

Abstract
Mechanistic studies on the effects of MeHg in the central nervous system (CNS) have been limited to morphology, substrate uptake and macromolecular synthesis, differentiation, and changes in gene expression during development and adulthood, but its primary site of action has yet to be identified. Proper functioning of the nitric oxide synthase (NOS)-cyclic GMP and the cyclooxygenase (COX)-prostaglandin (PG) signaling pathways in the CNS depend on post-translational modifications of key enzymes by chaperone proteins. The ability of MeHg to alter or inhibit chaperone-client protein interactions is hitherto unexplored, and potentially offers an upstream unifying mechanism for the plethora of MeHg effects, ranging from reactive species generation (ROS) generation, mitochondrial dysfunction, changes in redox potential, macromolecule synthesis, and cell swelling. In view of the prominent function of astrocytes in the maintenance of the extracellular milieu and their critical role in mediating MeHg neurotoxicity, they afford a relevant and well-established experimental model. The present review is predicated on (a) the remarkable affinity of mercurials for the anionic form of sulfhydryl (-SH) groups, (b) the essential role of thiols in protein biochemistry, and (c) the role of molecular chaperone proteins, such as heat shock protein 90 (Hsp90) in the regulation of protein redox status by facilitating the formation and breakage of disulfide bridges. We offer potential sites where MeHg may interfere with cellular homeostasis and advance a novel mechanistic model for MeHg-induced neurotoxicity.
INTRODUCTION
Methylmercury: A Ubiquitous Environmental Threat
Mercury (Hg) is a global pollutant which knows no environmental boundaries. Even the most stringent control of man-made sources of mercury pollution will not eliminate human exposure to potentially toxic quantities, given its ubiquitous presence in the environment. Environmental exposure to mercury occurs primarily via the food chain due to accumulation of MeHg in fish. Latest statistics in the US indicate that 46 states have fish consumption advisories covering 40% of the nation’s rivers, lakes and streams. In addition, mercury is a common pollutant in hazardous waste sites in the nation (EPA 2001) [1]. It is estimated that 3–4 million children live within one mile of at least one of the 1,300+ active hazardous waste sites in the USA [1].
The effects of prenatal MeHg exposure associated with maternal fish consumption on intellectual function in children have been investigated in two large, prospective, longitudinal studies; one in the Seychelles Islands [2] and the other in the Faroes Islands [3]. A National Academy of Sciences (NAS) expert panel reviewed the studies, concluding that the weight of the evidence supported MeHg’s adverse health effects [4] and recommended that levels of mercury not exceed 5.0 μg/L in whole blood or 1.0 μg/g in hair, corresponding to a reference dose (RfD) of 0.1 μg/kg body weight/day.
Evidence that Astrocytes Mediate MeHg-Induced Neuro-Toxicity
Astrocytes are the most numerous non-neuronal cell type in the CNS. They make up ~50% of human brain volume [5]. Astrocytes perform several functions that are essential for normal neuronal activity, including glutamate uptake (80% of synaptic glutamate), glutamate release, K+ and H+ buffering, and water transport [6]. Interactions between axons and astrocytes (radial glia) furnish the cues for axon guidance of migrating neurons. Understanding of the molecular signals that regulate neuron-glia interactions has increased greatly with the advent of molecular and cellular biological techniques and genetically modified mice. Cell ablation studies using genetically targeted ectopic gene expression and gene knockout with single cell specificity have established the distinct roles played by astrocytes during development [7].
There is abundant evidence in support of the pivotal role of astrocytes in mediating neurotoxicity, establishing astrocytes as a unique and relevant experimental model for the assessment of mechanisms underlying MeHg-induced cytotoxicity.
Chronic exposure to MeHg in primates is associated with preferential accumulation of MeHg in astrocytes (and to some extent in microglia) [8–10].
MeHg inhibits astrocytic glutamate uptake and stimulates its efflux [11–15], increasing in vivo glutamate concentrations in the extra-cellular fluid (ECF) and sensitizing neurons to excitotoxic injury [16]. Central nervous system damage associated with MeHg correlates spatially with brain areas that have dense glutamatergic innervation [17]. The ionotropic glutamate receptor N-methyl-D-aspartate (NMDA) antagonist, dizocilpine (MK801), protects against neuronal damage induced by in vivo MeHg [17–19].
MeHg selectively inhibits astrocytic (but not neuronal) uptake systems for cystine and cysteine transport [20–22] compromising glutathione (GSH) synthesis and the CNS redox potential [13]. Compared with astrocytes, neurons have lower levels of GSH and of a second putative antioxidant, metallothionein (MT) [23,24], making them more susceptible to the effects of increased ROS. Since GSH synthesis is dependent upon precursors derived from astrocytes [25, 26], MeHg-induced inhibition of cystine transport and astrocytic GSH production would ultimately lead to decreased neuronal GSH levels and increased glutamate toxicity.
MeHg-induced ROS formation in astrocytes can be attenuated by antioxidants [27], reversing its functional effects on glutamate uptake inhibition [14]. Most recently, we have established MeHg-induced ROS generation using measurements of the sensitive lipid peroxidation biomarkers, isoprostanes [28].
Co-application of non-toxic concentrations of mercury with glutamate results in the appearance of typical neuronal lesions associated with excitotoxic stimulation [29].
In human and non-human primates chronic in vivo exposure to MeHg is associated with swelling of astrocytes [8, 9, 30, 31], a process associated with release of glutamate into the extracellular fluid [32].
In the absence of glutamate, neurons are unaffected by exposure to mercury, suggesting that neuronal dysfunction is secondary to disturbances in astrocytes [33].
Additional data invoke astrocytic cytosolic phospholipase A2 (cPLA2) as a target for MeHg toxicity, supporting the notion that cPLA2-stimulated hydrolysis and release of arachidonic acid (AA) play a role in MeHg-induced neurotoxicity [27].
MERCURY AND THIOLS
Thiol groups play fundamental structural and functional roles in protein chemistry, being located mainly within the active catalytic sites of many enzymes. Mercury compounds react specifically with active sulfhydryl (-SH) groups forming complexes of defined stoichiometry, -S-Hg-R. The high affinity of MeHg for the anionic form of -SH groups is responsible for the toxicological behavior of this compound [34, 35]. The affinity of MeHg for the anionic form of -SH groups (log K, where K is the association constant) is extremely high, on the order of 15–23, whereas its affinity constants for oxygen-, chloride-, or nitrogen-containing ligands such as carboxyl or amino groups are about 10 orders of magnitude lower [36]. Indeed, wherever a MeHg compound has been identified in biological media, it has been complexed to -SH-containing ligands. Complexes with cysteine and GSH have been identified in blood [37, 38], and complexes with GSH have been identified in brain [39], liver [40], and bile [41]. Armed with this knowledge, a search for thiol-containing proteins present a number of intriguing molecular targets that might explain the vast array of cellular disruptions caused by MeHg exposure. The class of proteins known as molecular chaperones or heat shock proteins represents one attractive target.
Hsp90
The 90-kDa heat shock protein (Hsp90) is one of the most abundant proteins in cells, constituting 1–2% of total intracellular protein [42, 43]. It is constitutively and ubiquitously expressed and is the most abundant molecular chaperone of the eukaryotic cytoplasm. Chaperones help to achieve and maintain the conformational status of cellular proteins and enzyme complexes. By influencing higher order protein structure, Hsp90 is involved in the conformational regulation of key proteins in multiple signaling pathways, including nitric oxide synthases (NOS), kinases, phosphatases and steroid hormone receptors.
