

Abstract
An understanding of the mechanisms underlying acquired resistance to cetuximab is urgently needed to improve cetuximab efficacy in patients with head and neck squamous cell carcinoma (HNSCC). Here, we present a clinical observation that MET pathway activation constitutes the mechanism of acquired resistance to cetuximab in a patient with HNSCC. Specifically, RNA sequencing and mass spectrometry analysis of cetuximab-sensitive (CetuxSen) and cetuximab-resistant (CetuxRes) tumors indicated MET amplification and overexpression in the CetuxRes tumor compared to the CetuxSen lesion. Stimulation of MET in HNSCC cell lines was sufficient to reactivate the MAPK pathway and to confer resistance to cetuximab in vitro and in vivo. In addition to the direct role of MET in reactivation of the MAPK pathway, MET stimulation abrogates the well-known cetuximab-induced compensatory feedback loop of HER2/HER3 expression. Mechanistically, we showed that the overexpression of HER2 and HER3 following cetuximab treatment is mediated by the ETS homologous transcription factor (EHF), and is suppressed by MET/MAPK pathway activation. Collectively, our findings indicate that evaluation of MET and HER2/HER3 in response to cetuximab in HNSCC patients can provide the rationale of successive line of treatment.
Keywords: head and neck cancer, MET, signaling, drug resistance, cetuximab
Introduction
The mortality rate of patients with metastatic head and neck squamous cell carcinoma (HNSCC) is as high as 50–80% within 24 months of diagnosis.1 The treatment regimens for this disease are aggressive, as reflected in poor patient quality of life, which is characterized by daily dysfunctions in speech, chewing, swallowing and facial expressions.2 These unfavorable outcomes underscore the shortcomings of the therapeutic options available for patients with advanced HNSCC. The FDA-approved treatment of these patients comprises a combination of cetuximab—an antibody that blocks the receptor tyrosine kinase (RTK) epithelial growth factor receptor (EGFR) —with either radiation for advanced local disease or with chemotherapy for metastatic disease.3,4 Although cetuximab significantly prolongs the median overall survival of HNSCC patients, the clinical response rate is limited to a duration of 10 months for advanced local disease3 or 2–3 months in the metastatic setting.4,5 For patients treated with cetuximab monotherapy, the pattern of clinical response indicates that 87% of HNSCC patients are intrinsically (de novo) resistant, while the remaining 13% initially respond to cetuximab but develop (acquired) resistance over time.5 Therefore, there is an urgent clinical need to identify both the molecular determinants of sensitivity to cetuximab and the mechanisms of resistance to cetuximab in HNSCC, as such findings may have an immediate impact on patient prognosis.
EGFR overexpression or gene amplification is common in cancer, especially in HNSCC, lung and colorectal cancers.6,7 EGFR triggers multiple downstream signaling pathways linked to cell survival and proliferation, such as the STAT, SRC, PI3K and MAPK pathways [reviewed in Refs. 7–11], and therefore blocking EGFR has been intensely pursued as a target for cancer therapy, including treatment for HNSCC.3–5,12–14 Cetuximab inhibits tumor cell growth by preventing ligand binding to the extracellular domain of EGFR, thereby preventing the dimerization and auto-phosphorylation of this receptor and the subsequent activation of the signaling pathways downstream of EGFR.15 Resistance to cetuximab is thought to be, at least in some cases, the consequence of the persistent activation of the survival signaling that is mediated by mutations in the EGFR gene (such as EGFRVIII)16–18 or in genes downstream of EGFR, such as HRAS or KRAS.19–22 An additional common resistance mechanism to cetuximab is reactivation of signaling pathways due to upregulation and activation of alternative RTKs, which termed as a compensatory activation loop [reviewed in Ref. 23]. One of the major compensatory feedback loops is the upregulation and activation of the tyrosine kinases, human epidermal growth factor receptors 2 and 3 (HER2 and HER3, respectively). As a result, the PI3K and MAPK pathways are reactivated and bypass cetuximab inhibition.24–26 The activation of the HER2/HER3 feedback loop in response to anti-EGFR therapies has also been reported in other cancers such as lung, breast and colorectal cancers.27–29
The molecular machinery that underlies the compensatory feedback loop in response to anti-cancer therapies has been well characterized in breast cancer. Specifically, transcription upregulation of HER2/HER3 were observed in breast cell lines treated with AKT and mTOR inhibitors.30,31 Nonetheless, despite the intensive work showing HER2/HER3 activation of a compensatory feedback loop in response to EGFR inhibitors,24–29 the molecular machinery that regulates their expression in this context has not yet been identified.
In HNSCC, in addition to the upregulation of HER2/HER3 in response to cetuximab, other RTKs have been shown to be upregulated or activated, including MET and AXL.24,26,32 Activation of the MET oncogene regulates cell proliferation and differentiation either by receptor dimerization or by stimulation by its ligand, hepatocyte growth factor (HGF) [reviewed in Ref. 33]. A number of studies on preclinical models of HNSCC have shown that activation of MET—in either a ligand-dependent or a ligand-independent manner— is sufficient to confer resistance to cetuximab.34–37 In agreement with these in-vitro and patient-derived xenograft studies, a single investigation has shown a correlation between MET expression and the response to cetuximab in patient samples.38 The importance of MET in the acquisition of resistance to anti-EGFR treatment has been demonstrated in other cancers, such as colorectal and lung cancers.39–41 However, there are no reports in the literature providing clinical evidence that MET plays a key role in the acquisition of resistance to cetuximab in HNSCC patients.
Materials and Methods
RNA sequencing
RNA-seq libraries were prepared using the TruSeq RNA Sample Preparation kit (Illumina) according to the manufacturer’s protocol. Libraries were sequenced on the Illumina HiSeq2500 platform at Memorial Sloan Kettering Cancer Center to produce, on average, 70 million paired-end sequences (50 bp) per sample. Reads were then quality trimmed using the fastx toolkit to remove bases with a base quality score of less than 10, and reads shorter than 50 bp were discarded. Alignment was based on a two-pass mapping procedure, namely, reads were first mapped with RNAStar,42 and then reads that did not map in the first pass were re-mapped using the BWA MEM method. The output BAM files from these two steps were merged. Thereafter, the gene expression level was counted with htseq-count (–s yes -m intersection-strict), and the exon expression level was quantitated with DEXSeq. Only coding genes were included in the analysis. After quantification of gene expression, duplicate quality was assessed in the different samples (‘10124_1’, ‘10124_1’, ‘21549_1’, ‘21549_1’). The differential analysis was performed using the DESeq2 package in R. DESeq2, which performs differential analysis using raw read counts, under a negative binomial noise model. Model fitting was followed by FDR (BH) correction for multiple hypothesis testing. Our list of upregulated genes was determined by requiring a log2 fold change >1, baseMean >200 (parental expression level), and adjusted p-value (p.adj) < 0.01; similarly, for the downregulated genes, we required a log2 fold change < –1, baseMean >200, and p.adj < 0.01.
Targeted laser microdissection—mass spectrometry proteomics
This analytic technique was performed as previously described.43 Briefly, 10-μm tissue sections were cut from formalin-fixed paraffin-embedded (FFPE) blocks. Following hematoxylin staining, tumor areas (12 mm2) were marked out by a pathologist for microdissection. Selected reaction monitoring-mass spectrometry (SRM/MS) analysis (with a stable isotope-labeled internal standard) was used for accurate quantitation of the analytical targets. Mass spectra were obtained on a TSQ series (Vantage or Quantiva) triple quadrupole mass spectrometer (Thermo Scientific, San Jose, CA). MS and chromatography conditions were as previously described.44
Fluorescent in situ hybridization
Fluorescent in situ hybridization (FISH) analysis was performed on FFPE sections using a home-brew 2-color MET/-Cen7 probe. The probe mix consisted of BAC clones containing the full-length MET gene (clones RP11–39 K12, and RP11–163C9; labeled with red/orange dUTP), and a centromeric repeat plasmid for chromosome 7 served as the control (clone p7t1; labeled with green dUTP). Probe labeling, tissue processing, hybridization, post-hybridization washing and fluorescence detection were performed according to standard laboratory procedures. Slides were scanned using a Zeiss Axioplan 2i epifluorescence microscope equipped with a megapixel CCD camera (CV-M4 + CL, JAI) controlled by Isis 5.5.9 imaging software (MetaSystems Group Inc, Waltham, MA). For each slide, the entire section was scanned under a 63× or 100× objective, and representative regions were imaged throughout the depth of the tissue (compressed/-merged stack of 12 z-section images taken at 0.5-μm intervals under the green and red filter). At least 2 images per representative region were captured, and a minimum of 20 discrete interphase nuclei were analyzed. Amplification was defined as a MET:Cen7 (control) ratio of ≥2.2, >10 copies of MET (independent of the control locus), or at least one small cluster of MET (≥4 signals resulting from tandem repeat/duplication). In cells with large clusters of MET (HSR type amplification), signals beyond 20 cannot be accurately counted and were therefore given a score/count of 20. Cells with 3–5 and 6–10 discrete copies of MET/Cen7 were considered to be polysomic and high-polysomic, respectively.
