

Abstract
The cGAS-STING pathway plays an important role in pathogen-induced activation of the innate immune response. The 29-kDa amino-terminal fibronectin fragment (29-kDa FN-f) found predominantly in the synovial fluid of osteoarthritis (OA) patients increases the expression of catabolic factors via the toll-like receptor-2 (TLR-2) signaling pathway. In this study, we investigated whether 29-kDa FN-f induces inflammatory responses via the cyclic GMP-AMP synthase (cGAS)/stimulator of interferon gene (STING) pathway in human primary chondrocytes. The levels of cGAS and STING were elevated in OA cartilage compared with normal cartilage. Long-term treatment of chondrocytes with 29-kDa FN-f activated the cGAS/STING pathway together with the increased level of gamma-H2AX, a marker of DNA breaks. In addition, the expression of pro-inflammatory cytokines, including granulocyte-macrophage colony-stimulating factor (GM-CSF/CSF-2), granulocyte colony-stimulating factor (G-CSF/CSF-3), and type I interferon (IFN-α), was increased more than 100-fold in 29-kDa FN-f-treated chondrocytes. However, knockdown of cGAS and STING suppressed 29-kDa FN-f-induced expression of GM-CSF, G-CSF, and IFN-α together with the decreased activation of TANK-binding kinase 1 (TBK1), interferon regulatory factor 3 (IRF3), and inhibitor protein κBα (IκBα). Furthermore, NOD2 or TLR-2 knockdown suppressed the expression of GM-CSF, G-CSF, and IFN-α as well as decreased the activation of the cGAS/STING pathway in 29-kDa FN-f-treated chondrocytes. These data demonstrate that the cGAS/STING/TBK1/IRF3 pathway plays a critical role in 29-kDa FN-f-induced expression of pro-inflammatory cytokines.
Keywords: cGAS, Cytokine, Fibronectin fragments, Osteoarthritis, STING
INTRODUCTION
Osteoarthritis (OA) is a joint disease prevalent among the elderly and is caused by abnormal mechanical stress as well as altered biochemical factors associated with aging. OA patients exhibit joint pain, stiffness, and disability, accompanied by loss of articular cartilage and synovial inflammation (1, 2). Chondrocytes, the only cells embedded in cartilage tissues, regulate the synthesis of catabolic factors, such as pro-inflammatory cytokines and matrix-degrading enzymes, and anabolic factors leading to formation of extracellular matrix (ECM) components (3). Degradation products of ECM components, including fibronectin (FN) fragments (FN-fs), have been reported to induce matrix-degrading enzymes, including matrix metalloproteinases (MMPs) (4, 5). Previously, we demonstrated that 29-kDa FN-f-mediated cartilage catabolism signals through MyD88-dependent toll-like receptor (TLR)-2 pathway, suggesting that the modulation of TLR-2 signaling activated by damage-associated molecular patterns (DAMP), including 29-kDa FN-f, may be used as a therapeutic strategy for the regulation of cartilage damage in OA (6). In addition to increased catabolic signaling, 29-kDa FN-f was also found to decrease the expression of xylosyltransferase-1, which is a rate-limiting enzyme in matrix anabolic response, in chondrocytes via TLR-2 (7).
TLRs recognize bacterial products such as pathogen-associated molecular patterns (PAMPs) and molecules released upon tissue injury such as DAMP, and therefore, TLRs have been implicated in innate immunity, an immediate host defense system against infections, as well as in autoimmune and inflammatory diseases (8, 9). Recently, DNA damage response, the activation of the host immune system by damaged DNA resulting from genomic instability has attracted considerable interest. Cyclic guanosine monophosphate (GMP)-adenosine monophosphate (AMP) synthase (cGAS) plays a pivotal role in eliciting effective immunity against microbial pathogens (10). It also acts as a DNA sensor in cytoplasm to detect DNA nuclear and mitochondrial DNA, and pathogen infections (11). This activity, in turn, triggers innate immune responses via production of the second messenger cyclic GMP-AMP (cGAMP), which binds and activates the adaptor protein, the stimulator of interferon gene (STING) on the endoplasmic reticulum membranes (12, 13). Because TLRs are reported to induce type I interferons (IFNs) via activation of nuclear factor kappa B (NF-κB) and interferon regulatory factors (IRFs), it is plausible that TLRs mediate the upstream signaling in cGAS-STING pathway (14). Previous reports show that OA articular cartilage shows increased oxidative DNA damage compared with non-OA, and mitochondrial DNA damage accumulates in OA chondrocytes indicating the role of DNA damage response in the pathogenesis of OA (15, 16).