Reactive cysteines are usually found in the vicinity of the ATP binding site of chaperone proteins [44]. Hsp90 regulates the redox status of other proteins by assisting in the formation and breakage of disulfide bridges. The cysteine groups participate in the binding of Hsp90 with its client proteins and with molybdate, a stabilizer of Hsp90-client protein interactions [44]. The high redox reactivity of its cysteines (cysteines 521 and 589/590) provides the mechanism by which this molecular chaperone helps maintain the redox status of the cytosol. Oxidizing conditions impair the chaperone activity of Hsp90, further proof of the active participation of sulfhydryl groups in the function of Hsp90 [44].
Hsp90 AND CYTOCHROME C
The primary function of the mitochondrial electron transport chain (ETC) is ATP synthesis. The ETC, present in the inner mitochondrial membrane, is grouped into four enzyme complexes. Damage to one or more of the ETC complexes may lead to impairment of cellular ATP synthesis [45]. The electron transport protein, cytochrome c (MW 12,000 kDa), serves as the electron carrier from complex III (ubiquinol:cytochrome c oxidoreductase) to complex IV (cytochrome c oxidase). Hsp90 reduces cytochrome c, an effect that is mediated by the -SH groups of Hsp90. The effects of Hsp90 can be blocked by the sulfhydryl reagents, arsenite and cadmium, indicating the involvement of the highly conserved vicinal cysteine pair, Cys589/590, in the reduction of cytochrome c [44]. The high reactivity of Hsp90 cysteine groups toward cytochrome c may indicate a role of this chaperone in modulation of the redox status of the cytosol in resting and apoptotic cells.
Although classically considered the cell powerhouse, mitochondria are also “gatekeepers” that ultimately determine whether a cell lives or dies [46]. Upon exposure to oxidizing species and thiol-reactive agents, mitochondria undergo a permeability transition whereby inner membrane proteins form a non-specific pore [47]. Pore opening causes a loss of the mitochondrial membrane potential and impairs ATP synthesis. Pore opening also results in an inability to sequester calcium. The resulting energy depletion and calcium overload are important factors in necrotic cell death. Pore opening also leads to release of cytochrome c from the outer surface of the inner mitochondrial membrane to the cytosol. Cytochrome c release is an important early event in apoptotic cell death [47].
Several heavy metals, including MeHg, induce neuronal apoptosis in whole animals and cultured cells [48]. Possible triggering mechanisms include calcium overload and generation of ROS [49]. Calcium binds directly to the divalent metal binding site on the matrix side of the mitochondrial permeability transition pore and induces its opening [47]. MeHg may also open the pore by oxidizing GSH and/or NADH resulting in the oxidation of the sensitive vicinal dithiol present in the S-site and P-site of the permeability transition pore, respectively, or by cross-linking the dithiol in the S-site [47]. It has been shown that exposure to mercurials partially inactivates cytochrome i oxidase [50]. It is notable that cytochrome c oxidase has 7 cysteine residues as sulfhydryl groups which are subject to modification by hydrophobic mercurials, such as MeHg. It is also noteworthy that apoptosis and the stress response are highly interrelated and altered expression of heat shock proteins exerts great influence on the development of apoptosis [44]. Studies directed at the effect of MeHg on the ability of Hsp90 to reduce cytochrome c or inactive cytochrome c oxidase may shed light on the mechanisms of MeHg-induced cell death.
NITRIC OXIDE (·NO) IN THE CNS
·NO is a gaseous messenger molecule with diverse biological roles. In the brain, ·NO has a number of important biochemical and physiological functions, such as neurotransmission and learning, regulation of glycolytic enzymes, pain perception, immune function, and vascular regulation. ·NO is formed from L-arginine by NO synthase (NOS). In the brain, astrocytes may be a major source of ·NO, because these cells have the highest concentration of the ·NO precursor, Larginine [51, 52]. At least three isoforms of NOS have been identified, namely, neuronal NOS (nNOS, NOS1), inducible NOS (iNOS, NOS2), and endothelial NOS (eNOS, NOS3). All three NOS isoforms use NADPH as an electron donor and employ five enzyme cofactors to catalyze oxidation of arginine to ·NO with stoichiometric formation of citrulline [53]. The calcium/calmodulin (CaM) complex is the common key cofactor that triggers NOS activation. Activation of NOS enzyme activity is ultimately determined by the binding affinity of CaM to NOS. At resting calcium levels, nNOS and eNOS are inactive. Agonists, such as glutamate, initiate nNOS or eNOS activation by raising intracellular Ca2+ concentrations sufficiently to maintain CaM binding; it is the high binding affinity of iNOS for CaM that renders iNOS fully active under basal calcium levels in quiescent cells. The production of ·NO must be modulated precisely because too much or too little ·NO formation will perturb cellular homeostasis. Each of the three major NOS isoforms is regulated at multiple levels, including transcriptional and post-translational steps (See Hsp90 and NOS below).
In the CNS, all three NOS isoforms are expressed constitutively or inducibly [52, 54]. nNOS is localized to discrete regions of the brain where constitutive expression is exclusive to neurons [55]. However, astrocytes express nNOS after induction by LPS and/or cytokines [56]. The expression of iNOS is below detectable levels in the normal brain, but is induced in neurons, astrocytes, and other cells in various pathological conditions [57–61]. For example, iNOS expression increases in astrocytes after transient global ischemia and after LPS and cytokine exposures [55]. eNOS is also induced in rat brain by ip injections of LPS. eNOS protein expression is localized to astrocytes of both grey and white matter as well as blood vessels [57]. Several groups have reported that eNOS is expressed preferentially in astrocytes rather than in neurons [62–65].
While it is now apparent that ·NO has many physiological roles, it is also clear that excessive ·NO generation is cytotoxic. Overactivation of glutamate receptors associated with cerebral ischemia and other excitotoxic processes results in massive release of ·NO [53]. ·NO mediates cellular toxicity by damaging critical metabolic enzymes and by reacting with O2 to form an even more potent oxidant, per-oxynitrite [45]. High levels of ·NO are associated with inflammatory, neurodegenerative, and cardiovascular/ischemic pathologies. Several in vitro studies show that ·NO produced by iNOS in astrocytes mediates neuronal cell death after excitotoxic injury [46, 66, 67].
·NO AND CYTOCHROME C OXIDASE
There is clear-cut evidence that ·NO controls cell respiration [68] and that the neurotoxicity of ·NO is mediated though mitochondrial dysfunction. Low nanomolar levels of ·NO rapidly and reversibly inhibit mitochondrial respiration by binding to the oxygen binding site of cytochrome c oxidase (complex IV). Such inhibition will lead to increased generation of O2−upstream of complex IV. Reversible inhibition of respiration by ·NO has been observed in intact cells, including astrocytes in culture [69]. This inhibition can be reversed by NOS inhibitors and by binding ·NO with hemoglobin [70]. Higher concentrations of ·NO can produce irreversible modifications of proteins and lipids and impairment of mitochondrial respiration. Cultured astrocytes activated to express iNOS produce up to 1 μM ·NO and strongly inhibit their own cellular respiration by inhibiting complex IV [71]. The generation of the extremely potent oxidizing agent, per-oxynitrite (ONOO−), formed from the reaction of ·NO with ·O2−, is a major mediator of these effects and is a strong inducer of the permeability transition pore [69]. Mitochondria are a major site of production of ·O2− under normal and pathological conditions and are a likely source of this anion for the generation of ONOO− [67]. These results suggest that any cell producing high levels of ·NO will inhibit its own respiration and that of surrounding cells. There is increasing evidence that defects in mitochondrial energy metabolism may underlie the pathology of neurodegenerative diseases such as Parkinson’s disease or Alzheimer’s disease [72].
