovarian cancer cell lines (n = 42). (b) Immunoblots showing expressions of SNAI1 & SNAI2 in 37 ovarian cancer cell lines representing four phenotypes of the EMT spectrum. GAPDH was used as a loading control.
SNAI1 represses SNAI2 expression
To confirm whether the correlation was an actual causal relationship, ovarian cancer cell line with epithelia-like phenotype, OVCA429 was engineered to overexpress SNAI1 with a stable GFP-tagged construct or a Tet-inducible system. Constitutively SNAI1 overexpressing OVCA429 (SNAI1-OVCA429) cells showed a complete EMT phenotype (Fig. 2a) with a spindle-shaped morphology compared to the respective empty vector (EV) control cells. Immunofluorescence imaging of SNAI1-OVCA429 cells completely lost cell-cell adhesion protein E-cadherin expression and showed a profound increase in mesenchymal marker Vimentin expression, denoting the acquisition of dispersed fibroblastic-like morphology. In addition, downregulation of SNAI2 in SNAI1-OVCA429 cells was also evident at the protein level (Fig. 2b). Tet-induced SNAI1 overexpressing OVCA429 cells displayed time-dependent morphological changes following doxycycline induction, towards more mesenchymal-like phenotypes (Fig. 2c). Quantitative PCR (qPCR) was performed to validate the overexpression of SNAI1 and to investigate the cross-regulation of other EMT-TFs comparing to the 0 h controls. qPCR results confirmed that after Tet induction, OVCA429 cells consistently demonstrated SNAI2 downregulation upon SNAI1 overexpression (Fig. 2d,e). Interestingly, TWIST1 and ZEB1/2 were also upregulated upon SNAI1 induction (Fig. 2d). TWIST1 overexpression in OVCA429 cells did not consistently cause SNAI2 repression (data not shown). Taken together, SNAI1 directly represses SNAI2 expression while other EMT transcription factors TWIST1 and ZEB1/2 had no major role in this system.
Figure 2.

SNAI1 overexpression leads to downregulation of SNAI2. (a) Phase contrast images showing morphology of OVCA429 cells stably expressing control (EV) and full-length SNAI1 (SNAI1). Bottom panels showing immunofluorescence staining of E-cadherin and Vimentin in control and SNAI1 overexpressing cells. Nuclei were stained blue (DAPI), Scale = 50 μm. (b) Immunoblots showing expression of SNAI1 & SNAI2 in control and SNAI1 overexpressing cells. α-Tubulin was used as a loading control. (c) Phase contrast images of OVCA429 cells expressing control and SNAI1 cells at different time points after the addition of doxycycline (2 μg/ml), Scale = 50 μm. (d,e) Expression levels of indicated genes upon different time points after doxycycline treatment as analysed by qRT-PCR (d) and western blotting (e).
SNAI1 represses SNAI2 via recruiting HDAC to its proximal promoter region
To further elucidate how SNAI1 repressed SNAI2, promoter assays were developed. A 2-kb SNAI2 promoter sequences containing four putative SNAI1 binding E-box sequences (Fig. 3a) were cloned into pGL3 vectors and transfected into both EV- and SNAI1-OVCA429. The full-length 3′UTR sequence of SNAI2 (Supplementary Fig. 2) was cloned into a pGL3 vector as a negative control. An E-cadherin promoter containing the SNAI1 binding E-box sequences (Supplementary Fig. 3) was used as a positive control of the transcriptional repression function of SNAI1. From the promoter luciferase activity results (Fig. 3b), the SNAI2 promoter luciferase activity was significantly lower in SNAI1-OVCA429 compared to EV-OVCA429. There was no significant change in the SNAI2 3′UTR luciferase activity. Consistent with its transcription repression role for E-cadherin, the E-cadherin promoter luciferase activity was significantly lower in SNAI1-OVCA429 compared to EV-OVCA429. There were five E-box sequences identified at the SNAI2 5′ promoter site (Fig. 3a). Chromatin immunoprecipitation (ChIP)-qPCR was utilized to identify which E-box sequence would be the putative SNAI1 binding site for SNAI2 regulation. The SNAI1 binding site for E-cadherin was used as a positive control. Two of the five E-box sequences displayed enrichments of SNAI1 binding compared to the IgG controls (Fig. 3c). These two E-boxes are located at −1744 and −1354 upstream from the transcription start site (TSS) of SNAI2. Our results thus suggested that SNAI1 repressed SNAI2 transcription through direct binding at its proximal promoter.