In the present study, we investigated whether 29-kDa FN-f induces the release of DNA to activate the cGAS-STING signaling pathway, and whether the expression of pro-inflammatory cytokines by 29-kDa FN-f may be affected by the cross-regulation between the cGAS-STING and TLR-2 signaling pathways in articular chondrocytes.
RESULTS
The cGAS/STING pathway is significantly activated by 29-kDa FN-f
We investigated whether the cGAS/STING signaling pathway was activated in OA cartilage. The levels of cGAS and STING mRNA were significantly higher in OA than in normal cartilage (Fig. 1A), indicating that increased activation of the cGAS/STING pathway may be associated with OA pathogenesis. Next, to elucidate the role of cGAS/STING pathway in 29-kDa FN-f-induced catabolic response, the levels of cGAS and STING expression were measured in 29-kDa FN-f-treated chondrocytes using western blot analysis. Following treatment with 29-kDa FN-f for 24 h, the expression of cGAS and STING, and an inducer of senescence, etoposide, was strongly elevated, and a pro-inflammatory cytokine IL-1β also increased it (Fig. 1B). On the other hand, a strong band of γH2AX, an indicator of cytoplasmic chromatin fragments, was detected only after incubation with etoposide (Fig. 1B). On the other hand, prolonged treatment with 29-kDa FN-f for 5 days increased the expression of γH2AX as well as cGAS and STING (Fig. 1C). The expression of γH2AX was attenuated on day 7. Fluorescence microscopy revealed small cytoplasmic chromatin fragments stained with DAPI and γH2AX in 29-kDa FN-f-treated cells, together with nuclei stained with only 4′,6′-diamidino-2-phenylindole (DAPI) (Fig. 1D). These data demonstrated that 29-kDa FN-f induced DNA damage and induced cytoplasmic DNA fragmentation leading to the activation of cGAS-STING pathway.
Fig. 1.

The cGAS/STING signaling pathway is activated by 29-kDa FN-f in primary chondrocytes. (A) The expression of cGAS and STING was increased in osteoarthritis (OA) compared to normal cartilage. The relative expression of cGAS and STING in human normal and OA cartilage was measured using SYBR Green-based real-time polymerase chain reaction (qPCR). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as an endogenous control. ***P < 0.001 and ****P < 0.0001 vs. normal cartilage. Data are presented as the mean ± standard deviation (SD) based on duplicate experiments using cartilages derived from different donors (normal cartilage, n = 6 and OA cartilage, n = 8). (B, C) The expression of γH2AX, cGAS, and STING in 29-kDa FN-f-, IL-1β-, and etoposide (ET)-treated chondrocytes. Chondrocytes were treated with 29-kDa FN-f (300 nM), IL-1β (1 ng/ml), and etoposide (200 μM) for (B) 24 h and (C) 3, 5, and 7 days. Cell lysates were subjected to western blot analysis. (D) Activation of γH2AX by 29-kDa FN-f. After treatment of primary chondrocytes with 29-kDa FN-f for 5 days, the cellular level of γH2AX was determined using fluorescence microscopy. Nuclei were stained with DAPI.
29-kDa FN-f-induced expression of pro-inflammatory cytokines is mediated via cGAS/STING/TBK1/IRF3 and NF-κB signaling pathways
Stimulation of TLRs induces pro-inflammatory cytokines and type I IFN expression via activation of the transcription factors IRF3 and NF-κB, which overlaps with the cGAS-STING pathway (14, 17). We thus investigated whether 29-kDa FN-f affects pro-inflammatory cytokine expression induced by cGAS-STING. The expression of type I IFN and pro-inflammatory cytokines, including GM-CSF and G-CSF, was significantly increased by 29-kDa FN-f (Fig. 2).
Fig. 2.