HSP90 AND NOS
Beyond CaM, nNOS and eNOS are also regulated by various scaffolding proteins through protein-protein interactions. Several laboratories have shown that both eNOS and nNOS activity are upregulated by Hsp90 binding [73–75]. Bender et al. [74] demonstrated that nNOS exists as a molybdate-stabilized heterocomplex with Hsp90 in the cytosolic fraction of human embryonic kidney 293 cells stably transfected with rat nNOS. Furthermore, nNOS activity is reduced by the Hsp90 inhibitors, geldanamycin [73] and radicicol [76]. Song AJp et al. [74] showed that Hsp90 directly augments nNOS catalytic activity mediated, at least in part, by enhanced CaM binding. Billecke et al. [76] proposed that Hsp90 facilitates functional heme entry into apo-nNOS by opening the hydrophobic heme-binding cleft in the protein. Using electron paramagnetic resonance spectroscopy, Song et al. [75] directly measured ·NO signals from purified nNOS and demonstrated that Hsp90 augmented ·NO formation from nNOS in a concentration-dependent manner.
Besides ·NO, all NOS isoforms can also produce ·O2−. Again, using electron paramagnetic resonance spectroscopy [77] showed that Hsp90 directly inhibits ·O2− formation from nNOS, particularly in the absence of L-arginine. This inhibition was not due to ·O2− scavenging because Hsp90 did not affect the ·O2− generated by xanthine oxidase. Significant ·O2− production was detected from nNOS even at normal intracellular levels of L-arginine, an effect that was prevented by the addition of Hsp90. These results support the hypothesis that normal coupling between Hsp90 and nNOS is important for normal nNOS function. Disruption of this coupling may result in ·O2− overproduction, which could in turn lead to nNOS dysfunction and CNS pathology. The importance of Hsp90/nNOS interactions in the promulgation of neurotoxicity is incompletely understood. Furthermore, (as has been shown for the glucocorticoid receptor), association of nNOS with Hsp90 opens the thiol moieties in the ligand binding domain to attack by thiol-derivatizing agents [73].
OXIDANTS AND ANTIOXIDANTS IN ASTROCYTES
The generation of ROS begins with univalent transfer of an unpaired electron to O2, yielding ·O2−. Some ROS, including ·O2−, H2O2, NO, and ONOO− are important signaling molecules that regulate cell function. There is also a growing body of evidence to implicate excessive or inappropriate generation of ROS in neuropathology [44]. Potential sources of ·O2− in the CNS include NOS, cOx, lipoxygenase, NAD(P)H oxidase, xanthine oxidase, and mitochondria [78]. NOS-dependent O2− formation has been implicated in multiple neuropathological conditions. The dominant effect of increased ROS production by astrocytes appears to be an attenuation of ·NO signaling by ·O2−. Studies suggest that inactivation of ·NO by ·O2− contributes to impaired cell function and that ·O2− scavengers are effective in restoring optimal cell function [79–81]. Local cellular levels of ·O2− reflect both the rate of ·O2− formation and the rate of its removal by endogenous antioxidants (primarily superoxide dismutases (SODs) and GSH). GSH is an important intracellular antioxidant in astrocytes. Reduced GSH can react with reactive nitrogen species (RNS) and hence can limit mitochondrial damage. The reactions of RNS with GSH provide a mechanism to explain the loss of cellular GSH that can occur following ·NO exposure. Furthermore, these reactions will divert RNS away from critical cellular targets, such as the ETC, thereby explaining why GSH status appears to be so critical in dictating susceptibility to ·NO and ONOO−. Loss of GSH may be an important factor in the neurotoxicity associated with excessive ·NO formation and mitochondrial damage [54, 82].
While MeHg is known to induce oxidative stress, it has yet to be determined whether this is due to a change in the balance of ·NO and other ROS. Studies on molecular teratogenic mechanisms of MeHg in early developing rat embryos have noted significant induction of iNOS mRNA and protein as potential mechanisms of aberrant neurulation [83]. In mature rats, MeHg treatment results in significant increases in nNOS activities in the cerebrum and cerebellum, notably two areas that are preferentially affected in the adult animal [84].
MeHg causes ROS generation in nerve cells [85, 86] and MeHg exposure in vivo elevates ROS in brain regions sensitive to MeHg, but not in regions less sensitive to MeHg [87, 88]. Consistent with the oxidative stress findings above, studies in cultured primary rat astrocytes suggest that a multitude of MeHg effects (cell swelling, glutamate release, inhibition of glutamate uptake) can be attenuated by antioxidants [14, 89], whilst astrocytes depleted of GSH or those isolated from metallothionein knockout mice (MT−/−) are significantly more sensitive to the effects MeHg [90, 91]. The impact of MeHg on Hsp90 chaperone function, NOS isoform expression and activity, NOS uncoupling and region-specific ROS formation should prove to be a profitable area for future study.
ROS AND GLUTAMATE NEUROTOXICITY
The excitatory neurotransmitter glutamate has been shown to play a crucial role in neuronal damage associated with trauma, ischemia, toxins and chronic neurodegenerative disorders. Glutamate neurotoxicity (GNT) takes place as a result of glutamate binding to the N-methyl-D-aspartic acid (NMDA) receptor and to a minor extent to other receptor subtypes. GNT is marked by acute neuronal swelling and depends on extracellular Na+ and Cl− uptake by the cell that causes plasma membrane depolarization. This causes Ca2+ channel opening that triggers massive influx of extracellular Ca2+ and release from intracellular Ca2+ stores, resulting in calcium overload that initiates a cascade-like effect leading to cell death [92]. A causal relationship between GNT and ROS production was first proposed by Dykens [93] who observed that neuronal damage caused by the excitotoxin, kainate, is prevented by SOD. Conversely, the depletion of the antioxidant GSH was found to exacerbate GNT [94]. Electron paramagnetic resonance spectroscopy provided direct evidence that NMDA receptor activation leads to the generation of ·O2− [19]. Glutamate stimulates production of ·O2− via both NOS- and AA-dependent mechanisms [95]. Schulz et al. [96] providing the first in vivo evidence that excitotoxic neuronal injury is linked to free radical generation from mitochondria.