Immunohistochemical staining
Immunohistochemistry (IHC) staining of MET and HGF was performed at the Molecular Cytology Core Facility of Memorial Sloan Kettering Cancer Center using Discovery XT processor (Ventana Medical Systems). IHC staining of KI67 and pERK was performed at Ben-Gurion University of the Negev. For MET and HGF staining, the tissue sections were deparaf-finized in EZPrep buffer (Ventana Medical Systems); antigen retrieval was performed in CC1 buffer (Ventana Medical Systems); and sections were blocked for 30 min with Background Buster solution (Innovex), followed by avidin-biotin blocking (Ventana Medical Systems) for 8 min. Sections were incubated with anti-MET (Abcam, cat#ab51067, 3.8 μg/mL) antibodies for 5 h, followed by 60 min of incubation with biotinylated goat anti-rabbit IgG (Vector labs, cat# PK6101) at a 1:200 dilution. For KI67 and pERK T202/Y204 staining Antibodies were detected with a ABC kit (Vectastain) and DAB detection kit (Zytome), used according to the manufacturer’s instructions. Slides were counterstained with hematoxylin (Leica) and coverslipped with Surgipath Micromount (Leica) and were scanned using the Panoramic MIDI II scanner, 3DHISTECH.
Cell lines and chemical compounds
The HNSCC cell lines SNU1076 (KCLB), HSC4 (HSRRB), CAL33 (DSMZ), HSC2 (HSRRB) and FaDu), and Lung Cancer cell line H1975 (ATCC) were maintained at 37°C in a humidified atmosphere at 5% CO2. Cetuximab (Erbitux, Merck) was used throughout at a concentration of 12.5 μg/mL. The MET inhibitor PHA-665752 (MedChemExpress) was dissolved in DMSO at a working concentration of 1 μM. Recombinant hepatocyte growth factor (rHGF) (GenScript) was dissolved in doubly distilled water at a working concentration 50 ng/mL. The MEK inhibitor PD-0325901 (Selleckchem) was dissolved in DMSO at a working concentration of 25 nM. The STAT3 inhibitor CAS 1041438–69–9 (Santa Croz) was dissolved in DMSO at a working concentration of 25 nM. The AKT inhibitor MK2206 (Selleckcheme) was dissolved in DMSO at a working concentration of 1 μM. The HER2/HER3 inhibitor lapatinib (Selleckcheme) was dissolved in DMSO at a working concentration of 500 nM. The HER1/HER2/HER3 inhibitor Afatinib (Selleckcheme) was dissolved in DMSO at a working concentration of 500 nM.
Cell proliferation
For ligand-induced proliferation experiments, 20,000 cells of the relevant HNSCC cell line were seeded in 24-well plates and treated for 5 days with cetuximab (12.5 μg/mL) in the presence of 50 ng/mL rHGF and/or 1 μM PHA-665752. Cells were fixed with TCA (0.6 M, Sigma) and stained with crystal violet (1 g/l). The crystal violet was dissolved in 10% acetic acid, and quantified by spectrophotometry (Epoch™, BioTech™) at OD 570 nm. The percentage of rescue was normalized for the effect of growth factors on the proliferation of tumor cells.
Mice and establishment of patient-derived xenografts
NOD.CB17-Prkdc-scid/NCr Hsd (Nod.Scid) were purchased from Envigo. NOD.Cg-Prkdc Il2rg/SzJ (NSG) mice were purchased from Jackson labs. Patient-derived xenografts (PDXs) were established from HNSCC patients treated in the Ear, Nose and Throat Unit, Soroka Medical Center, Beer-Sheva, Israel. All patients signed informed consent forms. All PDXs were first transplanted subcutaneously into the flanks of 6-week-old NSG mice. Upon successful tumor engraftment tumor were expanded and re-transplanted into Nod.Scid mice for drug efficacy experiments.
In-vivo experiments
To obtain cell-line-derived xenografts, CAL33 cells were injected subcutaneously into NOD.SCID mice. For PDX, we implanted 3mm3 pieces of tumors. When the tumor volume had reached 70 to 120 mm3, the animals were randomly divided into 5 groups, each receiving a different treatment. Each group contained 5 mice harboring 2 tumors (n = 10). The mice were treated with vehicle; cetuximab (10 mg/kg/5 days via intra-peritoneal injection) alone; cetuximab in combination with PHA-665752 (30 mg/kg/day by gavage); cetuximab plus PHA-665752 plus rHGF of (20 μg/kg/2 days by intratumoral injection); or PHA-665752 alone. Tumors were measured with digital caliper, and tumor volumes were determined with the formula: length × width2 × (π/6).
Mice were maintained and treated in accordance with the institutional guidelines of Ben-Gurion University of the Negev. Animal experiments were approved by the Institutional Animal Care and Use Committee (IL.80–12-2015). Mice were housed in air-filtered laminar flow cabinets with a 12-h light/dark cycle and food and water ad libitum. At the end of the experiment, animals were euthanized with CO2. Tumor volumes are plotted as means ± SEM.
Western blotting
Cells were washed with ice-cold PBS, scraped off the culture plates and spun for 10 min at 5000 rpm at 4 ° C. Cells were resuspended in ice-cold lysis buffer supplemented with phosphatase inhibitor cocktails (BiotoolB15001A/B) and protease inhibitor (Sigma-Aldrich P2714–1BTL) and placed on ice for 30 min, followed by 3 min of ultrasonic cell disruption. Lysates were cleared by centrifugation at 14,000 rpm for 10 min at 4 °C, and supernatants were removed and assayed for protein concentration using the Bradford Assay Kit (BioRad #500–0006). Twenty-five micrograms of total lysate were resolved on NuPAGE 10% Bis-Tris gels and electropho-retically transferred to Immobilon ® transfer membranes (BioRad trans blot® TurboTM transfer pack #1704157). Membranes were blocked for 1 h in 5% BSA (Amresco 0332-TAM) in Tris-buffered saline (TBS)-Tween (0.1%), and then hybridized with primary antibodies in 5% BSA and 0.1% of Tween. Mouse and rabbit horseradish peroxidase (HRP)-conjugated secondary antibodies (1:10,000, Jackson) were diluted in 5% BSA in TBS-Tween. Protein-antibody complexes were detected by chemiluminescence with ECL (Westar Supernova, Cyanagen XLS3.0100 and Westar Nova 2.0 Cyanagen XLS071.0250), and images were captured with an Azure bio-systems camera system. For full details of the antibodies, see Supporting Information.
Protein arrays
Proteins were determined according to manufacturer’s instructions for Proteome Profiler™ Antibody Arrays (R&D systems) and Pathscan® cell signaling technology. Pixel density quantification was performed by GelCount express and AzureSpot analysis programs.
Real-time PCR
Total RNA was isolated from SNU1076 and HSC4 cell lines with a PureLink™ RNA Mini Kit (Invitrogen, Thermo Fisher Scientific) according to the manufacturer’s instructions. Total RNA was reverse transcribed using a qScript™ cDNA synthesis kit (Quanta bioscience, 95047–100) according to the manufacturer’s instructions. All samples within an experiment were reverse transcribed at the same time. Real-time PCR was performed (Roche LightCycler® 480 II) using TaqMan® Universal Mastermix II (ThermoFisher Scientific) with matching probes (Thermo-fisher Hs02758991_g1 GAPDH, Hs00176538_m1 ERBB3, Hs01001580_m1 ERBB2). Analysis was performed with the LightCycler® 480 Gene Scanning Software version 1.5.1. Fold change was calculated by the ΔΔCt method.
RNA silencing
siRNAs were purchased from IDT (EHF: 73715511, FOXO3: 73715511). The relevant tumor cell lines were transfected using GenMute™ siRNA Transfection Reagent (SignaGen laboratories) according to manufacturer’s instructions. Cells were washed 4 h after transfection and incubated for 24 h before treatment.
For the production of shRNAs FaDu cell line, we created lentiviruses by transfecting HEK293 cells with the viral plasmids psPAX2, pMD2.G, and PLKO with shRNAs—a control scrambled sequence (shCT) or 2 different sequences for the silencing of MET expression (shMET1 and shMET2 obtained from Sigma) using PolyJet transfection reagent (SignaGen, Cat. SL100688) according to the manufacturer’s protocol. Viruses were collected after 48–72 h and used for cell infection. Cells were seeded in a 6-well plates (150,000 cells per well) and infected with the lentiviruses in the presence of Polybrene (Sigma Aldrich Cat. 5G-H9268). Cells were selected with puromycin (Gibco Cat. A11138–03).
Statistical analysis
Statistical analysis was performed using GraphPad Prism software, presented as mean ± SEM. All cellular experiments were repeated at least three times. A two-tailed Student’s unpaired t test was performed to compare control vs. treated group. p Values of 0.05 (*), 0.01 (**), 0.001 (***) and 0.0001 (****) were considered statistically significant. For experiment with more than two groups, one-way ANOVA was calculated using Turkey’s multiple comparison test. In vivo experiments were preformed with indicated n and permutation tests were used to compare growth curves using the Walter& Elisa Hall bioinformatics—Institute of Medical Research - http://bioinf.wehi.edu.au/software/compareCurves/index.html. For pathological analysis, IHC images were analyzed by Histoquant software (3D Histech) and one-way ANOVA test was performed to compare control vs. treated groups.