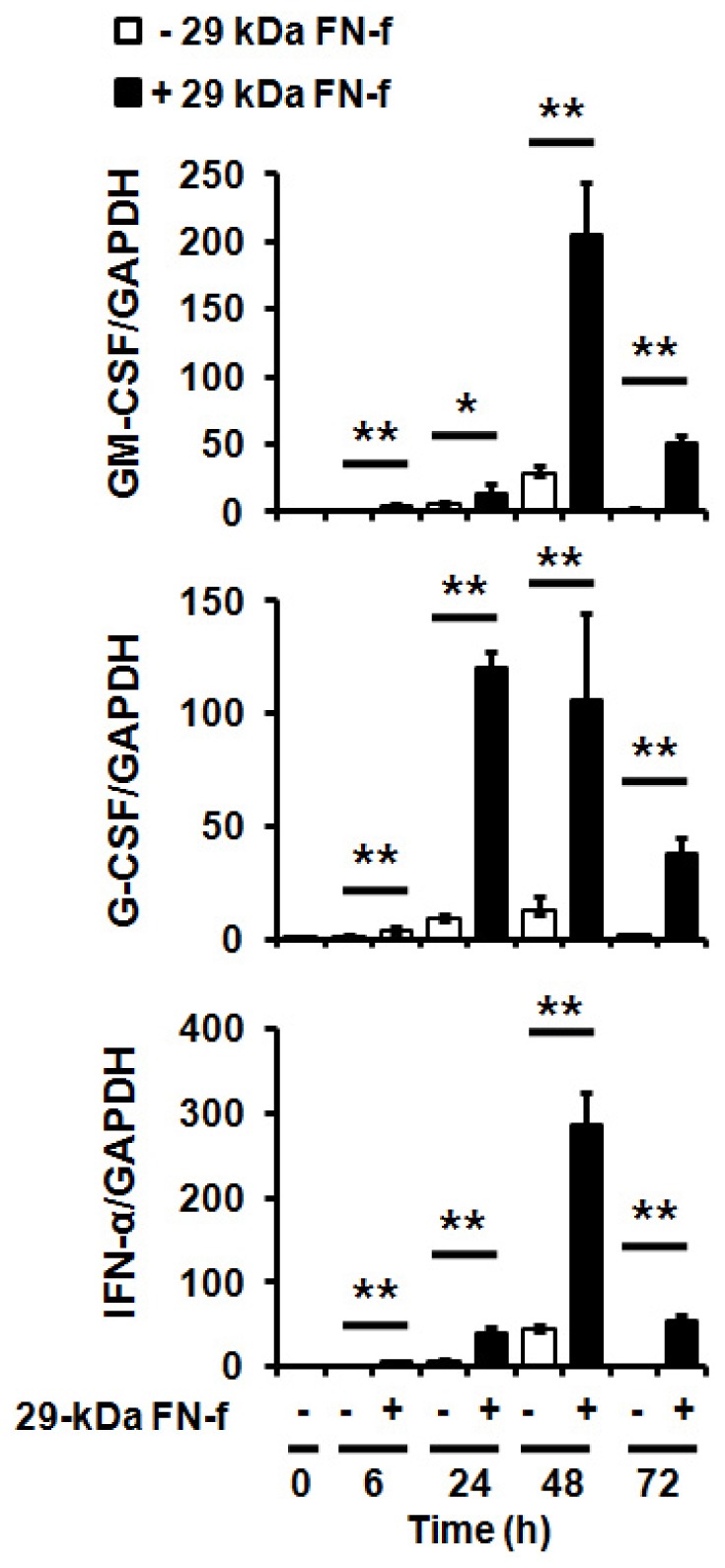
Expression of pro-inflammatory cytokines is significantly increased by 29-kDa FN-f. Chondrocytes were treated with 29-kDa FN-f (300 nM) for 6, 24, 48, and 72 h. The mRNA levels of GM-CSF (CSF-2) and G-CSF (CSF-3) and type I interferon (IFN-α) were measured using qRT-PCR analysis. GAPDH was used as an endogenous control. Data are presented as the mean ± standard deviation (SD) of duplicate data obtained from three different donors. *P < 0.05 and **P < 0.01 vs. untreated control.
We investigated the activation of IRF3 and NF-κB in primary human chondrocytes by 29-kDa FN-f. Incubation of chondrocytes with 29-kDa FN-f for 6 h led to a significant increase in the phosphorylation of IκBα, TBK1, and IRF3 (Fig. 3A). Silencing of cGAS or STING significantly suppressed phosphorylation of IκBα, TBK1, and IRF3. Furthermore, the knockdown of cGAS or STING inhibited 29-kDa FN-f-induced expression of pro-inflammatory cytokines, including GM-CSF and G-CSF, and IFN-α (Fig. 3B). These results demonstrate that 29-kDa FN-f modulated the expression of pro-inflammatory cytokines and type I IFN via activation of cGAS/STING/TBK1/IRF3 pathway.
Fig. 3.