Almeida et al. [97] showed that GNT is associated with ·NO-mediated mitochondrial dysfunction and GSH depletion. ·NO is synthesized via activation of Ca2+-dependent nNOS after glutamate receptor stimulation. Furthermore, glutamate-receptor stimulation in neurons causes a burst of ·O2− formation, which can combine with ·NO to form ONOO−. In rat neurons in primary culture, glutamate caused a dose-dependent increase in cGMP, delayed neurotoxicity and ATP depletion. GNT was prevented by NOS inhibition and by NMDA receptor antagonism [97, 98]. In striatal astrocytes from mouse embryos, glutamate evokes release of AA [99]. Released AA increases the accumulation of extracellular glutamate by blocking its reuptake and in turn glutamate acts on glutamate receptors to increase neuronal intracellular calcium and release additional AA [99]. Thus, AA inhibits glutamate reuptake by astrocytes and potentiates NMDA- evoked current in neurons. Interestingly AA and ROS were found to inhibit glutamate uptake in astrocytes via two distinct and additive mechanisms [100]. Ciani et al. [101] demonstrated that cPLA2 and NOS activation give rise to ROS, whose deleterious action can be counteracted either by inhibiting these enzymes or by scavenging the excess of free radicals produced by them. More recently it has been also demonstrated that cPLA2-stimulated hydrolysis and release of AA are potential mediators of MeHg-induced neurotoxicity [102].
DIFFERENTIAL SUSCEPTIBILITIES OF NEURONS AND ASTROCYTES
Within the CNS, the susceptibility of different brain cell types to ·NO and ONOO− exposure may be dependent on the intracellular reduced GSH concentration and an ability to increase glycolytic flux in the face of mitochondrial damage [44]. Acute exposure to ONOO− selectively damages neurons, whereas astrocytes remain unaffected [103]. ONOO− reacts with thiol-containing compounds and multiple lines of evidence support a key role for GSH in dictating cellular susceptibility to ONOO−. In particular, astrocytes have high reduced GSH concentrations [23, 104]. ONOO− causes a pronounced loss of neuronal GSH concentrations, without an effect on GSH levels in astrocytes [82]. This apparent preservation of GSH may reflect the greater activity, in astrocytes, of γ-glutamylcysteine synthetase, which is the rate limiting enzyme for GSH synthesis [105]. GSH depletion in neurons may be due to ·NO-dependent inhibition of cysteine uptake or from the direct reaction of ·NO/ONOO− with intracellular GSH. The importance of GSH in protecting the ETC from oxidizing species such as ONOO− is further illustrated by experiments with GSH depleted astrocytes. Under such conditions marked damage to the ETC and cell death occurs following ONOO− exposure [106]. Conversely, the apparent increase in resistance of neurons to ·NO when co-cultured with astrocytes can be explained by up-regulation of neuronal GSH as a result of the astrocytic release of GSH followed by cleavage, via γ-glutamyltranspeptidase (GABA), to dipeptides that can be utilized by the neuron for GSH synthesis [25, 104].
Susceptibility of different brain regions or cell types to MeHg [107] may also be dependent on factors such as the intracellular reduced GSH concentration and the ability to increase glycolytic flux in the face of mitochondrial damage. These observations are consistent with morphological observations in which astrocytes that accumulate MeHg appear normal, whilst neurons that are found in their proximity and are void of MeHg undergo cell death [10]. In view of the importance of antioxidants, in particular GSH, it is possible that agents that are capable of increasing cellular concentrations of GSH may prove to be of therapeutic importance.
PGES/p23
Prostaglandin (PG) biosynthesis can be divided into two kinetically distinct responses, the immediate and delayed responses, involving recruitment of distinct biosynthetic enzymes whose expression and activation are differentially regulated [107]. The immediate response occurs within minutes after stimulation by an agonist that increases cytoplasmic calcium. cPLA2 is a prerequisite for supplying AA to constitutive COX-1 [108]. Delayed PG synthesis proceeds gradually over a long period after a proinflammatory stimulus and is accompanied by de novo induction of COX-2. The preference for COX-2 over COX-1 in the delayed response is partially explained by the ability of COX-2 to metabolize lower levels of AA to PGH2 than is required for COX-1- mediated catalysis [108].
cPGES is a terminal enzyme of the COX-1-mediated PGE2 biosynthetic pathway. Peptide microsequencing of purified enzyme revealed that it is identical to p23, a putative chaperone molecule that binds to the ATP-dependent conformation of Hsp90 and acts as a cofactor in the molecular chaperone function of Hsp90 [109]. cPGES/p23 is expressed ubiquitously and in abundance in a wide variety of tissues and cell types. Its expression is constitutive and not affected by proinflammatory stimuli, with the exception of rat brain, where LPS treatment increases cPGES levels several fold. cPGES/p23 is functionally linked with COX-1 in marked preference to COX-2 to produce PGE2 from exogenous and endogenous AA. The cPLA2/COX-1/cPGES/p23 cascade is crucial for the production of PGE2 that is required for maintenance of tissue homeostasis [109]. cPGES/p23 is highly conserved among animal species (>95%) and requires GSH for optimal activity. Enzyme activity is also stimulated by other -SH reducing agents, such as dithiothreitol (DTT) and 2-mercaptoethanol [110]. cPGES may be regulated by Hsp90 in that the Hsp90-bound form is functionally active [108].
In contrast, membrane-associated PGES (mPGES) is a microsomal GST-like 1 (MGST-L1) enzyme in the MAPEG (membrane-associated proteins involved in eicosanoid and GSH metabolism) superfamily [108]. mPGES is an inducible perinuclear enzyme preferentially coupled with inducible COX-2 to promote delayed PGE2 generation. It is strongly induced following exposure to interleukin-1 and LPS in several cell types. The main PG species produced during the “delayed response” is PGE2 associated with COX-2 expression. COX-2 inhibitors reduce PGE2 more profoundly than other PGs. Whether cofactors assist the functional linkage between COX-2 and mPGES remains unclear. The small size and lipophilic properties of PGE2 allow it to pass across the blood-brain barrier (BBB) and to diffuse into CNS neurons.
Astrocytes release glutamate through a calcium dependent process mediated by PGs [111, 112]. Pharmacological inhibition of PG synthesis prevents glutamate release, whereas application of PGE2 promotes calcium-dependent glutamate release from cultured astrocytes and brain slices [111]. As shown by our lab, glutamate is released as part of regulatory volume decrease (RVD) and 80% of the released glutamate is taken up by astrocytes [6]. MeHg induces astrocytic swelling and inhibits RVD in part by inhibiting reuptake of glutamate [32]. It is not known if the mechanism by which MeHg inhibits glutamate uptake in astrocytes is mediated by PGE2. Distinct distribution of different receptor subtypes for PGE2 (of which there are 4) may account for the regional specificity of the cellular response triggered by MeHg in discrete brain structures, such as calcarine cortex, and the cerebellum [107].
Tying together the remarkable affinity of MeHg for -SH groups, the essential role of thiols in chaperone protein/client interactions, the crucial role of redox status and GSH sufficiency in cellular homeostasis and susceptibility to MeHg toxicity, we have identified Hsp90, cytochrome c, NOS and PGE2 as potential upstream molecular targets of MeHg’s cytotoxic effects. Fig. (1) depicts a working model where MeHg is posited to affect the cross talk between astrocytes and neurons. Panel A represents the normal interrelationship between astrocytes and neurons and Panel B depicts the alterations in astrocytic and neuronal cell signaling that occur with MeHg toxicity. Please refer to the figure legend for details.
Fig. (1).