Figure 3.

SNAI1 functionally represses SNAI2. (a) Schematic representation of the promoter region of human SNAI2, indicating putative SNAI1 binding E-box sequences. Numbers indicate positions in bps on chromosomal DNA relative to the transcription start site (+1). Nucleotide positions (in bps) cloned in to a luciferase construct for reporter assays were also indicated. (b) Luciferase activity of SNAI2 promoter, SNAI2 3′UTR & CDH1 (E-cad) promoter showing reduced activity in SNAI1-overexpressing OVCA429 cells compared to control cells. (c) ChIP-qPCR analysis of IgG (control) and SNAI1 in SNAI1-overexpressing OVCA429 cells showing enhanced enrichment of SNAI1 binding in 2′ and 3′ E-box sequences (indicated in A) of the SNAI2 promoter. SNAI1 binding E-box region of CDH1 (E-cad) promoter was used as a positive control. Signals were normalized to input DNA and plotted as enrichments relative to its respective IgG control (d) Fold change of SNAI2 expression after inhibitors of HDAC, EZH2, LSD1 and DNMT1 corepressor complexes in SNAI1-overexpressing OVCA429 cells. (e) Luciferase activity of SNAI2 and CDH1 (E-cad) promoter regions (containing E-boxes) with or without HDAC inhibitor treatments in control and SNAI1-overexpressing OVCA429 cells. (f) Luciferase activity of SNAI2 promoter with or without EZH2 and DNMT1 inhibitor treatments in control and SNAI1-overexpressing OVCA429 cells.
SNAI1 was known to repress target gene expression through recruiting corepressors including HDACs, Polycomb repressive complex 2 (PRC2), Lysine-specific demethylase 1 (LSD1) and DNA methyltransferase 1 (DNMT1)21,22. Therefore, we utilized a series of inhibitors of these known corepressors to further elucidate the mechanism of SNAI2 repression. SNAI1-OVCA429 was treated with HDAC inhibitors (TSA and SAHA), EZH2 inhibitors (GSK126 and GSK343), LSD1 inhibitors (Pargyline and TCP), and a DNMT1 inhibitor (5-AZA) to test the effects on SNAI2 expression. Only the DNMT1 inhibitor (5-AZA), one of the EZH2 inhibitors (GSK126), and both HDAC inhibitors (TSA and SAHA), showed partial rescue of SNAI2 expression (Fig. 3d). Among them, TSA showed the highest fold (up to 50%) of SNAI2 rescue expression. The LSD1 inhibitors had no effect to restore SNAI2 expression in SNAI1-OVCA429. We utilized the SNAI2 promoter activity to further confirm our results. Only HDAC inhibitor-treated SNAI1-OVCA429 showed a significant increase of SNAI2 and E-cadherin promoter activities (Fig. 3e). EZH2 and DNMT1 inhibitors showed no effect on the SNAI2 promoter activity (Fig. 3f). These results indicated that the recruitment of HDAC by SNAI1 might be directly responsible for the SNAI2 repression. The recruitment of other corepressors such as DNMT1 and EZH2 to the SNAI2 site might be secondary.