29-kDa FN-f induces pro-inflammatory cytokine expressions via the cGAS/STING/IRF3 signaling pathways. (A) Blockade of cGAS or STING inhibited 29-kDa FN-f-induced activation of TBK1 and IRF3. Chondrocytes were transfected with si-cGAS or si-STING and incubated with 29-kDa FN-f for 24 h. Levels of phosphorylated TBK1, IRF3 and IκBα were measured by western blot analysis. Western blot data are representative of three independent experiments from different donors. β-actin served as a loading control. (B) The 29-kDa FN-f-stimulated expression of GM-CSF, G-CSF, and IFN-α was decreased by silencing cGAS or STING. Chondrocytes were transfected with si-cGAS or si-STING and incubated with 29-kDa FN-f for 6 h. The expression of CSFs and IFN-α was measured using qRT-PCR analysis. GAPDH was used as an endogenous control. Data are presented as the mean ± standard deviation (SD) of duplicate data obtained from four different donors. *P < 0.05, **P < 0.01, ***P < 0.001 vs. si-control-transfected cells.
TLR-2 and NOD2 signaling pathway regulates the activation of the cGAS–STING pathway in the presence of 29-kDa FN-f
The 29-kDa FN-f induces expression of catabolic factors via the TLR-2 or NOD2 signaling pathways (6). We investigated whether 29-kDa FN-f-induced activation of TLR-2 or NOD2 signaling pathways influences the activation of cGAS/STING pathways. Silencing of TLR-2 or NOD2 suppressed the 29-kDa FN-f-induced activation of the cGAS/STING signaling pathway, and decreased the expression of cGAS and STING as well as the phosphorylation of TBK1 and IRF3 (Fig. 4A). Down-regulation of cGAS, STING and phosphorylated forms of TBK1 and IRF3 by si-TLR-2 and si-NOD2 was observed even in untreated chondrocytes. In addition, up-regulation of GM-CSF, G-CSF, and IFN-α by 29-kDa FN-f was significantly suppressed by the knockdown of TLR-2 or NOD2 (Fig. 4B). These results demonstrate that 29-kDa FN-f may affect the cGAS/STING signaling pathway via TLR-2 or NOD2 and subsequently up-regulate the pro-inflammatory cytokines.
Fig. 4.

29-kDa FN-f-activated TLR-2 and NOD2 signaling pathway modulates the cGAS/STING signaling pathway. (A) The cGAS/STING pathway was modulated by TLR-2 and NOD2 activated by 29-kDa FN-f. Chondrocytes were transfected with si-TLR-2 or si-NOD2 and incubated with 29-kDa FN-f for 24 h. The levels of phosphorylated TBK1 and IRF3 were measured using western blot analysis. The protein levels were measured by western blot analysis. Western blot data are representative of three independent experiments using different donors. β-actin served as a loading control. (B) GM-CSF (CSF-2), G-CSF (CSF-3), and IFN-α expressions were regulated by 29-kDa FN-f-modulated TLR-2 and NOD2. Chondrocytes were transfected with si-TLR-2 or si-NOD2 and incubated with 29-kDa FN-f for 6 h. Expression of GM-CSF, G-CSF, and IFN-α was measured by using qRT-PCR analysis. GAPDH was used as an endogenous control. Data are expressed as the mean ± standard deviation (SD) of duplicate data derived from three different donors. *P < 0.05 and **P < 0.01 vs. si-control-transfected cells. ns, not significant.
DISCUSSION
In this study, we investigated the role of the cGAS/STING signaling pathway in 29-kDa FN-f-induced synthesis of pro-inflammatory cytokines in chondrocytes. An increased level of the cGAS/STING pathway was found in the OA cartilage. The 29-kDa FN-f-induced expression of pro-inflammatory cytokines mediated via cGAS/STING/TBK1/IRF3 signaling was affected by TLR-2 or NOD2, suggesting that the cGAS/STING pathway may play a role in cartilage damage induced by DAMP.
Innate immunity has evolved as the first-line defense system against microbial invasion and has been conserved across a wide variety of living organisms. Innate immune responses are initiated by the recognition of conserved pathogen structures as well as molecules derived from host tissue damage, and are mediated via diverse pattern recognition receptors (PRRs) (18). PRRs consist of various cell surface receptors as well as receptors within the cellular compartments. They include TLRs, nucleotide binding and oligomerization, leucine-rich proteins (NLRs), retinoic acid-like receptors (RLRs), absent in melanoma 2 (AIM2)-like receptors (ALRs), and cytosolic DNA receptors (19, 20). In addition to the expression of PRRs, such as TLRs and NOD, in chondrocytes, numerous reports suggest that their expression aggravates degradation of the cartilage matrix (6, 21–24). Thus, PRR signaling may represent a vicious cycle in OA pathogenesis via generation of self-perpetuating catabolic signals induced by matrix degradation products.