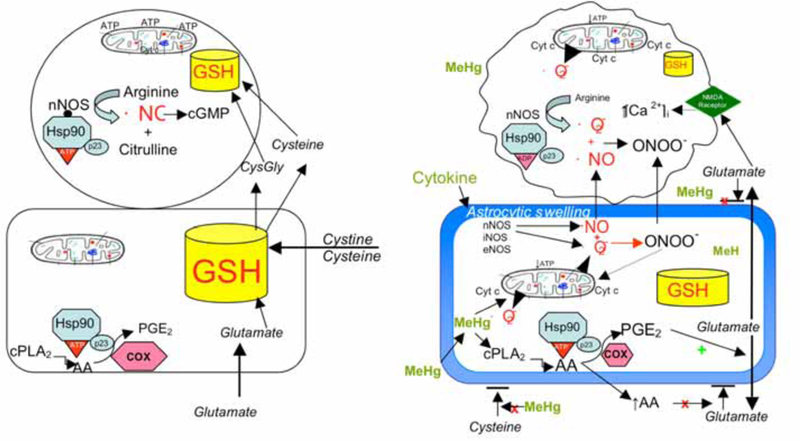
Panel A represents the normal interrelationship between astrocytes and neurons, in which astrocytes contain a larger pool of GSH (and other antioxidants) than is found in neurons. Astrocytes synthesize GSH from the uptake of cystine, cysteine and glutamate. Neuronal GSH concentrations are dependent largely on release of precursors from astrocytes. Healthy neurons express nNOS which, when bound to Hsp90 in the proper ATP conformation, produces ·NO which activates guanylate cyclase to produce cGMP for maintenance of important biological functions. Astrocytes do not express NOS but do produce PGE2, which is important for normal tissue homeostasis. Intact mitochondrial function and intracellular GSH levels in both cell types are important for normal ATP production and cell bioenergetics. Panel B depicts the alterations in astrocytic and neuronal cell signaling that occur with MeHg toxicity. MeHg induces astrocytic swelling triggering glutamate release, and inhibits uptake of cystine and cysteine, reducing astrocytic ability to synthesize GSH. MeHg also stimulates cPLA2 and AA release which stimulates increased production of PGE2 via PGES/p23 bound to Hsp90. PGE2 stimulates glutamate release and AA blocks reuptake, resulting in markedly increased levels of extracellular glutamate. Glutamate stimulates NMDA receptors on neurons increasing [Ca2+]I which causes activation of nNOS and mitochondrial dysfunction. MeHg (perhaps by induction of cytokines) induces astrocytes to express nNOS (and other NOS isoforms) which produce both ·NO and ·O2−. These ROS combine to form ONOO−, which together with NO diffuse to adjacent neurons increasing oxidant stress. This will markedly reduce intracellular GSH, especially in neurons. Increased ·NO, ONOO− and Ca2+ overload damages the mitochondrial ETC, resulting in reduced ATP formation, increased ·O2− formation, and cyt c release, all of which initiate the cascades leading to neuronal death.
CONCLUSIONS
In summary, we point to novel sites that are likely sensitive to interference by MeHg. Our theory is predicated upon a number of observation: (a) the remarkable affinity of mercurials for the anionic form of sulfhydryl (-SH) groups, (b) the essential role of thiols in protein biochemistry, and (c) the role of molecular chaperone proteins, such as heat shock protein 90 (Hsp90), in the regulation of protein redox status by facilitating the formation and breakage of disulfide bridges. Molecular chaperones help to achieve and maintain the conformational homeostasis of cellular proteins. The 90-kDa heat shock protein, Hsp90, is the most abundant molecular chaperone in the eukaryotic cytoplasm, helping to organize efficient multiprotein complexes for the execution of essential cellular processes. It has been shown that cysteine groups of Hsp90 participate in its interactions with client proteins and that oxidizing conditions impair the chaperone function of Hsp90. This appears to be a fruitful area that merits future research.
ACKNOWLEDGEMENT
This review was supported by PHS NIEHS 07331 to MA.
REFERENCES
- [1].Environmental Protection Agency; Water Quality Criterion for the Protection of Human Health: Methylmercury, Office of Science and Technology, Office of Water, Environmental Protection Agency; Washington, DC: 20460, EPA-823-R-01–001, 2001. [Google Scholar]
- [2].Davidson PW, Myers GJ, Cox C, et al. Effects of prenatal and postnatal methylmercury exposure from fish consumption on neurodevelopment: outcomes at 66 months of age in the Seychelles Child Development Study. JAMA 1998; 280: 701–707. [DOI] [PubMed] [Google Scholar]
- [3].Grandjean P, Weihe P, White RF, et al. Cognitive deficit in 7-year-old children with prenatal exposure to methylmercury. Neurotoxi col Teratol 1997; 19: 417–428. [DOI] [PubMed] [Google Scholar]
- [4].National Academy of Science, Committee on the Toxicological Effects of methylmercury, Board on Environmental Studies and Toxicology, national research Council Toxicological Effect of Methylmercury, National Academies Press, Washington, DC, 2000. [Google Scholar]
- [5].Chen Y, Swanson RA. The glutamate transporters EAAT2 and EAAT3 mediate cysteine uptake in cortical neuron cultures. J Neu-rochem 2003; 84: 1332–1339. [DOI] [PubMed] [Google Scholar]
- [6].Kimelberg HK, Aschner M. Functions of astrocytes In: Astrocytes in brain aging and neurodegeneration, Neuroscience Intelligence Unit Series. Schipper HM, Ed. RG Landes Co, Montreal, 1998; pp. 15–39. [Google Scholar]
- [7].Aschner M, Sonnewald U, Tan KH. Astrocyte modulation of neu-ropathologic damage. Brain Pathol 2002; 12: 476–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Charleston JS, Bolender RP, Mottet NK, Body RL, Vahter ME, Burbacher TM. Increases in the number of reactive glia in the visual cortex of Macaca fascicularis following subclinical long-term methyl mercury exposure. Toxicol Appl Pharmacol 1994; 129: 196–206. [DOI] [PubMed] [Google Scholar]
- [9].Charleston JS, Body RL, Bolander RP, Mottet NK, Vahter ME, Burbacher TM. Changes in the number of astrocytes and microglia in the thalamus of the monkey Macaca fascicularis following long-term subclinical methylmercury exposure. Neurotoxicology 1996; 17: 127–138. [PubMed] [Google Scholar]
- [10].Garman RH, Weiss B, Evans HL. Alkylmercurial encephalopathy in the monkey: a histopathologic and autoradiographic study. Acta Neuropathol (Berlin) 1975; 32: 61–74. [DOI] [PubMed] [Google Scholar]
- [11].Aschner M, Du YL, Gannon M, Kimelberg HK. Methylmercury-induced alterations in excitatory amino acid transport in rat primary astrocytic cultures. Brain Res 1993; 602: 181–186. [DOI] [PubMed] [Google Scholar]
- [12].Aschner M Methylmercury in astrocytes-what possible significance? Neurotoxicology 1996; 17: 93–106. [PubMed] [Google Scholar]
- [13].Aschner M, Yao CP, Allen JW, Tan KH. Methylmercury alters glutamate transport in astrocytes. Neurochem Intl 2000; 37: 199–206. [DOI] [PubMed] [Google Scholar]
- [14].Allen JW, Mutkus LA, Aschner M. Methylmercury-mediated inhibition of 3H-D-aspartate transporter in cultured astrocytes is reversed by the antioxidant catalase. Brain Res 2001; 902: 92–100. [DOI] [PubMed] [Google Scholar]