Results
Exceptional clinical response to cetuximab monotherapy followed by MET-induced acquired resistance
A 72-year-old woman was diagnosed with squamous cell carcinoma of the left lateral tongue in January 2012 (Fig. S1, Supporting Information). Initial treatment with pre-operative carboplatin + paclitaxel gave a transient tumor response (Fig. 1a). In April 2012, she underwent a total glossectomy (primary tumor resection). The patient was then treated with radiotherapy together with weekly cisplatin for two months. PET-scan imaging in August 2012 indicated a recurrent primary tumor and the emergence of bilateral lung metastasis. Treatment was initiated with cetuximab as a single agent (500 mg/kg q2W) and continued for 23 months with near complete response: The recurrent primary tumor disappeared, and the metastatic sites regressed significantly (monitored by CT in September 2013) (Figs. 1b and 1c). In light of this clinical response, we termed the primary tumor as cetuximab sensitive (CetuxSen). Despite the patient’s exceptionally good response, in January 2014 progression of a lung lesion was observed by PET-CT (Fig. 1d). A biopsy from the cetuximab-resistant lung lesion (CetuxRes) was taken for pathological analysis and molecular characterization. Following the aggressive progression on cetuximab treatment (multiple new bilateral pulmonary metastases and an increase in size of a mass in the right kidney, detected by PET/CT), combination treatment with cetuximab and the anti-hHER3 monoclonal antibody, LJM716, was initiated in August 2014. However, no clinical response was observed, and the disease progressed rapidly; the patient died in October 2014. In summary, 2 tumor samples obtained from an HNSCC patient who exhibited an exceptionally good response to single-agent cetuximab treatment were available for molecular analysis, namely, the primary CetuxSen lesion and the paired metastatic CetuxRes lesion.
Figure 1.
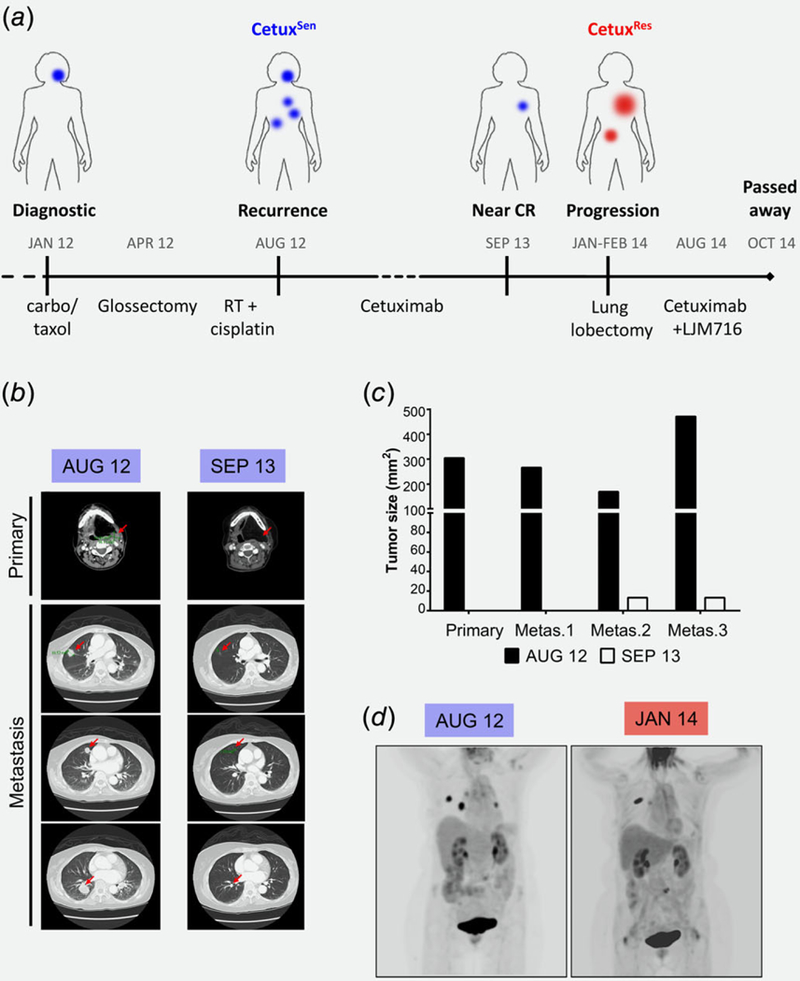
Exceptional clinical response to a single agent of cetuximab is limited by MET amplification and overexpression. (a) Time line describing treatments and responses in the case study. Blue indicates a CetuxSen tumor and red indicates a CetuxRes tumor. (b) CT scans fromthe time of disease recurrence (August 2012) and the best response (September 2013). Red arrows indicate lesions. (c) Tumor size (mm2) according to CT scans of the disease recurrence (August 2012) and the best response (September 2013). (d) PET scans from the time of disease recurrence (August 2012) and the first sign of progression under cetuximab treatment (January 2014).
To study the molecular changes that occurred in the tumor during the development of resistance to cetuximab, we performed in-depth molecular characterizations of the CetuxRes and CeutxSen lesions using RNA sequencing and targeted proteomics. Gene expression analysis of CetuxRes and CetuxSen tumors identified over 1,300 genes that were expressed differentially between the two samples. At the top of the list of upregulated genes, we found genes of the pulmonary-associated surfactant (SFRPA) family that are normally expressed in lung tissue.45 Among the non-lung specific genes, we found that MET was significantly overexpressed in the CetuxRes lesion (Fig. S2A, Supporting Information). Since MET is known to confer resistance to anti-EGFR therapies in different types of cancer,34–37,39,41,46–49and is expressed in HNSCC,48 we speculated that MET overexpression constituted may play a role in the resistance mechanism to cetuximab in our HNSCC patient. Locus analysis of the RNAseq data indicated that 50 genes located next to MET on chromosome 7-Q31 were also upregulated, suggesting copy number gain of the MET-containing chromosomal region (Fig. S2B, Supporting Information). Targeted laser-microdissecting–mass spectrometry (MS) proteomics50,51 of tumor cells obtained from FFPE CetuxRes and CetuxSen tumor tissues confirmed that MET was significantly overexpressed in the resistant lesion (Fig. 2a). The intense expression of MET supports our hypothesis of a functional role for MET in enhancing cell proliferation and limiting the sensitivity to cetuximab. Notable, as many other genes were differentially expressed between CetuxRes and CetuxSen tumors, it is possible that more than one resistance mechanisms co-existed in response to cetuximab.
Figure 2.
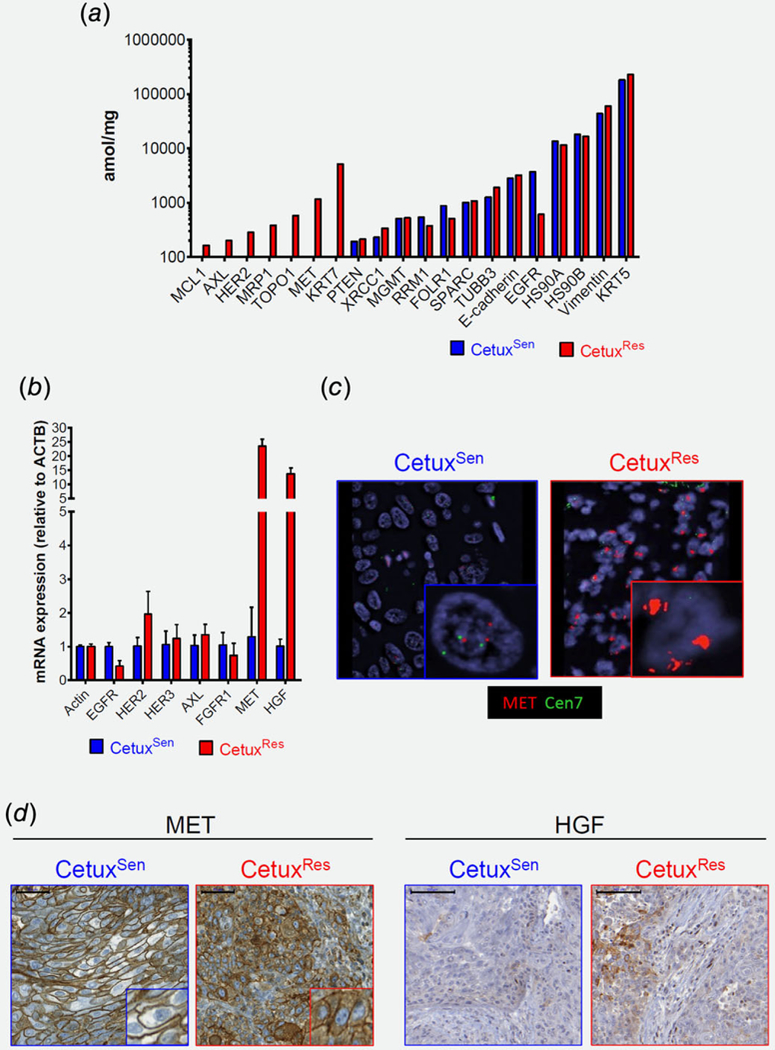
Molecular analysis comparing the CetuxSen and CetuxRes tumors. (a) Proteomic analysis by MS of targeted laser-microdissected cells obtained from formalin-fixed paraffin-embedded tumor tissue. (b) qPCR expression levels of the indicated gene. (c) Representative pictures (63X) of FISH assay using BAC clones for MET (red) and centromeric repeats for chromosome 7 (green) for control. (d) Representative pictures of IHC staining for MET (left, 40X, 50 μm) and rHGF (right, 20X, 100 μm).