Change of histone marks and chromatin structure at the SNAI2 promoter region in SNAI1 overexpressing cells
Knowing that HDAC activity is required for SNAI2 repression by SNAI1, the histone marks and chromatin structure at the SNAI2 locus following SNAI1 overexpression were further explored. Since SNAI1 was reported to modify chromatin histone marks, enrichments of histone acetylation mark H3K27Ac and methylation marks H3K27me3, H3K4me1 and H3K4me3 at the 2nd and 3rd E-box regions of SNAI2 promoter were investigated in the EV- and SNAI1-OVCA429 cells. Abundant enrichment of H3K27Ac (open promoter) was found in the EV-OVCA429 cells (Fig. 4a) compared to the SNAI1-OVCA429 cells, denoted that chromatin at these E-box positions could be deacetylated for transcriptional repression after SNAI1 overexpression. Among the HDACs, HDAC1 and HDAC2 were known to interact with SNAI1 to repress transcription of E-cadherin8. Accordingly, we observed significant enrichments of HDAC1 and HDAC2 binding in both E-box positions of SNAI2 promoter, exclusively in SNAI1-OVCA429 cells (Fig. 4b,c). In addition, there was a significant enrichment of repressive histone mark, H3K27me3 on both E-box regions of SNAI2 promoter only in SNAI1-OVCA429 cells (Fig. 4d). In the same cells, there were significant reductions in active H3K4 mono- and tri-methylation histone marks in both E-box segments, indicated that these regions were inaccessible after SNAI1 overexpression (Fig. 4e,f). Collectively, these data suggested that induction of SNAI1 expression in cells dramatically altered chromatin histone marks along the SNAI2 promoter region, which ultimately contributed to efficient repression of SNAI2 expression.
Figure 4.

Alterations of histone marks and chromatin landscape at the SNAI2 promoter. ChIP-qPCR analysis of IgG (control) and H3K27Ac (a), HDAC1 (b), HDAC2 (c), H3K27me3 (d), H3K4me3 (e) and H3K4me1 (f) in SNAI1 binding 2′ and 3′E-box sequences of the SNAI2 promoter. SNAI1 binding E-box region of CDH1 (E-cad) promoter was used as a positive control. Signals were normalized to input DNA and plotted as enrichments relative to its respective IgG control. Statistical significance were calculated by comparing the enrichments between EV (control) and SNAI-overexpressing OVCA429 cells.
Discussion
In this study, we showed that SNAI1 suppressed SNAI2 expression through direct promoter binding and this repressive activity required recruitment of transcriptional corepressor HDAC complex. In addition, we identified that SNAI1 association on SNAI2 promoter generated a dramatic change in chromatin organization that further influenced the expression on SNAI2 in cell-type specific states.
Our protein abundance profiling of SNAI1 and SNAI2 in the SGOCL panel showed that SNAI1 was predominantly expressed in cell lines with epithelial phenotype. In contrast, none of the cell lines with epithelial phenotype showed endogenous expression of SNAI2. At first, this mutually exclusive expression might have denoted that SNAI1 efficiently repressed SNAI2 expression. However, the expression of E-cadherin (regarded as primary target of SNAI1) in the same panel was unaffected23. This discrepancy could be partly explained by previous studies that showed repression of E-cadherin by SNAI1 was dependent on the activity of Polycomb repressive complex 2 (PRC2), histone methyltransferase Suv39H1 and histone demethylase LSD110,11,24. Whether our panel of epithelial cancer lines lacked LSD1, PRC2 and Suv39H1 activity or expression remains to be investigated. Moreover, the observed negative correlation of SNAI1 and SNAI2 in the SGOCL panel was evident at the transcript level, that led us to further investigate the mechanism of SNAI2 repression in SNAI1 expressing cells.