- [15].Brookes N, Kristt DA. Inhibition of amino acid transport and protein synthesis by HgCl2 and methylmercury in astrocytes: selectivity and reversibility. J Neurochem 1989; 53: 1228–1237. [DOI] [PubMed] [Google Scholar]
- [16].Juarez BI, Martinez ML, Montante M, Dufour L, Garcia E, Jime-nez-Capdeville ME. Methylmercury increases glutamate extracellular levels in frontal cortex of awake rats. Neurotoxicol Teratol 2002; 24: 767–771. [DOI] [PubMed] [Google Scholar]
- [17].Miyamoto K, Nakanishi H, Moriguchi S, et al. Involvement of enhanced sensitivity of N-methyl-D-aspartate receptors in vulnerability of developing cortical neurons to methylmercury neurotoxicity. Brain Res 2001; 901: 252–258. [DOI] [PubMed] [Google Scholar]
- [18].Miyamoto K, Murao K, Wakamiya J, et al. Protective effect of MK-801 in methylmercury-induced neuronal injury. In: Mercury as a Global Pollutant. 5th International Conference, 1999, pp. 376. [Google Scholar]
- [19].Lafon-Cazal M, Pietri S, Culcasi M, Boeckaert J. NMDA- dependent superoxide production and neurotoxicity. Nature 1993; 364: 535–537. [DOI] [PubMed] [Google Scholar]
- [20].Allen JW, Shanker G, Aschner M. Methylmercury inhibits the in vitro uptake of the glutathione precursor, cystine, in astrocytes, but not in neurons. Brain Res 2001; 894: 131–140. [DOI] [PubMed] [Google Scholar]
- [21].Shanker G, Allen JW, Mutkus LA, Aschner M. Methylmercury inhibits cysteine uptake in cultured primary astrocytes, but not in neurons. Brain Res 2001; 914: 159–165. [DOI] [PubMed] [Google Scholar]
- [22].Shanker G, Aschner M. Identification and characterization of uptake systems for cystine and cysteine in cultured astrocytes and neurons: evidence for methylmercury-targeted disruption of astrocytic transport. J Neurosci Res 2001; 66: 998–1002. [DOI] [PubMed] [Google Scholar]
- [23].Philbert MA, Beiswanger CM, Waters DK, Reuhl KR, Lowndes HE. Cellular and regional distribution of reduced glutathione in the nervous system of the rat: histochemical localization by mercury orange and o-phthaldialdehyde-induced histofluorescence. Toxicol Appl Pharmacol 1991; 107: 215–227. [DOI] [PubMed] [Google Scholar]
- [24].Maret M Oxidative metal release from metallothionein via zinc-thiol/disulfide interchange. Proc Natl Acad Sci USA 1994; 91: 237–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Dringen R, Hirrlinger J. Glutathione pathways in the brain. Biol Chem 2003; 384; 505–516. [DOI] [PubMed] [Google Scholar]
- [26].Wang XF, Cynader MS. Astrocytes provide cysteine to neurons by releasing glutathione. J Neurochem 2000; 74: 1434–1442. [DOI] [PubMed] [Google Scholar]
- [27].Shanker G, Mutkus LA, Walker S, Aschner M. Methylmercury enhances arachidonic acid release and cytosolic phospholipase A2 expression in primary cultures of neonatal astrocytes. Mol. Brain Res 2002; 106: 1–11. [DOI] [PubMed] [Google Scholar]
- [28].Yin Z, Milatovic D, Aschner JL, et al. Methylmercury induces oxidative injury, alterations in permeability and glutamine transport in cultured astrocytes. Brain Res 2007; 1131: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Matyja E, Albrecht J. Ultrastructural evidence that mercuric chloride lowers the threshold for glutamate neurotoxicity in an organotypic culture of rat cerebellum. Neurosci Lett 1993; 158: 155–158. [DOI] [PubMed] [Google Scholar]
- [30].Oyake Y, Tanaka M, Kubo H, Chichibu M. Neuropathological studies on organic mercury poisoning with special reference to the staining and distribution of mercury granules. Shinkei Kenkyu No Shimpo 1966; 10: 744–750. [PubMed] [Google Scholar]
- [31].Vahter M, Mottet NK, Friberg L, Lind B, Shen DD, Burbacher T. Speciation of mercury in the primate blood and brain following long-term exposure to methyl mercury. Toxicol Appl Pharmacol 1994; 124: 221–229. [DOI] [PubMed] [Google Scholar]
- [32].Aschner M, Eberle N, Miller K, Kimelberg HK. Interaction of methylmercury with rat primary astrocyte cultures: effects on rubidium uptake and efflux and induction of swelling. Brain Res 1990; 530: 245–250. [DOI] [PubMed] [Google Scholar]
- [33].Brookes N In vitro evidence for the role of glutamate in the CNS toxicity of mercury. Toxicology 1992; 76: 245–256. [DOI] [PubMed] [Google Scholar]
- [34].Hughes WH. A physicochemical rationale for the biological activity of mercury and its compound. Ann NY Acad Sci 1957; 65: 454–460. [DOI] [PubMed] [Google Scholar]
- [35].Bramanti E, D’Ulivo A, Lampugnani L, Zamboni R, Raspi G. Application of mercury cold vapor atomic fluorescence spectrometry to the characterization of mercury-accessible -SH groups in native proteins. Analyt Biochem 1999; 274: 153–173. [DOI] [PubMed] [Google Scholar]
- [36].Carty AJ, Malone SF. The chemistry of mercury in biological systems In: The Biogeochemistry of Mercury in the Environment, Nrigau JO, Ed., Elsevier/North Holland Biomedical Press, Amsterdam, 1979, pp 433–479. [Google Scholar]
- [37].Naganuma A, Imura N. Methylmercury binds to a low molecular weight substance in rabbit and human erythrocytes. Toxicol Appl Pharmacol 1979; 47: 613–616. [DOI] [PubMed] [Google Scholar]
- [38].Rabenstein DL, Fairhurst MT. Nuclear magnetic resonance studies of the solution chemistry of metal complexes. J Am Chem Soc 1975; 97: 2086–2092. [DOI] [PubMed] [Google Scholar]
- [39].Thomas DJ, Smith CJ. Effects of coadministered low molecular weight thiol compounds on short term distribution of methylmer- cury in the rat. Toxicol Appl Pharmacol 1979; 62: 104–110. [DOI] [PubMed] [Google Scholar]
- [40].Omata S, Sakimura K, Ishii T, Sugano H. Chemical nature of a methylmercury complex with a low molecular weight in the liver cytosol of rats exposed to methylmercury chloride. Biochem Pharmacol 1978; 27: 333–335. [DOI] [PubMed] [Google Scholar]
- [41].Refsvik T, Norseth T. Methyl mercuric compounds in rat bile. Acta Pharmacol Toxicol 1975; 36: 67–78. [DOI] [PubMed] [Google Scholar]
- [42].Capla AJ. Hsp90’s secrets unfold: new insights from structural and functional studies. Trends Cell Biol 1999; 9: 262–268. [DOI] [PubMed] [Google Scholar]
- [43].Balligand JL. Heat shock protein 90 in endothelial nitric oxide synthase signaling: following the lead(er)? Circ Res 2002; 90: 838–841. [DOI] [PubMed] [Google Scholar]