The cGAS is a nucleotidyltransferase identified as a cytosolic DNA sensor linking the production of interferons and STING signaling via generation of cGAMP (25). In addition to its role in sensing cytosolic DNA, the cGAS–STING pathway plays a role in chronic inflammatory diseases including obesity, autoimmune disease and cancer (9, 10). For example, the cGAS pathway plays a pivotal role in Aicardi-Gourtières syndrome, an autoimmune disease characterized by elevated expression of genes induced by type I interferon (26), in which patients carry mutations in genes such as TREX1 and the cytosolic receptor MDA5. Mice lacking both DNase II and IFN-IR exhibit chronic polyarthritis similar to human rheumatoid arthritis (RA) induced by excessive tumor necrosis factor (TNF)-α production as a result of the accumulation of undigested DNA either from phagocytosed erythroid precursor cells and apoptotic cells or from damaged DNA in the nucleus (27, 28). Loss of STING abrogates self-DNA-mediated cytokine production in Dnase2a−/− embryos and polyarthritis, suggesting an essential role of STING in aberrant DNA-induced inflammatory diseases (29). In our result, the up-regulation of cGAS-STING pathway was observed in human OA cartilage, indicating the activation of cytoplasmic DNA damage response in OA chondrocytes. This pathway was activated in vitro by IL-1 and FN-fs, two well-known mediators in catabolic signaling of the cartilage. It is plausible that DNA damage response occurs in OA chondrocytes due to increased rates of apoptosis and mitochondrial damage, which lead to the release of cytoplasmic DNA (30, 31). The cGAS/STING pathway activates two downstream pathways, such that pro-inflammatory cytokines are produced via NF-kB and type I IFN through IRF3 (11, 12, 18). In line with previous studies showing that cGAS-STING pathway stimulates pro-inflammatory genes, we showed that FN-f upregulates G-CSF, GM-CSF and INF, the typical intermediates in this pathway.
GM-CSF and G-CSF are hematopoietic growth factors induced by cytokines including interleukin-1 and TNF in fibroblasts and chondrocytes (32–35). Although CSFs play a more important role in inflammatory arthritis than OA, GM-CSF has been reported to function as a key mediator in OA and related pain. Furthermore, GM-CSF modulates inflammation via induction of the chemokine (c-c motif) ligand 17 (CCL17) (36, 37). Depletion and neutralization of GM-CSF in mice demonstrated that GM-CSF induces cartilage degradation, synovitis, and pain in rodent models of collagenase-induced OA and inflammatory arthritis (38–40). The expression of IFNs was significantly increased in the synovial tissues of patients with RA, and blockade of type 1 IFN using anti IFN antibody induced a significant decrease in the development of arthritis (41). Elevated type I IFN persistently up-regulated the expression of inflammation-related genes via activation of the autocrine loop (42). On the other hand, studies using collagen-induced arthritis models showed that an injection of IFN-expressing fibroblasts into joints reduced the severity of arthritis (43). Although a previous report showed that IFN-α treatment resulted in an increase in both IL-1Ra and soluble TNF receptor (sTNFR) production in OA synovial fluid and tissues, its role in the regulation of chondrocyte function has yet to be elucidated (44). Our results suggest that the cross-regulation between type I IFNs and pro-inflammatory cytokines induced by the DNA damage response may contribute to the pathogenesis of cartilage degeneration in OA.
We also found that the activation of cGAS-STING pathway and up-regulation of GM-CSF, G-CSF, and IFN-α by 29-kDa FN-f was mediated via TLR-2 and NOD2 signaling pathways in chondrocytes. This finding may be attributed to the elimination of TLR-2 and NOD2 receptor signaling in response to FN-fs, because of the TLR-independent cGAS-STING signaling response to intracellular DNA (45). However, it is also possible that the cross-regulation between PRRs and cGAS/STING pathways plays a key role since much of the signaling mechanism overlaps TBK1 and IRF3.
Taken together, our results demonstrate that 29-kDa FN-f significantly activates the cGAS/STING pathway, together with increase in γH2AX, an indicator of cytoplasmic chromatin fragmentation in human chondrocytes. The 29-kDa FN-f-induced increase in pro-inflammatory cytokines may be affected by both cGAS/STING and TLR-2 signaling pathways. The cGAS/STING pathway represents a novel therapeutic target against damage response due to cartilage degradation that may aggravate cartilage degeneration.
MATERIALS AND METHODS
Materials, western blot analysis, transfection with siRNA and overexpression vectors, qRT-PCR, and statistical analyses are described in the Supplementary Materials.