Recent findings clearly highlighted that EMT in cancer was a dynamic process with acquisition of molecular and phenotypical changes along multiple distinct intermediate phases20,25,26. Supporting to these findings, we observed that overexpression of SNAI1 in a epithelia-like cell line (OVCA420) generated only a partial EMT like changes (intermediate epithelial phenotype) and retained cell-cell adhesion (data not shown). Intriguingly, SNAI1 overexpression in intermediate epithelial cell line (OVCA429) driven the cells to a near-complete EMT like phenotype that completely lost cell-cell adhesion, leading to drastic rearrangement of cellular morphology. This clearly demonstrated that even a strong EMT inducing transcription factor such as SNAI1 operates differentially in each cell type belonging to different EMT status. Inducible expression of mouse Snail (mSnail) in MDCK cells modulated the levels of specific claudins but the epithelial tight junction organization remained unaltered27. In contrast, E-cadherin localization at the cell-cell contact became undetectable after the expression of mSnail in mouse epithelial cell line, Eph428. Therefore, it was evident that EMT induced through SNAI1 in cells were context-dependent and possibly have driven the cells to the next intermediary state close to a mesenchymal phenotype.
Moreover, completion of EMT could be attained through cooperative functioning of different transcription factors such as SNAI1/2, TWIST and ZEB1/2. Among which, SNAI1 was reported to express at the onset of EMT and subsequently other EMT factors were induced at later time points to strengthen a mesenchymal state3,29,30. Accordingly, in our Tet-inducible system, induction of SNAI1 at regular intervals displayed a consistent upregulation of TWIST1, ZEB1/2 and simultaneous downregulation of SNAI2 expression. The observed regulation potentially driven the cells towards a mesenchymal (near complete EMT) morphology. Similar observation was reported using human mammary epithelial cells, where induction of SNAI1 led to repression of SNAI2 and simultaneous induction of ZEB1/2 transcripts at an early stage, highlighting that execution of this functional circuit preceded other molecular and phenotypical changes that led to EMT31.
Earlier studies reported that SNAI1 and SNAI2 displayed a reciprocal expression in oral, breast cancer cells or during reprogramming of induced pluripotent stem cells32–34. In addition, another study showed that during mouse chondrogenic differentiation, endogenous SNAI1 and SNAI2 bind to their own and each other’s promoter35. However, none of these studies looked into the SNAI1 occupancy in SNAI2 promoter and delineated the molecular mechanism that controlled this repressive regulation. Through luciferase reporter activity and chromatin immunoprecipitation assays we clearly showed that SNAI1 predominantly occupied two of the E-box sequences in SNAI2 promoter region for transcriptional repression. Epigenetic repression of SNAI1 on its target genes have been carried out through recruitment of HDAC1/2 and corepressor Sin3A on its SNAG domain8,36. Supporting to these studies, our SNAI2 promoter assays, HDACi treatments and chromatin immunoprecipitation results indicated the involvement of HDACs in SNAI1 mediated SNAI2 suppression. However, further evidence has to be provided to elucidate whether HDACs were recruited to the SNA12 promoter directly or not. In addition, the alterations of related acetylation markers of Histone H3 at the SNAI1 binding sites would be required.
In addition to HDACs, SNAI1 also recruited other chromatin modifying enzymes including DNMT1, PRC2, LSD1, G9a and Suv39H1 at the E-cadherin promoter for transcriptional silencing21,37. Our results showed that the dissociation of active methylation marks and enhancement of repressive methylation marks along the SNAI1 binding SNAI2 promoter region. However, only DNMT1 inhibitor treatment could partially rescue the re-expression of SNAI2, indicated that epigenetic regulation of SNAI1 at the SNAI2 promoter region is primarily executed through the histone deacetylation complexes.
The expression of HDAC family members along the different histological subtypes of ovarian cancer showed a wide heterogeneity38. Therefore, utilization of HDAC inhibitors with or without conventional chemotherapy to treat ovarian cancer needs further investigations. Recently, ovarian cancer cells carrying ARID1A mutation showed higher sensitivity to SAHA and ACY1215 treatment, through inhibition of HDAC2 and HDAC6 activity respectively39,40. On a another note, SNAI1 expression was high in majority of ovarian cancer, except mucinous and clear cell carcinoma41. Therefore, a strategic method to segregate patients harbouring SNAI1 and HDAC dependency during tumour progression is essential to efficiently use existing HDAC inhibitors as cancer therapeutics.