The gene expression of MET and of its ligand HGF increased by 25- and 20-fold, respectively, in CetuxRes compared to CetuxSen (Fig. 2b). We also found changes in HER2 and HER3 expression, although these were less profound (Fig. 2b). FISH for MET indicated an amplification of over 10 copies in tumor cells of CetuxRes but not in the CetuxSen (Fig. 2 c). IHC staining of MET indicated that its expression was detectable in both CetuxRes and CetuxSen tumors. However, in the CetuxRes tumor, MET was also located in the intracellular compartment of the tumor cells, whereas in the CetuxSen it was expressed on the cell membrane. This localization of MET may indicate receptor activation.52,53 Consistent with the RNAseq and qPCR data, we observed an increase in HGF protein expression in CetuxRes compared to Cetux Sen (Fig. 2d). In keeping with other reports, it appears that HGF is expressed by non-tumor cells in the tumor microenvironment.34
In summary, we found that both MET amplification in the tumor cells and HGF overexpression in the stromal cells were associated with the acquisition of resistance to cetuximab in a HNSCC patient.
MET confers resistance to cetuximab in vitro and in vivo via MAPK pathway activation
To study the causative role of MET in inducing resistance to cetuximab, we tested the ability of MET activation to limit the efficacy of cetuximab in vitro using HNSCC cell lines. SNU1076, HSC4, CAL33, HSC2, and FaDu were chosen based their EGFR and MET expression (Cancer Cell Line Encyclopedia project, https://portals.broadinstitute.org/ccle, and Fig. S3A, Supporting Information), and their intrinsic sensitivity to cetuximab.54 Treatment of tumor cell lines with cetuximab in the presence or absence of recombinant HGF (rHGF) showed that ligand stimulation of MET is sufficient to limit the efficacy of cetuximab (Fig. 3a and Fig. S3B, Supporting Information). Reduction of MET expression using shRNA abolishes this rescue (Fig. S3C, Supporting Information). To understand the signaling downstream of MET that is responsible for limiting the efficacy of cetuximab, we initially characterized the phosphorylation status of several key signaling mediators by protein array in SNU1076 cell lines (Figs. S3D and S3E, Supporting Information). WB Analysis of two additional cell lines (HSC4 and CAL33) showed that stimulation of MET activated MAPK, indicated by an increase of pERK, despite cetuximab treatment. Moreover, blocking MET signaling with PHA-665752 in these tumor cell lines reversed MAPK pathway inhibition (decrease of pERK), indicating that MET is directly involved in MAPK pathway activation (Fig. 3b, Figs. S3E, S3F and S3G, Supporting Information). Finally, we confirmed that blocking the MAPK pathway in SNU1076 and HSC4 cell lines—using the MEK½ inhibitor PD-0325901—prevented the rHGF-induced rescue of tumor cells from cetuximab (Fig. 3c).
Figure 3.
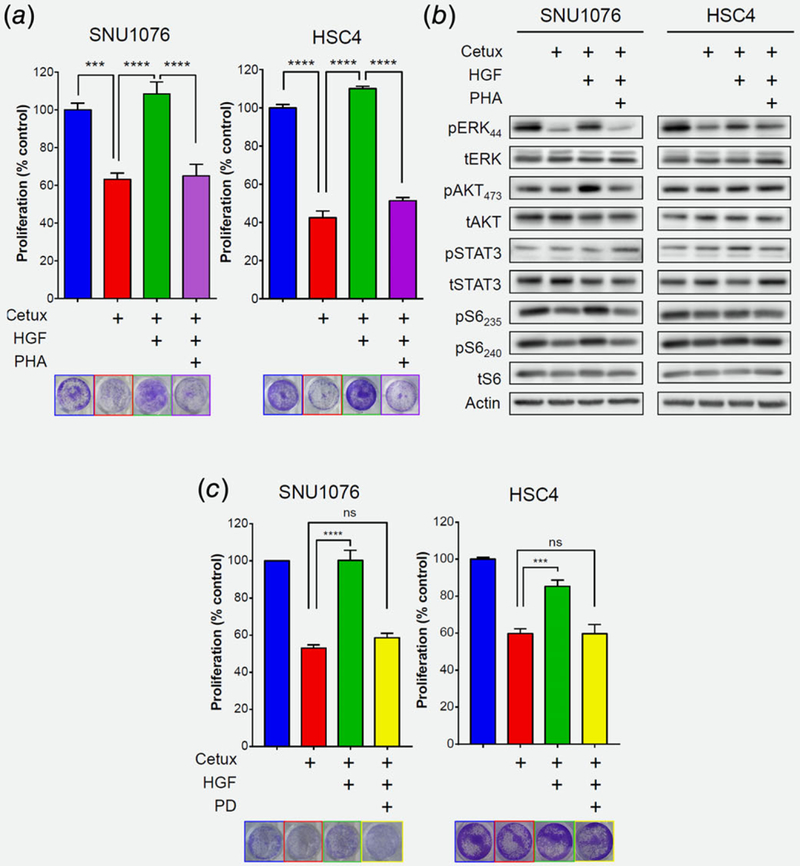
MET confers resistance predominantly through MAPK pathway activation. (a) 5-day proliferation assay testing cetuximab (12.5 μg/mL) efficacy in the SNU1076 and HSC4 HNSCC cell lines, with and without rHGF (50 ng/mL) and the MET inhibitor PHA-665752 (1 μM). Statistical significance was calculated from 3 to 5 independent experiments using one-way ANOVA test (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001). (b) Western blot for the indicated protein levels following 24 h treatment of cetuximab (12.5 μg/mL), with and without rHGF (50 ng/mL) and the MET inhibitor PHA-665752 (1 μM), in the SNU1076 and HSC4 HNSCC cell lines. Data represent a representative experiment (from three independent experiments) (c) 5-day proliferation assay testing cetuximab (12.5 μg/mL) efficacy in the SNU1076 and HSC4 HNSCC cell lines, with and without rHGF (50 ng/mL) and the MEK½ inhibitor PD-0325901 (25 nM). Statistical significance was calculated from three independent experiments using one-way ANOVA test (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
To explore whether MET activation is sufficient to confer resistance to cetuximab in vivo, we implanted the tumorigenic CAL33 cells into NOD.SCID mice. After the tumors developed, we assigned the mice randomly into 5 groups, each receiving a different treatment—vehicle, cetuximab, cetuximab with a local injection of rHGF, PHA-665752, or cetuximab + rHGF + PHA-665752. As expected, the CAL33 tumors were sensitive to cetuximab, as indicated by the arrest of tumor growth in 7 of 10 tumors that lasted for the 30 days of the experiment, while 3 of the 10 tumors regressed significantly. However, in the group of mice receiving a local injection of rHGF into the tumor site, the efficacy of cetuximab was partially limited, resulting in progression of all tumors (9 out of 9) and doubling of the average volume of the tumors (Fig. 4a, Figs. S4A and S4B, Supporting Information). However, blocking rHGF-induced MET activation with PHA-665752 abolished the effects of rHGF and restored the sensitivity to cetuximab, while a single-agent treatment with PHA-665752 had no effect on tumor growth (Fig. 4a, Figs. S4A and S4B, Supporting Information). In addition, IHC analysis of these tumors (6 days after of treatment) showed that local injection of rHGF enhanced tumor cell proliferation (KI67 staining) and re-activated MAPK (pERK) (Figs. 4b and 4c). To further support our in vivo results, we conducted a separate experiment in which we recapitulate the clinical scenario, using patient derived xenograft (PDX), and stimulating MET by supplementation of rHGF together with cetuximab, or after 21 days of chronic treatment with cetuximab. Local injection of rHGF resulted in increased tumor volume in both scenarios (Fig. 4d)
Figure 4.