Cartilage collection, primary chondrocyte culture, and cartilage explant culture
Normal cartilages were obtained from the femoral head of patients sustaining femoral neck fractures without a known history of OA or RA. The OA cartilage samples were obtained from the knee joints of OA patients at the time of total knee replacement surgery. Patient diagnoses were determined using the criteria developed by the American College of Rheumatology. The collection and use of human tissue samples was reviewed and approved by the Institutional Review Board of Hallym University Sacred Heart Hospital, Anyang, South Korea (approval number 2018-05-040). All patients provided written informed consent for the use of their discarded cartilage samples. Primary human chondrocytes were isolated from articular cartilage and cultured as described previously (6). Briefly, the articular cartilage dissected from a relatively lesion-free area was incubated with a protease (8 μg/ml) derived from Streptomyces griseus for 1 h and with collagenase (4 μg/ml) obtained from Clostridium histolyticum and hyaluronidase (0.2 μg/ml) from bovine testes for 2 h. Isolated primary chondrocytes were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS).
Immunofluorescence microscopy
For detection of γH2AX in chondrocytes, cells were seeded on glass coverslips and incubated with 29-kDa FN-f for 5 days. The cells were washed with PBS, fixed with 4% paraformaldehyde at room temperature for 10 min, and incubated with a primary antibody against γH2AX (1:200 dilution) and Alexa Fluor 588-conjugated secondary antibody (Invitrogen). Nuclei were stained for 30 min with DAPI (1 μg/ml Roche Applied Science, Basel, Switzerland).
SUPPLEMENTARY INFORMATION
ACKNOWLEDGEMENTS
This study was supported by the Basic Science Research Program through the National Research Foundation (NRF) of Korea funded by the Ministry of Education (2017R1A2B 2001881) and in part by Hallym University Research Fund.
Footnotes
CONFLICTS OF INTEREST
The authors have no conflicting interests.
REFERENCES
- 1.Goldring MB. The role of the chondrocyte in osteoarthritis. Arthritis Rheum. 2000;43:1916–1926. doi: 10.1002/1529-0131(200009)43:9<1916::AID-ANR2>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 2.Goldring MB, Goldring SR. Osteoarthritis. J Cell Physiol. 2007;213:626–634. doi: 10.1002/jcp.21258. [DOI] [PubMed] [Google Scholar]
- 3.Goldring SR, Goldring MB. The role of cytokines in cartilage matrix degeneration in osteoarthritis. Clin Orthop Relat Res. 2004:S27–36. doi: 10.1097/01.blo.0000144854.66565.8f. [DOI] [PubMed] [Google Scholar]
- 4.Rosenberg JH, Rai V, Dilisio MF, Agrawal DK. Damage-associated molecular patterns in the pathogenesis of osteoarthritis: potentially novel therapeutic targets. Mol Cell Biochem. 2017;434:171–179. doi: 10.1007/s11010-017-3047-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rosenberg JH, Rai V, Dilisio MF, Sekundiak TD, Agrawal DK. Increased expression of damage-associated molecular patterns (DAMPs) in osteoarthritis of human knee joint compared to hip joint. Mol Cell Biochem. 2017;436:59–69. doi: 10.1007/s11010-017-3078-x. [DOI] [PubMed] [Google Scholar]
- 6.Hwang HS, Park SJ, Cheon EJ, Lee MH, Kim HA. Fibronectin fragment-induced expression of matrix metalloproteinases is mediated by MyD88-dependent TLR-2 signaling pathway in human chondrocytes. Arthritis Res Ther. 2015;17:320. doi: 10.1186/s13075-015-0833-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hwang HS, Lee MH, Kim HA. Fibronectin fragment inhibits xylosyltransferase-1 expression by regulating Sp1/Sp3-dependent transcription in articular chondrocytes. Osteoarthritis Cartilage. 2019;27:833–843. doi: 10.1016/j.joca.2019.01.006. [DOI] [PubMed] [Google Scholar]
- 8.Kim HA, Cho ML, Choi HY, et al. The catabolic pathway mediated by Toll-like receptors in human osteoarthritic chondrocytes. Arthritis Rheum. 2006;54:2152–2163. doi: 10.1002/art.21951. [DOI] [PubMed] [Google Scholar]
- 9.Bobacz K, Sunk IG, Hofstaetter JG, et al. Toll-like receptors and chondrocytes: the lipopolysaccharide-induced decrease in cartilage matrix synthesis is dependent on the presence of toll-like receptor 4 and antagonized by bone morphogenetic protein 7. Arthritis Rheum. 2007;56:1880–1893. doi: 10.1002/art.22637. [DOI] [PubMed] [Google Scholar]