Methods
Generation of stable and inducible cell lines
For stable overexpression of SNAI1, plasmid encoding full length wide type SNAI1 cloned from pCMV-Entry-SNAI1 (Origene) into pLenti-GIII-CMV-GFP-2A-Puro vector (ABM). Empty vector with no inserts was used as negative control. Plasmids were mixed with Mission Lentiviral Packaging Mix (Sigma-Aldrich) before added to a mixture of transfection reagent Fugene 6 (Roche). After 15 minutes incubation at room temperature, plasmid mix was added to 293T cells and viral supernatants were harvested at 48 and 72 hours post-transfection. Cells infected with lentivirus were selected using puromycin at a proper concentration decided by their respective puromycin kill curve.
To generate Tet-inducible SNAI1 expressing cells, SNAI1 was cloned from pCMV6-Entry-SNAI1 vector using into pLVX-TRE3G vector (Clontech). pLVX-TRE3G-SNAI1 was cotransfected with pLVX-EF1Alpha-Tet3G (Clontech) to 293T cells to generate lentiviral supernatants. Cells infected with lentivirus were dually selected using puromycin and G418. Same primer pairs containing BamHI and EcoRI were used for generating both vectors and were listed in Supplementary Table S1.
Quantitative real-time PCR (qRT-PCR) and ChIP-qPCR
Total RNA from Tet-inducible SNAI1 overexpressing OVCA429 cells and corepressor inhibitor treated cells were extracted using RNeasy mini kit (SAbiosciences, Qiagen) according to manufacturer’s protocol. mRNA was reverse-transcribed to cDNA using RT2 first strand kit (SAbiosciences, Qiagen) and cDNA was mixed with SYBR green master mix (SAbiosciences, Qiagen) for qPCR analysis. Five housekeeping genes ACTB, B2M, GAPDH, HPRT1 and RPL13A were used for normalization and used as previously described23. mRNA expression level was presented as average fold change (2−∆∆Ct) with respect to control, from minimum two biological replicates. All qPCR experiments were done using ABI 7900HT (Life Technologies).
Chromatin immunoprecipitation (ChIP) were performed as described previously with following modifications23. Sheared chromatin was incubated with IgG (sc-2028, Santa Cruz), SNAI1 (sc-10432, Santa Cruz), H3K27Ac, H3K27me1, H3K3me1, H3K4me3, HDAC1 and HDAC2 antibodies. Crosslinked DNA was eluted using QIAquick PCR purification kit (Qiagen) following manufacturer’s protocol. Following DNA purification, primers spanning E-box regions of SNAI2 and E-cad promoters were used (listed in Supplementary Table S2) and were amplified by qRT-PCR. Fold enrichments were calculated relative to respective IgG controls, from at least two biological replicates.
Western blot analysis
An ovarian cell line library, referred as SGOCL, comprising of 43 different ovarian cancer cell lines were generated from different sources as described previously20. Whole cell protein cell lysates for 37 cell lines of the SGOCL were extracted as described previously23. Other cell lysates were harvested using RIPA buffer and resolved by standard reducing SDS-PAGE followed by blotting on PVDF membranes. Immunoblots were incubated with appropriate antibodies, anti-SNAI1 (C15D3, CST); anti-SNAI2 (C19G7, CST); anti-α-Tubulin (DM1A, Abcam) diluted in 2% BSA in PBS. Blots were scanned using the Odyssey Infrared Imaging System (Li-COR). Images were transferred to gray scale.
Immunofluorescence staining
Cells were fixed with 4% paraformaldehyde and blocked with 3% BSA/PBS. Fixed cells were incubated with primary antibodies: anti-E-cadherin (61018, BD Biosciences) and secondary antibody conjugated with Alexa Fluor 594 (Invitrogen). F-actin was stained by Rhodamine phalloidin (R415, Life Technologies). Stained cover slips were mounted onto glass slides using Vectashield mounting medium containing DAPI (#H-1200, Vector Laboratories).