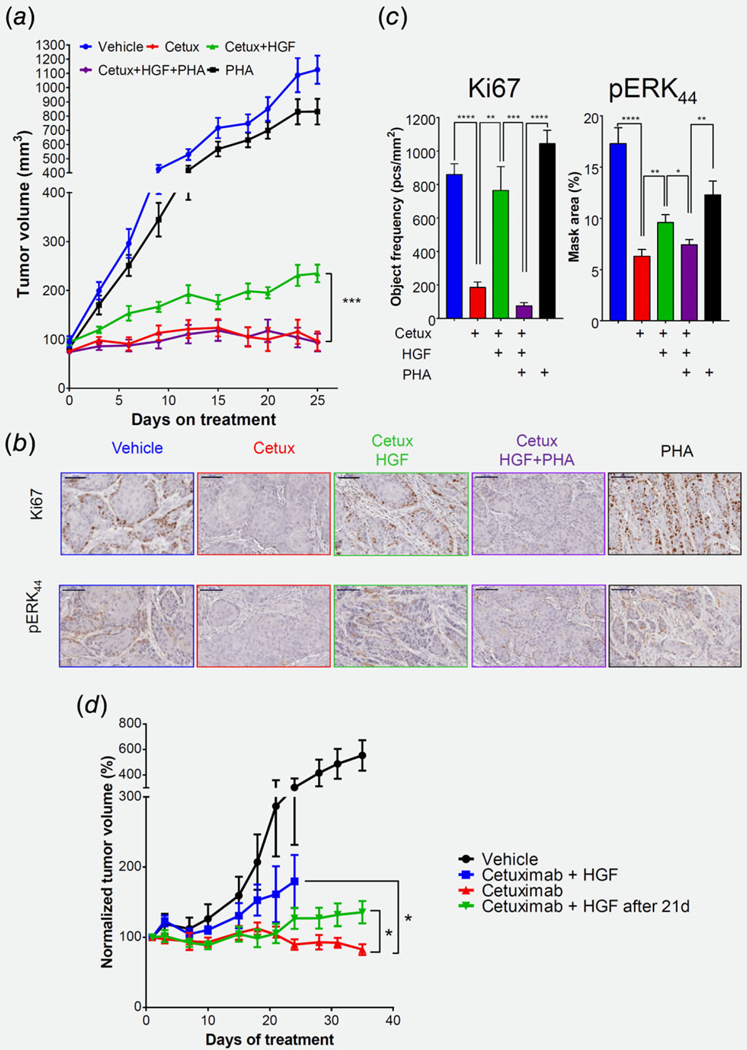
MET confers resistance to cetuximab in vivo via MAPK pathway activation. (a) Tumor volume of the CAL33 xerograph model in Nod. Scid mice. 2X106 CAL33 tumor cells were injected subcutaneously. Mice were randomized into 5 arms (n = 9–10): Mice were treated with cetuximab (10 mg/kg/5d) via intraperitoneal injection and/or MET inhibitor PHA-665752(30 mg/kg/d) by gavage and/or intratumoral injection of rHGF (20 μg/kg/2d). Statistical significance was calculated by permutation test (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001). (b) Representative pictures and (c) quantification of IHC staining for KI67 (left, 20X, 100 |^m) and pERK (right, 20X, 100 μm) after six days of treatments. (d) (left) Tumor volume of the PDX model in Nod.Scid mice. Tumor bearing mice were randomized into 4 arms (n = 9–10). Arm#1- Vehicle. Arm #2—cetuximab, Arm #3—cetuximab with rHGF, Arm #4—cetuximab for 21 days and then injection of rHGF. Statistical significance was calculated using permutation test (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001). (right) Change of tumor volume on day 31 compared to day 21.
MET/MAPK pathway activation abolishes cetuximab-induced HER2/HER3 upregulation
To determine how HGF limits the efficacy of cetuximab, we measured the expression levels of phospho-RTKs (using a Proteom Profiler™ protein array) in SNU1076 cells treated with DMSO, cetuximab, or cetuximab together with rHGF. As expected, we observed an increase of phosphorylated HER2 and HER3, which are known to be elevated in response to cetuximab in different types of cancer, including in HNSCC.24–29 However, in cetuximab-treated SNU1076 cells, MET activation reduced the phosphorylation levels of both HER2 and HER3 (Fig. S5A, Supporting Information). We then further characterized the effect of MET activation on HER2/HER3 expression and activation (by western blot analysis) in HSC4 and SNU1076 cell lines. The levels of phosphorylated HER2 and HER3 and the total levels of HER2 and HER3 together increased following cetuximab treatment, while stimulation of cells with rHGF reduced this activation and inhibition of MET enabled overexpression of HER2/HER3 (Fig. 5a). This reverse expression of HER2/HER3 and MET in HNSCC patients from the TCGA data was also confirmed (Fig. S5B, Supporting Information). To investigate the role of HER2/HER3 inhibition in enhancing the sensitivity to cetuximab and PHA600125, we tested the anti-tumor effects of lapatinib or afatinib in combination with cetuximab and cetuximab/PHA-665752. Five days proliferation assays showed that co-targeting of EGFR and HER2/HER3 with cetuximab and lapatinib or afanitib has potent anti-tumor activity, and triple treatment with PHA600125 has minimal superior effects. However, the importance of MET inhibition becomes critical when tumor cells are stimulated with rHGF, as activation of MET is sufficient to limit the sensitivity to the combinations of cetuximab/lapatinib and cetuximab/afatinib (Fig. S5C, Supporting Information).
Figure 5.
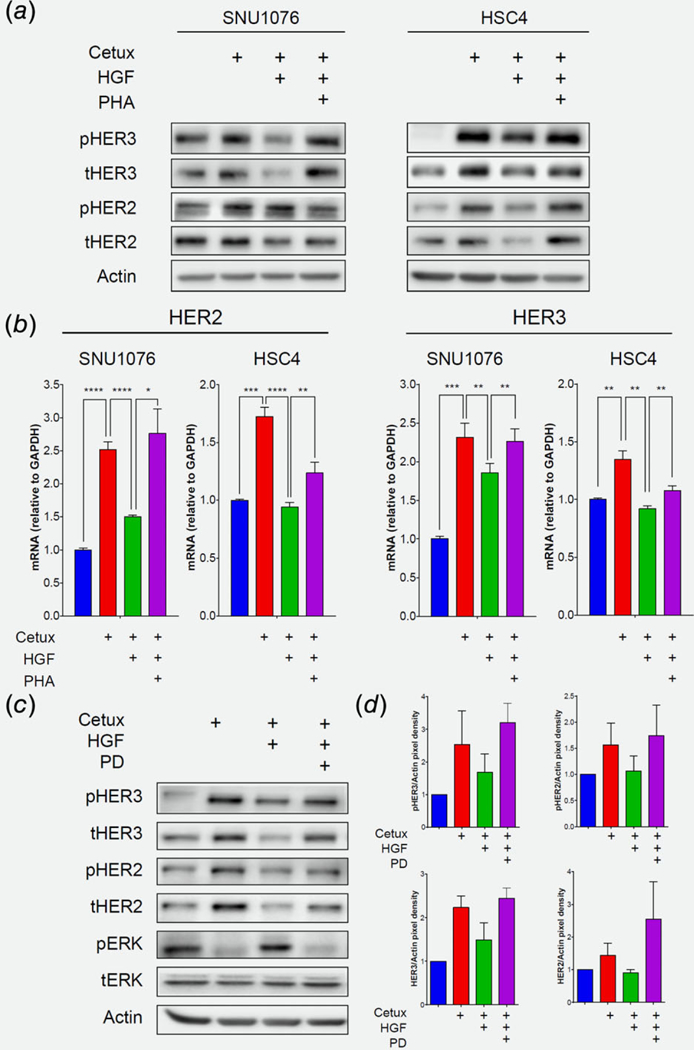
MET/MAPK pathway activation abolishes cetuximab-induced HER2/HER3 upregulation. (a) Western blot for the indicated protein levels fallowing 24 h treatment of cetuximab (12.5 μg/mL), with and without rHGF (50 ng/mL) and the MET inhibitor PHA-665752 (1 μM), in the SNU1076 and HSC4 HNSCC cell lines. Data represent a representative experiment (from three independent experiments). (b) mRNA levels of HER2 and HER3 following 24 h treatment of cetuximab (12.5 μg/mL), with and without rHGF (50 ng/mL) and the MET inhibitor PHA-665752 (1 μM), in the SNU1076 and HSC4 HNSCC cell lines. Statistical significance was calculated from 3 to 4 independent experiments using one-way ANOVA test (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001). (c) Western blot for the indicated protein levels following 24 h treatment of cetuximab (12.5 μg/mL), with and without rHGF (50 ng/mL) and the MEK½ inhibitor PD-0325901 (25 nM), in the SNU1076 HNSCC cell line. Data represent a representative experiment (from three independent experiments). (d) Densitometry of ERBBs normalized to actin.
To obtain further molecular insight into HER2 and HER3 expression in this setting, we determined the levels of their mRNAs (by qPCR). Our data indicated that 24 h of cetuximab treatment increased the expression levels of HER2 and HER3, and stimulation with rHGF prevented this induction of transcription (Fig. 5b). Blocking MET restored, at least in part, the upregulation of HER2 and HER3, thereby highlighting the role of MET in the transcriptional regulation of these genes in response to cetuximab. Lastly, inhibition of MEK½ reinstated MET blockage and hence allowed HER2/HER3 upregulation, indicating that the MET/MAPK pathway regulates this feedback loop (Fig. 5c and Fig. S5D, Supporting Information). Notably, blocking AKT using MK2206 was also sufficient to induce HER2/HER3 expression (Fig. S5E, Supporting Information). However, it is known that AKT inhibition increases HER2/HER3 expression via FOXO transcription factor,30,31,55 and given the fact that cetuximab has s minimal effect on AKT pathway inhibition, the overall data supports that MAPK activation downstream of MET plays the key role in determine HER2/HER3 expression.