- [44].Nardai G, Sass B, Eber J, Orosz G, Csermely P. Reactive Cysteines of the 90-kDa Heat Shock Protein, Hsp90. Arch Biochem Biophys 2000; 384: 59–67. [DOI] [PubMed] [Google Scholar]
- [45].Heales SJR, Bolanos JP, Stewart VC, Brookes PS, Land JM, Clark JB. Nitric oxide, mitochondria and neurological disease. Biochim Biophys Acta 1999; 1410: 215–228. [DOI] [PubMed] [Google Scholar]
- [46].Bolaños JP, Peuchen S, Heales SJR, Land JM, Clark JB. Nitric oxide-mediated inhibition of the mitochondrial respiratory chain in cultured astrocytes. J Neurochem 1994; 63: 910. [DOI] [PubMed] [Google Scholar]
- [47].He L, Poblenz AT, Medrano CJ, Fox DA. Lead and calcium produce photoreceptor cell apoptosis by opening the mitochondrial permeability transition pore. J. Biol Chem 2000; 275: 12175–12184. [DOI] [PubMed] [Google Scholar]
- [48].Castoldi AF, Coccini T, Ceccatelli S, Manzo L. Neurotoxicity and molecular effects of methylmercury. Brain Res Bull 2001; 55: 197–203. [DOI] [PubMed] [Google Scholar]
- [49].Atchison WD, Hare MF. Mechanisms of methylmercury-induced neurotoxicity. FASEB J 1994; 8: 622–629. [DOI] [PubMed] [Google Scholar]
- [50].Mann A, Auer HE. Partial inactivation of cytochrome c oxidase by nonpolar mercurial reagents. J Biol Chem 1980; 255: 454–458. [PubMed] [Google Scholar]
- [51].Aoki E, Semba R, Mikoshiba K, Kashiwamata S. Predominant localization in glial cells of free L-arginine. Immunocytochemical evidence. Brain Res 1991; 547: 190–192. [DOI] [PubMed] [Google Scholar]
- [52].Wiesinger H Arginine metabolism and the synthesis of nitric oxide in the nervous system. Prog Neurobiol 2001; 64: 365–391. [DOI] [PubMed] [Google Scholar]
- [53].Bredt DS. Endogenous nitric oxide synthesis: biological functions and pathophysiology. Free Radic Res 1999; 31: 577–596. [DOI] [PubMed] [Google Scholar]
- [54].Heales SJR, Bolanos JP. Impairment of brain mitochondrial function by reactive nitrogen species: the role of glutathione in dictating susceptibility. Neurochem Int 2002; 40: 469–474. [DOI] [PubMed] [Google Scholar]
- [55].Caggiano AO, Kraig RP. Neuronal nitric oxide synthase expression is induced in neocortical astrocytes after spreading depression. J Cereb Blood Flow Metab 1998; 18: 75–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Bolanos JP, Peuchen S, Land JM, Clark JB, Heales SJR. Evaluation of the efficacy of potential therapeutic agents at protecting against nitric oxide synthase-mediated mitochondrial damage in activated astrocytes. Brain Res Protoc 1997; 1: 258–262. [DOI] [PubMed] [Google Scholar]
- [57].Iwase K, Miyanaka K, Shimizu A, et al. Induction of endothelial nitric-oxide synthase in rat brain astrocytes by systemic lipopolysaccharide treatment. J Biol Chem 2000; 275: 11929–11933. [DOI] [PubMed] [Google Scholar]
- [58].Iadecola C, Xu X, Zhang F, el-Fakahany EE, Ross ME. Marked induction of calcium-independent nitric oxide synthase activity after focal cerebral ischemia. J Cereb Blood Flow Metab 1995; 15: 52–59. [DOI] [PubMed] [Google Scholar]
- [59].Niwa M, Inao S, Takayasu M, et al. Time course of expression of three nitric oxide synthase isoforms after transient middle cerebral artery occlusion in rats. Neurol Med Chir 2001; 41: 63–72. [DOI] [PubMed] [Google Scholar]
- [60].Knott C, Stern G, Wilkin GP. Inflammatory regulators in Parkinson’s disease: iNOS, lipocortin-1, cyclooxygenases-1 and −2. Mol Cell Neurosci 2000; 16: 724–739. [DOI] [PubMed] [Google Scholar]
- [61].Luth HJ, Holzer M, Gartner U, Staufenbiel M, Arendt T. Expression of endothelial and inducible NOS-isoforms is increased in Alzheimer’s disease, in APP23 transgenic mice and after experimental brain lesion in rat: evidence for an induction by amyloid pathology. Brain Res 2001; 913: 57–67. [DOI] [PubMed] [Google Scholar]
- [62].Barna M, Komatsu T, Reiss CS. Activation of type III nitric oxide synthase in astrocytes following a neurotropic viral infection. Virology 1996; 223: 331–343. [DOI] [PubMed] [Google Scholar]
- [63].Gabbott PL, Bacon SJ. Localization of NADPH diaphorase activity and NOS immunoreactivity in astroglia in normal adult rat brain. Brain Res 1996; 714: 135–144. [DOI] [PubMed] [Google Scholar]
- [64].Wiencken AE, Casagrande VA. Endothelial nitric oxide synthetase (eNOS) in astrocytes: another source of nitric oxide in neocortex. Glia 1999; 26: 280–290. [PubMed] [Google Scholar]
- [65].Shin T Enhanced expression of constitutive endothelial nitric oxide synthase by astrocytes in the spinal cords of rats with experimental autoimmune encephalomyelitis. Immunol Invest 1999;28: 381–90. [DOI] [PubMed] [Google Scholar]
- [66].Golde S, Chandran S, Brown GC, Compston A. Different pathways for iNOS-mediated toxicity in vitro dependent on neuronal maturation and NMDA receptor expression. J Neurochem 2002; 82: 269–82. [DOI] [PubMed] [Google Scholar]
- [67].Lecanu L, Verrecchia C, Margaill I, Boulu RG, Plotkine M. iNOS contribution to the NMDA-induced excitotoxic lesion in the rat striatum. J Pharmacol 1998; 125: 584–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Sarti P, Giuffre A, Barone MC, Forte E, Mastronicola D, Brunori M. Nitric oxide and cytochrome oxidase: reaction mechanisms from the enzyme to the cell. Free Radic Biol Med 2003; 34: 509–520. [DOI] [PubMed] [Google Scholar]
- [69].Brown GC. Nitric oxide and mitochondrial respiration. Biochim Biophys Acta 1999; 1411: 351–369. [DOI] [PubMed] [Google Scholar]
- [70].Brown GC, Bolanos JP, Heales SJ, Clark JB. Nitric oxide produced by activated astrocytes rapidly and reversibly inhibits cellular respiration. Neurosci Lett 1995; 193: 201–204. [DOI] [PubMed] [Google Scholar]
- [71].Brown GC. Nitric oxide inhibition of cytochrome oxidase and mitochondrial respiration: implications for inflammatory, neurodegenerative and ischaemic pathologies. Mol Cell Biochem 1997; 174: 189–192. [PubMed] [Google Scholar]
- [72].Beal MF. Does impairment of energy metabolism result in excito- toxic neuronal death in neurodegenerative illnesses? Ann Neurol 1992;31: 119–130. [DOI] [PubMed] [Google Scholar]
- [73].Bender AT, Silverstein AM, Demady DR, et al. Neuronal nitric- oxide synthase is regulated by the Hsp90-based chaperone system in vivo. J Biol Chem 1999; 274: 1472–1478. [DOI] [PubMed] [Google Scholar]