Molecular cloning
The 2569 bp length of SNAI2 promoter segment, 658 bp length of CDH1 promoter regions and were cloned in to pGL3 Luciferase reporter vector (Promega) using primers containing KpnI and XhoI restriction sites. Primers used for cloning were listed in Supplementary Table S1.
Promoter assay
Plasmid transfections were carried out using transfection reagent Fugene 6 (Roche) following manufacturer protocol. EV-OVCA429 and SNAI1-OVCA429 cells were transfected with 100 ng of pGL3-SNAI2-promoter/pGL3-SNAI2-3′UTR/pGL3-Ecadherin-promoter together with 1.5 ng of pCMV-renilla. 24 hours post transfection, Firefly and Renilla luciferase activities were measured using Dual-Glo Luciferase assay system (Promega).
Statistical analyzes
To analyze ChIP data, multi-group comparisons were performed using two-way ANOVA and Tukey’s post hoc test if necessary. For other analyzes, two-tailed Student’s t-test was used to access statistical significance. For correlation analysis, Pearson correlation was performed. *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; n.s. not-significant.
Supplementary information
Acknowledgements
We thank Dr. Veidehi Krishnan for providing TGFβ treated cells. This work is supported by the National Research Foundation Singapore and the Singapore Ministry of Education under its Research Centres of Excellence initiative to R.Y.H. and National Medical Research Council (NMRC) Clinician Scientist-Individual Research Grant (CS-IRG) (NMRC/CIRG/1449/2016) to R.Y.H. and National University Cancer Centre of Singapore (NCIS) Research Grant, Theme: EMT in Cancer to R.Y.H.
Author Contributions
M.T., J.P.T. and R.Y.H. planned the study; V.S., M.T. and J.Y. performed the experiments and made statistical analyses. T.Z.T. performed correlation analyses. V.S., J.P.T. and R.Y.H. analysed the data and wrote the manuscript. All authors read and approved the final manuscript.
Data Availability
Datasets used in the current study are available upon request from the corresponding author.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Vignesh Sundararajan and Ming Tan contributed equally.
Supplementary information
Supplementary information accompanies this paper at 10.1038/s41598-019-44826-8.
References
- 1.Lim J, Thiery JP. Epithelial-mesenchymal transitions: insights from development. Development. 2012;139:3471–3486. doi: 10.1242/dev.071209. [DOI] [PubMed] [Google Scholar]
- 2.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J. Clin. Invest. 2009;119:1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thiery JP, Acloque H, Huang RYJ, Nieto MA. Epithelial-Mesenchymal Transitions in Development and Disease. Cell. 2009;139:871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 4.Barrallo-Gimeno A, Nieto MA. The Snail genes as inducers of cell movement and survival: implications in development and cancer. Development. 2005;132:3151–3161. doi: 10.1242/dev.01907. [DOI] [PubMed] [Google Scholar]
- 5.Grau Y, Carteret C, Simpson P. Mutations and Chromosomal Rearrangements Affecting the Expression of Snail, a Gene Involved in Embryonic Patterning in Drosophila Melanogaster. Genetics. 1984;108:347–360. doi: 10.1093/genetics/108.2.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nieto MA, Sargent MG, Wilkinson DG, Cooke J. Control of cell behavior during vertebrate development by Slug, a zinc finger gene. Science. 1994;264:835–839. doi: 10.1126/science.7513443. [DOI] [PubMed] [Google Scholar]
- 7.Manzanares M, Locascio A, Nieto MA. The increasing complexity of the Snail gene superfamily in metazoan evolution. Trends Genet. 2001;17:178–181. doi: 10.1016/S0168-9525(01)02232-6. [DOI] [PubMed] [Google Scholar]