EHF regulates HER2 and HER3 gene expression following cetuximab treatment
Next, we investigated the transcriptional machinery that regulates the increased HER2/HER3 expression following treatment with cetuximab. There are several TFs known to regulate HER2/HER3 expression in cancer, such as AP-2,56 EHF,57,58 FOXO3,30,31,59 FOXO131 and ZNF217.60 To prioritize these TF candidates for validation, we first explored the correlation between each of them with HER2 and HER3 expression in the HNSCC datasets of the TCGA Data Portal. For both HER2 and HER3, the strongest correlation was found with EHF and FOXO3 (Fig. 6a and Fig. S6A, Supporting Information). We then tested the role of these two TFs in regulating HER2/HER3 expression in response to cetuximab, by knocking down each TF in t SNU1076 and HSC4 cell lines using siRNA. Knockdown of EHF prevented HER2 and HER3 upregulation in response to cetuximab in these cells (Fig. 6b). In contrast, knockdown of FOXO3a did not prevent the upregulation of HER2/HER3 upon treatment with cetuximab (Figs. S6B and S6C, Supporting Information). In addition, we confirmed the role of EHF in regulating HER2 and HER3 protein expression and activation following cetuximab treatment in SNU1076 cells, as knockdown of EHF was sufficient to reduce HER2/HER3 upregulation and HER3 activation after EGFR blockade (Figs. S6C and S6D, Supporting Information). Notably, this mechanism seems not to be specific for HNSCC, as EHF regulated HER2/HER3 expression also in the lung cancer cell line H1975 (Fig. S6E, Supporting Information).
Figure 6.
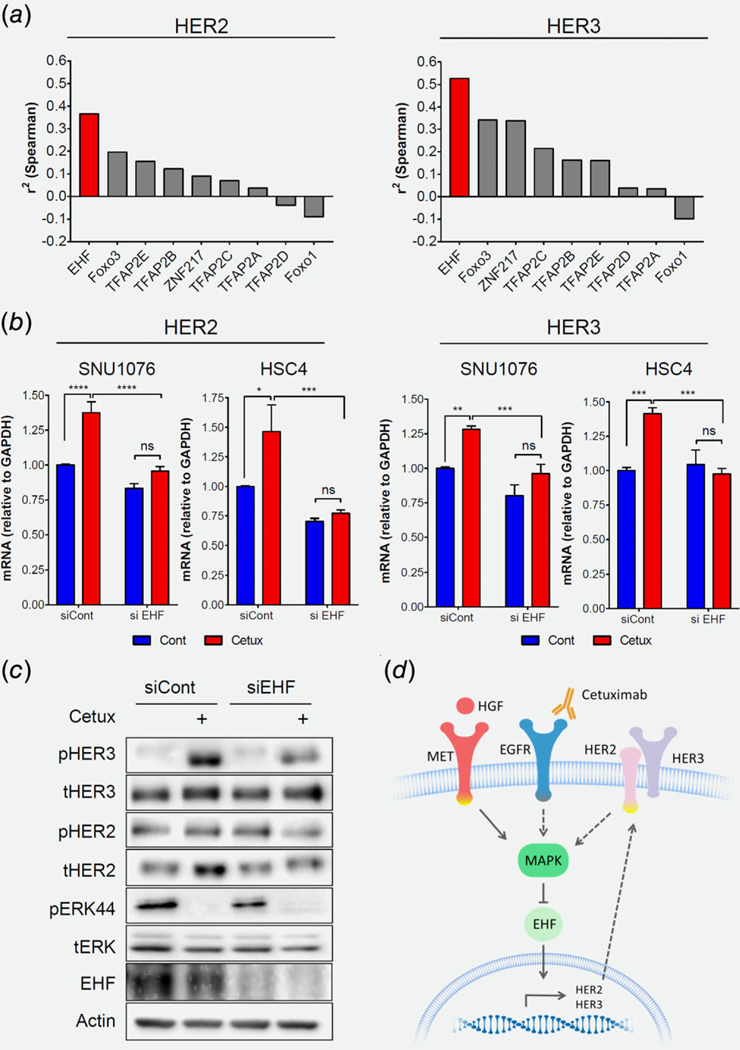
EHF regulates HER2 and HER3 gene expression following cetuximab treatment. (a) Correlation between TF candidates with HER2 and HER3 expression in the HNSCC datasets of TCGA. (b) mRNA levels of HER2 and HER3 following 24 h treatment of cetuximab (12.5 μg/mL), with either siCont or siEHF. Statistical significance was calculated from three independent experiments using one-way ANOVA (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001). (c) Western blot for the indicated protein levels following 24 h treatment of cetuximab (12.5 1 μg/mL), with either siCont or siEHF. Data represent a representative experiment (from two independent experiments). (d) Scheme summarizing the suggested model: MET bypasses cetuximab inhibition by reactivating the MAPK pathway. MET/MAPK activation abolishes HER2 and HER3, EHF dependence, and upregulation.
Discussion
In this work, we show that MET confers resistance via reactivation of MAPK pathway and that MET/MAPK activation abolishes the compensatory feedback loop of HER2/HER3. In addition, we identified EHF as the TF that regulates the expression of HER2/HER3 in response to cetuximab.
For HNSCC patients with advanced local or metastatic disease, the standard of care is treatment with cetuximab in combination with radiation or chemotherapy, but in most cases the patients do not respond or experience rapid acquisition of resistance to therapy.4 Unlike the situation for colorectal and lung cancers, for which biomarkers for treatment with cetuximab are available,61,62 the treatment regime for HNSCC patients is not determined on the basis of molecular determinants of response. The ‘discovery’ of such biomarkers can be facilitated by exploring the molecular mechanism/s of acquisition of resistance to cetuximab in HNSCC. While we show a single case of MET amplification as a resistance mechanism, our laboratory data suggest that MET amplification or over-expression can serve as a biomarker of response to cetuximab. Because copy number gain of MET in HNSCC is rare (1–2% TCGA cohort), this mechanism can explain only a small subgroup of the innate resistance cases to cetuximab. Nevertheless, MET/HGF may play a role in a larger portion of cases that acquired resistance to cetuximab.
MET amplification, over-expression and activation are well-described resistance mechanisms to anti-EGFR therapies in lung and colorectal cancers.39,41,63,64 In preclinical HNSCC models, MET expression and activation have been shown to be associated with resistance to anti-EGFR,34,36–38 and Wheeler et al demonstrated that MET overexpression was detected in HNSCC cell lines that acquired resistant to cetuximab in vitro (24). Moreover, a single retrospective study in HNSCC patients demonstrated the association between MET levels and poor response to cetuximab.38 Here, we report a clinical observation that MET amplification and overexpression is associated with progression to cetuximab therapy, and potentially plays a causative role in the development of resistance to cetuximab in HNSCC.
Activation of MET signaling is known to occur by ligand-dependent (stimulation of MET by HGF) or ligand-independent (genetic abnormalities, such as MET amplification and activating mutations) mechanisms.33 In HNSCC, MET is expressed by tumor cells,48 while HGF is secreted by stromal cells, mainly tumor-associated fibroblasts.34,37 In agreement with this accumulated knowledge, we show that acquisition of resistance to cetuximab is associated with both MET gene amplification in tumor cells and HGF expression by stromal cells (Fig. 2). The elevation of the MET receptor and its ligand HGF support our premise the possibility that the MET/HGF axis mediates the resistance to cetuximab. MET pathway activation facilitates the growth, survival and motility of tumor cells through stimulating downstream signaling of MAPK, STAT, SRC and PI3K-AKT.33,37 MAPK is a common pathway reported to be re-activated following MET/HGF-induced drug resistance.39,40 In HNSCC, the MET/HGF axis determines the activation of the SRC, AKT and MAPK pathways and thus affects the sensitivity of tumor cell lines to anti-EGFR therapies.34,36,65 In this study, we observed MET-induced MAPK re-activation in all tested HNSCC models. Blocking of MAPK with an MEK inhibitor resensitized the rHGF-stimulated tumor cells to cetuximab (Fig. 3).
It has previously been shown that upregulation and activation of HER2 and HER3 described as a mechanism of resistance to anti-EGFR therapies in HNSCC, colon and lung cancers.24–29 In HNSCC cell lines, chronic treatment with cetuximab induced HER2/HER3 overexpression, and cotargeting of EGFR and HER2 or HER3 resensitized the cells to cetuximab. This compensatory feedback loop provided the rationale for treating the patient discussed in this work with LJM716, an anti-HER3 antibody,66 which in retrospect was not effective. When we tested the effect of MET activation on the signaling of the tumor cells following treatment with cetuximab, we noticed that the expression of the compensatory feedback loop of HER2/HER3 expression was impaired. This finding may explain the minimal upregulation of HER2 and HER3 expression in the cetuximab-acquired resistant lesion. Moreover, CetuxRes tumor showed no response to drug combination of LJM716 and cetuximab. This lack of response can be explain by MET activation, as blockage of MET receptor become crucial only when HGF/MET pathway is active. These results may also explain why MET suppression showed poor clinical outcome in patients, as MET inhibition will improve anti-tumor effects only in a subset of HNSCC patients where the MET pathway is active, while HER2/HER3 inhibition is required for preventing tumor progression.
An important finding in this study was also the cross-talk between MET and HER2/HER3 expression in response to cetuximab, which has not been described previously. We showed that MET activation limits the cetuximab-induced HER2/HER3 mRNA levels. We further identified that EHF is the TF that regulates HER2/HER3 expression following cetuximab administration. EHF was recently described to regulate HER3 levels in thyroid57 and gastric cancer58 and highlights its role in tumor progression.67 However, here we also show that EHF plays a key role in the resistance to cetuximab by mediating the HER2/HER3 compensatory feedback loop. Alongside the elucidation of the above-described mechanism of HER2/HER3 regulation in response to cetuximab in HNSCC, we posit that the role played by EHF in the cetuximab-induced expression of HER2/HER3 in HNSCC cell lines may also apply in lung or colon cancer.
