
Fig. 1|. The substrate-overload liver injury model of NASH pathogenesis.
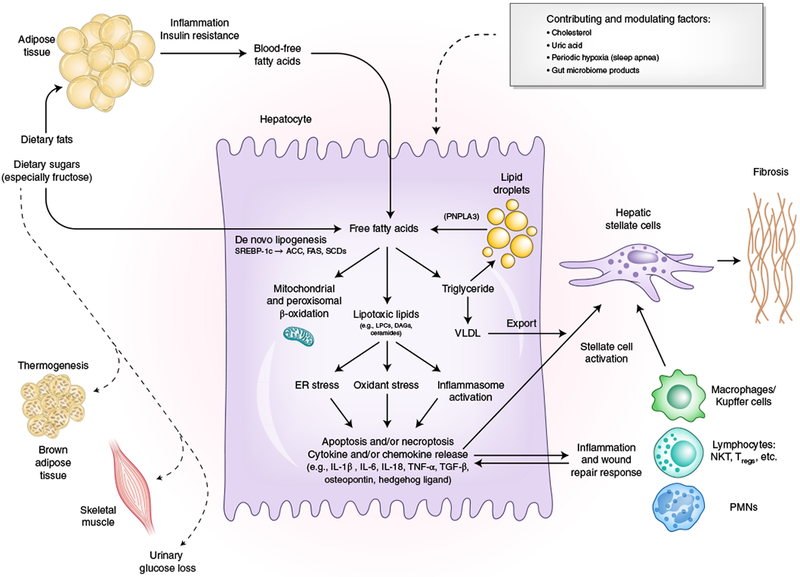
Free fatty adds are central to the pathogenesis of NASH. Free fatty adds that originate from lipolysis of triglyceride in adipose tissue are delivered through blood to the liver. The other major contributor to the free fatty acid flux through the liver is DNL, the process by which hepatocytes convert excess carbohydrates, especially fructose, to fatty acids. The two major fates of fatty acids in hepatocytes are mitochondrial beta-oxidation and re-esterification to form triglyceride. Triglyceride can be exported into the blood as VLDL or stored in lipid droplets. Lipid droplet triglyceride undergoes regulated lipolysis to release fatty acids back into the hepatocyte free fatty acid pool. PNPLA3 participates in this lipolytic process, and a single-nucleotide variant of PNPLA3 is strongly associated with NASH progression, underscoring the importance of the regulation of this lipolysis. When the disposal of fatty acids through beta-oxidation or formation of triglyceride is overwhelmed, fatty acids can contribute to the formation of lipotoxic species that lead to ER stress, oxidant stress and inflammasome activation. These processes are responsible for the phenotype of NASH with hepatocellular injury, inflammation, stellate cell activation and progressive accumulation of excess extracellular matrix. Lifestyle modifications that include healthy eating habits and regular exercise reduce the substrate overload through decreased intake and diversion of metabolic substrates to metabolically active tissues and can thereby prevent or reverse NASH. SCD, steroyl CoA-desaturase; FAS, fatty acid synthase; NKT, natural killer T cell; Tregs, regulatory T cells; PMNs, polymorphonuclear leukocytes. Credit: Marina Corral Spence/Springer Nature