- 10.Xiao TS, Fitzgerald KA. The cGAS-STING pathway for DNA sensing. Mol Cell. 2013;51:135–139. doi: 10.1016/j.molcel.2013.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bai J, Cervantes C, Liu J, et al. DsbA-L prevents obesity-induced inflammation and insulin resistance by suppressing the mtDNA release-activated cGAS-cGAMP-STING pathway. Proc Natl Acad Sci U S A. 2017;114:12196–12201. doi: 10.1073/pnas.1708744114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li T, Chen ZJ. The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J Exp Med. 2018;215:1287–1299. doi: 10.1084/jem.20180139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dou Z, Ghosh K, Vizioli MG, et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature. 2017;550:402–406. doi: 10.1038/nature24050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. 2011;34:637–650. doi: 10.1016/j.immuni.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 15.Chen AF, Davies CM, De Lin M, Fermor B. Oxidative DNA damage in osteoarthritic porcine articular cartilage. J Cell Physiol. 2008;217:828–833. doi: 10.1002/jcp.21562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grishko VI, Ho R, Wilson GL, Pearsall AWt. Diminished mitochondrial DNA integrity and repair capacity in OA chondrocytes. Osteoarthritis Cartilage. 2009;17:107–113. doi: 10.1016/j.joca.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barber GN. STING: infection, inflammation and cancer. Nat Rev Immunol. 2015;15:760–770. doi: 10.1038/nri3921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ma Z, Damania B. The cGAS-STING Defense Pathway and Its Counteraction by Viruses. Cell Host Microbe. 2016;19:150–158. doi: 10.1016/j.chom.2016.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Medzhitov R. TLR-mediated innate immune recognition. Semin Immunol. 2007;19:1–2. doi: 10.1016/j.smim.2007.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brubaker SW, Bonham KS, Zanoni I, Kagan JC. Innate immune pattern recognition: a cell biological perspective. Annu Rev Immunol. 2015;33:257–290. doi: 10.1146/annurev-immunol-032414-112240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heinhuis B, Koenders MI, van de Loo FA, et al. IL-32gamma and Streptococcus pyogenes cell wall fragments synergise for IL-1-dependent destructive arthritis via upregulation of TLR-2 and NOD2. Ann Rheum Dis. 2010;69:1866–1872. doi: 10.1136/ard.2009.127399. [DOI] [PubMed] [Google Scholar]
- 22.Hwang HS, Lee MH, Choi MH, Kim HA. NOD2 signaling pathway is involved in fibronectin fragment-induced pro-catabolic factor expressions in human articular chondrocytes. BMB Rep. 2019 doi: 10.5483/BMBRep.2019.52.6.165. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Joosten LA, Heinhuis B, Abdollahi-Roodsaz S, et al. Differential function of the NACHT-LRR (NLR) members Nod1 and Nod2 in arthritis. Proc Natl Acad Sci U S A. 2008;105:9017–9022. doi: 10.1073/pnas.0710445105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vieira SM, Cunha TM, Franca RF, et al. Joint NOD2/RIPK2 signaling regulates IL-17 axis and contributes to the development of experimental arthritis. J Immunol. 2012;188:5116–5122. doi: 10.4049/jimmunol.1004190. [DOI] [PubMed] [Google Scholar]
- 25.Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339:786–791. doi: 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Crow YJ. Type I interferonopathies: mendelian type I interferon up-regulation. Curr Opin Immunol. 2015;32:7–12. doi: 10.1016/j.coi.2014.10.005. [DOI] [PubMed] [Google Scholar]
- 27.Kawane K, Ohtani M, Miwa K, et al. Chronic polyarthritis caused by mammalian DNA that escapes from degradation in macrophages. Nature. 2006;443:998–1002. doi: 10.1038/nature05245. [DOI] [PubMed] [Google Scholar]
- 28.Lan YY, Londono D, Bouley R, Rooney MS, Hacohen N. Dnase2a deficiency uncovers lysosomal clearance of damaged nuclear DNA via autophagy. Cell Rep. 2014;9:180–192. doi: 10.1016/j.celrep.2014.08.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ahn J, Gutman D, Saijo S, Barber GN. STING manifests self DNA-dependent inflammatory disease. Proc Natl Acad Sci U S A. 2012;109:19386–19391. doi: 10.1073/pnas.1215006109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim HA, Lee YJ, Seong SC, Choe KW, Song YW. Apoptotic chondrocyte death in human osteoarthritis. J Rheumatol. 2000;27:455–462. [PubMed] [Google Scholar]