In summary, we present evidence that the MET pathway constitutes a clinically relevant resistance mechanism to cetuximab, and the development of MET inhibitors may be the way forward as a therapeutic strategy for selected patients who develop resistance to cetuximab via the MET/HGF pathway. The lack of selection of such patients in clinical trials combining cetuximab and anti-MET therapies may explain the limited efficacy of this combination therapy in HNSCC68 (reviewed in Ref. 37). Collectively, our findings underscore that the evaluation of MET, HER2 and HER3 adaptive response to cetuximab in HNSCC could provide the rationale for successive lines of treatment with FDA-approved agents.
Supplementary Material
What’s new?
Resistance to cetuximab is a major obstacle in the treatment of patients with head and neck squamous cell carcinoma (HNSCC), though the underlying mechanisms of resistance remain unclear. In the present study, analyses of tumor samples from an HNSCC patient with exceptional clinical response to cetuximab monotherapy implicate MET pathway activation as a causative factor in acquired cetuximab resistance. Experiments in vitro and in vivo show that MET confers resistance to cetuximab via activation of the MAPK pathway. MET stimulation further limits cetuximab-induced HER2/HER3 overexpression, highlighting the importance of HER2/HER3 and MET evaluation when monitoring patient reponse to cetuximab.
Acknowledgements
We would like to acknowledge the Memorial Sloan Kettering Cancer Center (MSKCC) Molecular Cytology Core Facility for the IHC staining of the human sample. Memorial Sloan Kettering Cancer Center (MSKCC) Genomics Core Laboratory for performing and analyzing the RNA-seq experiments. This work was funded by the Israel Cancer Association (ICA, 20170024) (to M.E.), the Israel Cancer Research Foundation (ICRF, 17–1693-RCDA) (to M.E), Israel Science Foundation (ISF, 700/16) (to M.- E.), United State - Israel Binational Science foundation (BSF, 2017323) (to M.E, J.B, M.S), and the National Institutes of Health (P30CA008748) (to J.B and M.S.). Fellowship: Alon fellowship to M.E, and Biotech BGU fellow to O.N.
Grant sponsor: Israel Cancer Research Foundation; Grant number: 17–1693-RCDA; Grant sponsor: United State - Israel Binational Science foundation; Grant number: 2017323; Grant sponsor: National Institutes of Health; Grant number: P30CA008748; Grant sponsor: Israel Science Foundation; Grant number: 700/16; Grant sponsor: Israel Cancer Association; Grant number: 20170024
F.C was NantOmics employee. J.B. is on the Board of Directors of Foghorn and is a past board member of Varian Medical Systems, Bristol-Myers Squibb, Grail, Aura Biosciences and Infinity Pharmaceuticals. He has performed consulting and/or advisory work for Grail, PMV Pharma, ApoGen, Juno, Lilly, Seragon, Novartis, and Northern Biologics. He has stock or other ownership interests in PMV Pharma, Grail, Juno, Varian, Foghorn, Aura, Infinity Pharmaceuticals, ApoGen, as well as Tango and Venthera, for which is a cofounder. He has previously received Honoraria or Travel Expenses from Roche, Novartis, and Eli Lilly. M.S. is in the Advisory Board of Bioscience Institute, received research funds from Puma Biotechnology, Daiichi-Sankio, Targimmune, Immunomedics and Menarini Ricerche, is a co-founder of Medendi Medical Travel and in the past two years he received honoraria from Menarini Ricerche and ADC Pharma. M.F is an employee and stock holder of Regeneron Pharmaceuticals.
Abbreviations:
- HNSCC
head and neck squamous cell carcinoma
- CetuxSen
cetuximab-sensitive
- CetuxRes
cetuximab-resistant
- EHF
ETS homologous transcription factor
- RTK
receptor tyrosine kinase
- EGFR
epithelial growth factor receptor
- HER2/HER3
human epidermal growth factor receptors 2 and 3
- HGF
hepatocyte growth factor
- rHGF
recombinant HGF
- MS
Targeted laser-microdissecting-mass spectrometry
- IHC
Immunohistochemistry
Footnotes
Data and materials availability
All data needed to evaluate the conclusions in the paper are present in the paper or the Supporting Information Materials.
Additional Supporting Information may be found in the online version of this article.
Conflict of interests: All other authors declare that they have no conflict of interests.
References
- 1.Wiegand S, Zimmermann A, Wilhelm T, et al. Survival after distant metastasis in head and Neck Cancer. Anticancer Res 2015;35:5499–502. [PubMed] [Google Scholar]
- 2.Trotti A, Bellm LA, Epstein JB, et al. Mucositis incidence, severity and associated outcomes in patients with head and neck cancer receiving radiotherapy with or without chemotherapy: a systematic literature review. Radiother Oncol 2003;66:253–62. [DOI] [PubMed] [Google Scholar]
- 3.Bonner JA, Harari PM, Giralt J, et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N Engl J Med 2006;354:567–78. [DOI] [PubMed] [Google Scholar]
- 4.Vermorken JB, Mesia R, Rivera F, et al. Platinum-based chemotherapy plus cetuximab in head and neck cancer. N Engl J Med 2008;359:1116–27. [DOI] [PubMed] [Google Scholar]
- 5.Vermorken JB, Trigo J, Hitt R, et al. Open-label, uncontrolled, multicenter phase II study to evaluate the efficacy and toxicity of cetuximab as a single agent in patients with recurrent and/or metastatic squamous cell carcinoma of the head and neck who failed to respond to platinum-based therapy. J Clin Oncol 2007;25:2171–7. [DOI] [PubMed] [Google Scholar]
- 6.Ciardiello F, Tortora G. EGFR antagonists in cancer treatment. N Engl J Med 2008;358:1160–74. [DOI] [PubMed] [Google Scholar]
- 7.The YY. EGFR family and its ligands in human cancer. Signalling mechanisms and therapeutic opportunities. European journal of cancer 2001;37 (Suppl 4):S3–8. [DOI] [PubMed] [Google Scholar]
- 8.Normanno N, De Luca A, Bianco C, et al. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene 2006;366:2–16. [DOI] [PubMed] [Google Scholar]
- 9.Yarden Y, Shilo BZ. SnapShot: EGFR signaling pathway. Cell 2007;131:1018. [DOI] [PubMed] [Google Scholar]
- 10.Harris TJ, McCormick F. The molecular pathology of cancer. Nat Rev Clin Oncol 2010;7:251–65. [DOI] [PubMed] [Google Scholar]
- 11.Scaltriti M, Baselga J. The epidermal growth factor receptor pathway: a model for targeted therapy. Clin Cancer Res 2006;12:5268–72. [DOI] [PubMed] [Google Scholar]
- 12.Hitt R, Irigoyen A, Cortes-Funes H, et al. Phase II study of the combination of cetuximab and weekly paclitaxel in the first-line treatment of patients with recurrent and/or metastatic squamous cell carcinoma of head and neck. Ann Oncol 2012;23:1016–22. [DOI] [PubMed] [Google Scholar]
- 13.Leemans CR, Braakhuis BJ, Brakenhoff RH. The molecular biology of head and neck cancer. Nat Rev Cancer 2011;11:9–22. [DOI] [PubMed] [Google Scholar]
- 14.Ang KK, Berkey BA, Tu X, et al. Impact of epidermal growth factor receptor expression on survival and pattern of relapse in patients with advanced head and neck carcinoma. Cancer Res 2002;62:7350–6. [PubMed] [Google Scholar]
- 15.Mendelsohn J The epidermal growth factor receptor as a target for cancer therapy. Endocr Relat Cancer 2001;8:3–9. [DOI] [PubMed] [Google Scholar]
- 16.Tinhofer I, Klinghammer K, Weichert W, et al. Expression of amphiregulin and EGFRvIII affect outcome of patients with squamous cell carcinoma of the head and neck receiving cetuximab-docetaxel treatment. Clin Cancer Res 2011;17: 5197–204. [DOI] [PubMed] [Google Scholar]
- 17.Sok JC, Coppelli FM, Thomas SM, et al. Mutant epidermal growth factor receptor (EGFRvIII) contributes to head and neck cancer growth and resistance to EGFR targeting. Clin Cancer Res 2006;12:5064–73. [DOI] [PubMed] [Google Scholar]
- 18.Arena S, Bellosillo B, Siravegna G, et al. Emergence of multiple EGFR extracellular mutations during Cetuximab treatment in colorectal Cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 2015; 21:2157–66. [DOI] [PubMed] [Google Scholar]
- 19.Pietrantonio F, Vernieri C, Siravegna G, et al. Heterogeneity of acquired resistance to anti-EGFR monoclonal antibodies in patients with metastatic colorectal cancer. Clin Cancer Res 2017;23: 2414–2422. [DOI] [PubMed] [Google Scholar]
- 20.Bertotti A, Papp E, Jones S, et al. The genomic landscape of response to EGFR blockade in colorectal cancer. Nature 2015;526:263–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bossi P, Bergamini C, Siano M, et al. Functional genomics uncover the biology behind the responsiveness of head and Neck squamous cell Cancer patients to Cetuximab. Clin Cancer Res 2016;22: 3961–70. [DOI] [PubMed] [Google Scholar]