- 8.Peinado H, Ballestar E, Esteller M, Cano A. Snail Mediates E-Cadherin Repression by the Recruitment of the Sin3A/Histone Deacetylase 1 (HDAC1)/HDAC2 Complex. Molecular and Cellular Biology. 2004;24:306–319. doi: 10.1128/MCB.24.1.306-319.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hou Z, et al. The LIM protein AJUBA recruits protein arginine methyltransferase 5 to mediate SNAIL-dependent transcriptional repression. Mol. Cell. Biol. 2008;28:3198–3207. doi: 10.1128/MCB.01435-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin Y, et al. The SNAG domain of Snail1 functions as a molecular hook for recruiting lysine‐specific demethylase 1. The EMBO Journal. 2010;29:1803–1816. doi: 10.1038/emboj.2010.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Herranz N, et al. Polycomb complex 2 is required for E-cadherin repression by the Snail1 transcription factor. Mol. Cell. Biol. 2008;28:4772–4781. doi: 10.1128/MCB.00323-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Molina-Ortiz P, et al. Characterization of the SNAG and SLUG domains of Snail2 in the repression of E-cadherin and EMT induction: modulation by serine 4 phosphorylation. PLoS One. 2012;7:e36132. doi: 10.1371/journal.pone.0036132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.De Craene B, van Roy F, Berx G. Unraveling signalling cascades for the Snail family of transcription factors. Cell. Signal. 2005;17:535–547. doi: 10.1016/j.cellsig.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 14.Kaufhold S, Bonavida B. Central role of Snail1 in the regulation of EMT and resistance in cancer: a target for therapeutic intervention. Journal of Experimental & Clinical Cancer Research. 2014;33:62. doi: 10.1186/s13046-014-0062-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu Y-N, et al. Critical and reciprocal regulation of KLF4 and SLUG in transforming growth factor β-initiated prostate cancer epithelial-mesenchymal transition. Mol. Cell. Biol. 2012;32:941–953. doi: 10.1128/MCB.06306-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Acloque H, et al. Reciprocal repression between Sox3 and snail transcription factors defines embryonic territories at gastrulation. Dev. Cell. 2011;21:546–558. doi: 10.1016/j.devcel.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Laffin B, et al. Loss of singleminded-2s in the mouse mammary gland induces an epithelial-mesenchymal transition associated with up-regulation of slug and matrix metalloprotease 2. Mol. Cell. Biol. 2008;28:1936–1946. doi: 10.1128/MCB.01701-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chakrabarti R, et al. Elf5 inhibits the epithelial-mesenchymal transition in mammary gland development and breast cancer metastasis by transcriptionally repressing Snail2. Nat. Cell Biol. 2012;14:1212–1222. doi: 10.1038/ncb2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ye X, et al. Distinct EMT programs control normal mammary stem cells and tumour-initiating cells. Nature. 2015;525:256–260. doi: 10.1038/nature14897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang RY-J, et al. An EMT spectrum defines an anoikis-resistant and spheroidogenic intermediate mesenchymal state that is sensitive to e-cadherin restoration by a src-kinase inhibitor, saracatinib (AZD0530) Cell Death & Disease. 2013;4:e915. doi: 10.1038/cddis.2013.442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin Y, Dong C, Zhou B. Epigenetic Regulation of EMT: The Snail Story. Current Pharmaceutical Design. 2014;20:1698–1705. doi: 10.2174/13816128113199990512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Serrano-Gomez SJ, Maziveyi M, Alahari SK. Regulation of epithelial-mesenchymal transition through epigenetic and post-translational modifications. Molecular Cancer. 2016;15:18. doi: 10.1186/s12943-016-0502-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chung, V. Y. et al. GRHL2-miR-200-ZEB1 maintains the epithelial status of ovarian cancer through transcriptional regulation and histone modification. Scientific Reports6 (2016). [DOI] [PMC free article] [PubMed]
- 24.Dong C, et al. Interaction with Suv39H1 is critical for Snail-mediated E-cadherin repression in breast cancer. Oncogene. 2013;32:1351–1362. doi: 10.1038/onc.2012.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pastushenko I, et al. Identification of the tumour transition states occurring during EMT. Nature. 2018;556:463–468. doi: 10.1038/s41586-018-0040-3. [DOI] [PubMed] [Google Scholar]