- 31.Kim J, Xu M, Xo R, et al. Mitochondrial DNA damage is involved in apoptosis caused by pro-inflammatory cytokines in human OA chondrocytes. Osteoarthritis Cartilage. 2010;18:424–432. doi: 10.1016/j.joca.2009.09.008. [DOI] [PubMed] [Google Scholar]
- 32.Leizer T, Cebon J, Layton JE, Hamilton JA. Cytokine regulation of colony-stimulating factor production in cultured human synovial fibroblasts: I. Induction of GM-CSF and G-CSF production by interleukin-1 and tumor necrosis factor. Blood. 1990;76:1989–1996. [PubMed] [Google Scholar]
- 33.Hamilton JA, Filonzi EL, Ianches G. Regulation of macrophage colony-stimulating factor (M-CSF) production in cultured human synovial fibroblasts. Growth Factors. 1993;9:157–165. doi: 10.3109/08977199309010831. [DOI] [PubMed] [Google Scholar]
- 34.Campbell IK, Novak U, Cebon J, Layton JE, Hamilton JA. Human articular cartilage and chondrocytes produce hemopoietic colony-stimulating factors in culture in response to IL-1. J Immunol. 1991;147:1238–1246. [PubMed] [Google Scholar]
- 35.Campbell IK, Ianches G, Hamilton JA. Production of macrophage colony-stimulating factor (M-CSF) by human articular cartilage and chondrocytes. Modulation by interleukin-1 and tumor necrosis factor alpha. Biochim Biophys Acta. 1993;1182:57–63. doi: 10.1016/0925-4439(93)90153-R. [DOI] [PubMed] [Google Scholar]
- 36.Achuthan A, Cook AD, Lee MC, et al. Granulocyte macrophage colony-stimulating factor induces CCL17 production via IRF4 to mediate inflammation. J Clin Invest. 2016;126:3453–3466. doi: 10.1172/JCI87828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee MC, Saleh R, Achuthan A, et al. CCL17 blockade as a therapy for osteoarthritis pain and disease. Arthritis Res Ther. 2018;20:62. doi: 10.1186/s13075-018-1560-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cook AD, Pobjoy J, Steidl S, et al. Granulocyte-macrophage colony-stimulating factor is a key mediator in experimental osteoarthritis pain and disease development. Arthritis Res Ther. 2012;14:R199. doi: 10.1186/ar4037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cook AD, Pobjoy J, Sarros S, et al. Granulocyte-macrophage colony-stimulating factor is a key mediator in inflammatory and arthritic pain. Ann Rheum Dis. 2013;72:265–270. doi: 10.1136/annrheumdis-2012-201703. [DOI] [PubMed] [Google Scholar]
- 40.Yang YH, Hamilton JA. Dependence of interleukin-1-induced arthritis on granulocyte-macrophage colony-stimulating factor. Arthritis Rheum. 2001;44:111–119. doi: 10.1002/1529-0131(200101)44:1<111::AID-ANR15>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 41.Miller JC, Ma Y, Bian J, et al. A critical role for type I IFN in arthritis development following Borrelia burgdorferi infection of mice. J Immunol. 2008;181:8492–8503. doi: 10.4049/jimmunol.181.12.8492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roelofs MF, Wenink MH, Brentano F, et al. Type I interferons might form the link between Toll-like receptor (TLR) 3/7 and TLR4-mediated synovial inflammation in rheumatoid arthritis (RA) Ann Rheum Dis. 2009;68:1486–1493. doi: 10.1136/ard.2007.086421. [DOI] [PubMed] [Google Scholar]
- 43.Triantaphyllopoulos KA, Williams RO, Tailor H, Chernajovsky Y. Amelioration of collagen-induced arthritis and suppression of interferon-gamma, interleukin-12, and tumor necrosis factor alpha production by interferon-beta gene therapy. Arthritis Rheum. 1999;42:90–99. doi: 10.1002/1529-0131(199901)42:1<90::AID-ANR12>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 44.Wong T, Majchrzak B, Bogoch E, Keystone EC, Fish EN. Therapeutic implications for interferon-alpha in arthritis: a pilot study. J Rheumatol. 2003;30:934–940. [PubMed] [Google Scholar]
- 45.Burdette DL, Vance RE. STING and the innate immune response to nucleic acids in the cytosol. Nat Immunol. 2013;14:19–26. doi: 10.1038/ni.2491. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.