- 22.Iida M, Brand TM, Starr MM, et al. Overcoming acquired resistance to cetuximab by dual targeting HER family receptors with antibody-based therapy. Mol Cancer 2014;13:242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brand TM, Iida M, Wheeler DL. Molecular mechanisms of resistance to the EGFR monoclonal antibody cetuximab. Cancer Biol Ther 2011;11: 777–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wheeler DL, Huang S, Kruser TJ, et al. Mechanisms of acquired resistance to cetuximab: role of HER (ErbB) family members. Oncogene 2008;27:3944–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang D, Qian G, Zhang H, et al. Her3 targeting sensitizes hnscc to cetuximab by reducing her3 activity and her2/her3 dimerization: evidence from cell line and patient-derived xenograft models. Clin Cancer Res 2017;23:677–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leonard B, Brand TM, O’Keefe RA, et al. BET inhibition overcomes receptor tyrosine kinasemediated cetuximab resistance in HNSCC. Cancer research 2018;78:4331–4343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sergina NV, Rausch M, Wang D, et al. Escape from HER-family tyrosine kinase inhibitor therapy by the kinase-inactive HER3. Nature 2007; 445:437–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yonesaka K, Zejnullahu K, Okamoto I, et al. Activation of ERBB2 signaling causes resistance to the EGFR-directed therapeutic antibody cetuximab. Sci Transl Med 2011;3:99ra86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou B-BS, Peyton M, He B, et al. Targeting ADAM-mediated ligand cleavage to inhibit HER3 and EGFR pathways in non-small cell lung cancer. Cancer Cell 2006;10:39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chandarlapaty S, Sawai A, Scaltriti M, et al. AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell 2011;19:58–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chakrabarty A, Sanchez V, Kuba MG, et al. Feedback upregulation of HER3 (ErbB3) expression and activity attenuates antitumor effect of PI3K inhibitors. Proc Natl Acad Sci USA 2012;109: 2718–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brand TM, Iida M, Stein AP, et al. AXL mediates resistance to cetuximab therapy. Cancer Res 2014; 74:5152–64. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 33.Gherardi E, Birchmeier W, Birchmeier C, et al. Targeting MET in cancer: rationale and progress. Nat Rev Cancer 2012;12:89–103. [DOI] [PubMed] [Google Scholar]
- 34.Knowles LM, Stabile LP, Egloff AM, et al. HGF and c-met participate in paracrine tumorigenic pathways in head and neck squamous cell cancer. Clin Cancer Res 2009;15:3740–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krumbach R, Schuler J, Hofmann M, et al. Primary resistance to cetuximab in a panel of patient-derived tumour xenograft models: activation of MET as one mechanism for drug resistance. Eur J Cancer 2011;47:1231–43. [DOI] [PubMed] [Google Scholar]
- 36.Stabile LP, He G, Lui VW, et al. C-Src activation mediates erlotinib resistance in head and neck cancer by stimulating c-met. Clin Cancer Res 2013;19:380–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hartmann S, Bhola NE, Grandis JR. HGF/met signaling in head and Neck Cancer: impact on the tumor microenvironment. Clin Cancer Res 2016;22:4005–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Madoz-Gurpide J, Zazo S, Chamizo C, et al. Activation of MET pathway predicts poor outcome to cetuximab in patients with recurrent or metastatic head and neck cancer. J Transl Med 2015;13:282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Engelman JA, Zejnullahu K, Mitsudomi T, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007;316:1039–43. [DOI] [PubMed] [Google Scholar]
- 40.Straussman R, Morikawa T, Shee K, et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature 2012;487:500–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bardelli A, Corso S, Bertotti A, et al. Amplification of the MET receptor drives resistance to anti- EGFR therapies in colorectal cancer. Cancer Dis-cov 2013;3:658–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hembrough T, Thyparambil S, Liao WL, et al. Selected reaction monitoring (SRM) analysis of epidermal growth factor receptor (EGFR) in formalin fixed tumor tissue. Clin Proteomics 2012;9:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Catenacci DV, Liao W-L, Thyparambil S, et al. Absolute quantitation of met using mass spectrometry for clinical application: assay precision, stability, and correlation with MET gene amplification in FFPE tumor tissue. PloS one 2014;9:e100586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Giordano TJ, Shedden KA, Schwartz DR, et al. Organ-specific molecular classification of primary lung, colon, and ovarian adenocarcinomas using gene expression profiles. Am J Pathol 2001;159:1231–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kwak EL, Ahronian LG, Siravegna G, et al. Molecular heterogeneity and receptor Coamplification drive resistance to targeted therapy in MET-amplified Esophagogastric Cancer. Cancer Discov 2015;5:1271–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Misale S, Di Nicolantonio F, Sartore-Bianchi A, et al. Resistance to anti-EGFR therapy in colorectal cancer: from heterogeneity to convergent evolution. Cancer Discov 2014;4:1269–80. [DOI] [PubMed] [Google Scholar]
- 48.Seiwert TY, Jagadeeswaran R, Faoro L, et al. The MET receptor tyrosine kinase is a potential novel therapeutic target for head and neck squamous cell carcinoma. Cancer Res 2009;69:3021–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Siravegna G, Mussolin B, Buscarino M, et al. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat Med 2015;21:795–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hembrough T, Thyparambil S, Liao WL, et al. Application of selected reaction monitoring for multiplex quantification of clinically validated biomarkers in formalin-fixed, paraffin-embedded tumor tissue. J Mol Diagn 2013;15:454–65. [DOI] [PubMed] [Google Scholar]
- 51.Nuciforo P, Thyparambil S, Aura C, et al. High HER2 protein levels correlate with increased survival in breast cancer patients treated with anti- HER2 therapy. Mol Oncol 2016;10:138–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Eder JP, Vande Woude GF, Boerner SA, et al. Novel therapeutic inhibitors of the c-met signaling pathway in cancer. Clin CancerRes 2009;15:2207–14. [DOI] [PubMed] [Google Scholar]
- 53.Ponzetto C, Bardelli A, Zhen Z, et al. A multifunctional docking site mediates signaling and transformation by the hepatocyte growth factor/scatter factor receptor family. Cell 1994;77:261–71. [DOI] [PubMed] [Google Scholar]
- 54.Elkabets M, Pazarentzos E, Juric D, et al. AXL mediates resistance to PI3Kalpha inhibition by activating the EGFR/PKC/mTOR axis in head and neck and esophageal squamous cell carcinomas. Cancer Cell 2015;27:533–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Muranen T, Selfors LM, Worster DT, et al. Inhibition of PI3K/mTOR leads to adaptive resistance in matrix-attached cancer cells. Cancer Cell 2012; 21:227–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhu CH, Huang Y, Oberley LW, et al. A family of AP-2 proteins down-regulate manganese superoxide dismutase expression. J Biol Chem 2001;276: 14407–13. [DOI] [PubMed] [Google Scholar]
- 57.Lv Y, Sui F, Ma J, et al. Increased expression of EHF contributes to thyroid tumorigenesis through transcriptionally regulating HER2 and HER3. Oncotarget 2016;7:57978–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shi J, Qu Y, Li X, et al. Increased expression of EHF via gene amplification contributes to the activation of HER family signaling and associates with poor survival in gastric cancer. Cell Death Dis 2016;7:e2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Garrett JT, Olivares MG, Rinehart C, et al. Transcriptional and posttranslational up-regulation of HER3 (ErbB3) compensates for inhibition of the HER2 tyrosine kinase. Proc Natl Acad Sci 2011; 108:5021–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Krig SR, Miller JK, Frietze S, et al. ZNF217, a candidate breast cancer oncogene amplified at 20q13, regulates expression of the ErbB3 receptor tyrosine kinase in breast cancer cells. Oncogene 2010;29:5500–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lievre A, Bachet JB, Le Corre D, et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res 2006;66:3992–5. [DOI] [PubMed] [Google Scholar]
- 62.Pirker R, Pereira JR, von Pawel J, et al. EGFR expression as a predictor of survival for first-line chemotherapy plus cetuximab in patients with advanced non-small-cell lung cancer: analysis of data from the phase 3 FLEX study. Lancet Oncol 2012;13:33–42. [DOI] [PubMed] [Google Scholar]
- 63.Diaz LA Jr, Williams RT, Wu J, et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature 2012;486:537–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sequist LV, Waltman BA, Dias-Santagata D, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Science translational medicine 2011;3:75ra26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xu H, Stabile LP, Gubish CT, et al. Dual blockade of EGFR and c-met abrogates redundant signaling and proliferation in head and neck carcinoma cells. Clin Cancer Res 2011;17:4425–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Garner AP, Bialucha CU, Sprague ER, et al. An antibody that locks HER3 in the inactive conformation inhibits tumor growth driven by HER2 or neuregulin. Cancer Res 2013;73:6024–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Brenne K, Nymoen DA, Hetland TE, et al. Expression of the Ets transcription factor EHF in serous ovarian carcinoma effusions is a marker of poor survival. Hum Pathol 2012;43:496–505. [DOI] [PubMed] [Google Scholar]
- 68.Seiwert T, Sarantopoulos J, Kallender H, et al. Phase II trial of single-agent foretinib (GSK1363089) in patients with recurrent or metastatic squamous cell carcinoma of the head and neck. Invest New Drugs 2013;31:417–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.