- 26.Yu M, et al. Circulating Breast Tumor Cells Exhibit Dynamic Changes in Epithelial and Mesenchymal Composition. Science. 2013;339:580–584. doi: 10.1126/science.1228522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carrozzino F, et al. Inducible expression of Snail selectively increases paracellular ion permeability and differentially modulates tight junction proteins. American Journal of Physiology-Cell Physiology. 2005;289:C1002–C1014. doi: 10.1152/ajpcell.00175.2005. [DOI] [PubMed] [Google Scholar]
- 28.Ikenouchi J, Matsuda M, Furuse M, Tsukita S. Regulation of tight junctions during the epithelium-mesenchyme transition: direct repression of the gene expression of claudins/occludin by Snail. Journal of Cell Science. 2003;116:1959–1967. doi: 10.1242/jcs.00389. [DOI] [PubMed] [Google Scholar]
- 29.Huang RY-J, Guilford P, Thiery JP. Early events in cell adhesion and polarity during epithelial-mesenchymal transition. Journal of Cell Science. 2012;125:4417–4422. doi: 10.1242/jcs.099697. [DOI] [PubMed] [Google Scholar]
- 30.Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nature Reviews Cancer. 2007;7:415–428. doi: 10.1038/nrc2131. [DOI] [PubMed] [Google Scholar]
- 31.Javaid S, et al. Dynamic Chromatin Modification Sustains Epithelial-Mesenchymal Transition following Inducible Expression of Snail-1. Cell Reports. 2013;5:1679–1689. doi: 10.1016/j.celrep.2013.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ganesan R, Mallets E, Gomez-Cambronero J. The transcription factors Slug (SNAI2) and Snail (SNAI1) regulate phospholipase D (PLD) promoter in opposite ways towards cancer cell invasion. Molecular. Oncology. 2016;10:663–676. doi: 10.1016/j.molonc.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gingold JA, et al. A Genome-wide RNAi Screen Identifies Opposing Functions of Snai1 and Snai2 on the Nanog Dependency in Reprogramming. Molecular Cell. 2014;56:140–152. doi: 10.1016/j.molcel.2014.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nakamura R, et al. Reciprocal expression of Slug and Snail in human oral cancer cells. Plos One. 2018;13:e0199442. doi: 10.1371/journal.pone.0199442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen Y, Gridley T. The SNAI1 and SNAI2 proteins occupy their own and each other’s promoter during chondrogenesis. Biochemical and Biophysical Research Communications. 2013;435:356–360. doi: 10.1016/j.bbrc.2013.04.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.von Burstin J, et al. E-Cadherin Regulates Metastasis of Pancreatic Cancer In Vivo and Is Suppressed by a SNAIL/HDAC1/HDAC2 Repressor Complex. Gastroenterology. 2009;137:361–371.e5. doi: 10.1053/j.gastro.2009.04.004. [DOI] [PubMed] [Google Scholar]
- 37.Christofori G. Snail1 links transcriptional control with epigenetic regulation. The EMBO Journal. 2010;29:1787–1789. doi: 10.1038/emboj.2010.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yano M, et al. Association of histone deacetylase expression with histology and prognosis of ovarian cancer. Oncol Lett. 2018;15:3524–3531. doi: 10.3892/ol.2018.7726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bitler BG, et al. ARID1A-mutated ovarian cancers depend on HDAC6 activity. Nature Cell Biology. 2017;19:962–973. doi: 10.1038/ncb3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fukumoto T, et al. Repurposing Pan-HDAC Inhibitors for ARID1A -Mutated Ovarian Cancer. Cell Reports. 2018;22:3393–3400. doi: 10.1016/j.celrep.2018.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim, M.-K., Kim, M. A., Kim, H., Kim, Y.-B. & Song, Y.-S. Expression Profiles of Epithelial-Mesenchymal Transition-Associated Proteins in Epithelial Ovarian Carcinoma. Biomed Res Int2014 (2014). [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Datasets used in the current study are available upon request from the corresponding author.