- [74].Song Y, Zweier JL, Xia Y. Heat-shock protein 90 augments neuronal nitric oxide synthase activity by enhancing Ca2+/calmodulin binding. Biochem J 2001; 355: 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Song Y, Zweier JL, Xia Y. Determination of the enhancing action of HSP90 on neuronal nitric oxide synthase by EPR spectroscopy. Am J Physiol Cell Physiol 2001; 281: C1819–C1824. [DOI] [PubMed] [Google Scholar]
- [76].Billecke SS, Bender AT, Kanelakis KC, et al. Hsp90 is required for heme binding and activation of apo-neuronal nitric-oxide synthase. J Biol Chem 2002; 277: 20504–20509. [DOI] [PubMed] [Google Scholar]
- [77].Song Y, Cardounel AJ, Zweier JL, Xia Y. Inhibition of superoxide generation from neuronal nitric oxide synthase by heat shock protein 90: Implications in NOS regulation. Biochemistry 2002; 41: 10616–10622. [DOI] [PubMed] [Google Scholar]
- [78].Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res 2000; 87: 840–844. [DOI] [PubMed] [Google Scholar]
- [79].Rubanyi GM, Vanhoutte PM. Superoxide anions and hyperoxia inactivate endothelium-derived relaxing factor. Am J Physiol 1986; 250: H822–H827. [DOI] [PubMed] [Google Scholar]
- [80].Tarpey MM, Fridovich I. Methods of detection of vascular reactive species: nitric oxide, superoxide, hydrogen peroxide, peroxynitrite. Circ Res 2001; 89: 224–236. [DOI] [PubMed] [Google Scholar]
- [81].Didion SP, Ryan MJ, Didion LA, Fegan PE, Sigmund CD, Faraci FM. Increased superoxide and vascular dysfunction in CuZnSOD- deficient mice. Circ Res 2002; 91: 938–944. [DOI] [PubMed] [Google Scholar]
- [82].Bolanos JP, Heales SJ, Peuchen S, Barker JE, Land JM, Clark JB. Nitric oxide-mediated mitochondrial damage: a potential neuroprotective role for glutathione. Free Radic Biol Med 1996; 21: 995–1001. [DOI] [PubMed] [Google Scholar]
- [83].Li Y, Zhang T, Wang L, Li Z. Studies on molecular teratogenic mechanism of methylmercury in early developing rat embryos. Wei Sheng Yan Jiu 1998; 27: 306–308 [PubMed] [Google Scholar]
- [84].Shinyashiki M, Kumagai Y, Nakajima H, et al. Differential changes in rat brain nitric oxide synthase in vivo and in vitro by methylmercury. Brain Res 1998; 798: 147–155. [DOI] [PubMed] [Google Scholar]
- [85].Sarafian TA, Verity MA. Oxidative mechanisms underlying methyl mercury neurotoxicity. Int J Dev Neurosci 1991; 9: 147–153. [DOI] [PubMed] [Google Scholar]
- [86].Sarafian TA, Vartavarian L, Kane DJ, Bredesen DE, Verity MA. bcl-2 expression decreases methyl mercury-induced free-radical generation and cell killing in a neural cell line. Toxicol Lett 1994; 74: 149–155. [DOI] [PubMed] [Google Scholar]
- [87].LeBel CP, Ali SF, McKee M, Bondy SC. Organometal-induced increases in oxygen reactive species: the potential of 2’,7’- dichlorofluorescin diacetate as an index of neurotoxic damage. Toxicol Appl Pharmacol 1990; 104: 17–24. [DOI] [PubMed] [Google Scholar]
- [88].LeBel CP, Ali SF, Bondy SC. Deferoxamine inhibits methyl mercury-induced increases in reactive oxygen species formation in rat brain. Toxicol Appl Pharmacol 1992; 112: 161–165. [DOI] [PubMed] [Google Scholar]
- [89].Mullaney KJ, Fehm MN, Vitarella D, Wagoner DE Jr, Aschner M. The role of -SH groups in methylmercuric chloride-induced D- aspartate and rubidium release from rat primary astrocyte cultures. Brain Res 1994; 641: 1–9. [DOI] [PubMed] [Google Scholar]
- [90].Yao CP, Allen JW, Conklin DR, Aschner M. Transfection and overexpression of metallothionein-I in neonatal rat primary astrocyte cultures and in astrocytoma cells increases their resistance to methylmercury-induced cytotoxicity. Brain Res 1999; 818: 414–420. [DOI] [PubMed] [Google Scholar]
- [91].Yao CP, Allen JW, Mutkus LA, Xu SB, Tan KH, Aschner M. Foreign metallothionein-I expression by transient transfection in MT-I and MT-II null astrocytes confers increased protection against acute methylmercury cytotoxicity. Brain Res 2000; 855: 32–38. [DOI] [PubMed] [Google Scholar]
- [92].Atlante A, Calissano P, Bobba A, Giannattasio S, Marra E, Pas-sarella S. Glutamate neurotoxicity, oxidative stress and mitochondria. FEBS Lett 2001; 497: 1–5. [DOI] [PubMed] [Google Scholar]
- [93].Dykens JA, Stern A, Trenkner E. Mechanism of kainate toxicity to cerebellar neurons in vitro is analogous to reperfusion tissue injury. J Neurochem 1987; 49: 1222–1228. [DOI] [PubMed] [Google Scholar]
- [94].Bridges RJ, Stanley MS, Anderson MW, Cotman CW, Chamberlin AR. Conformationally defined neurotransmitter analogues. Selective inhibition of glutamate uptake by one pyrrolidine-2,4- dicarboxylate diastereomer. J Med Chem 1991; 34: 717–725. [DOI] [PubMed] [Google Scholar]
- [95].Culcasi M, Lafon-Cazal M, Pietri S, Bockaert J. Glutamate receptors induce a burst of superoxide via activation of nitric oxide synthase in arginine-depleted neurons. J Biol Chem 1994; 269: 12589–12593. [PubMed] [Google Scholar]
- [96].Schulz JB, Lindenau J, Seyfried J, Dichgans J. Glutathione, oxidative stress and neurodegeneration. Eur J Biochem 2000; 267: 4904–4911. [DOI] [PubMed] [Google Scholar]
- [97].Almeida A, Heales SJR, Bolanos JP, Medina JM. Glutamate neurotoxicity is associated with nitric oxide-mediated mitochondrial dysfunction and glutathione depletion. Brain Res 1998; 790: 209–216. [DOI] [PubMed] [Google Scholar]
- [98].Dawson VL, Dawson TM, London ED, Bredt DS, Snyder SH. Nitric oxide mediates glutamate neurotoxicity in primary cortical cultures. Proc Natl Acad Sci USA 1991; 88: 6368–6371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Stella N Glutamate-evoked release of arachidonic acid from mouse brain astrocytes. J Neurosci 1994; 14: 568–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Volterra A, Trotti D, Racagni G. Glutamate uptake is inhibited by arachidonic acid and oxygen radicals via two distinct and additive mechanisms. Mol Pharmacol 1994; 46: 986–992. [PubMed] [Google Scholar]
- [101].Ciani E, Groneng L, Voltattorni M, Rolseth V, Contestabile A, Paulsen RE. Inhibition of free radical production or free radical scavenging protects from the excitotoxic cell death mediated by glutamate in cultures of cerebellar granule neurons. Brain Res 1996; 728: 1–6. [PubMed] [Google Scholar]
- [102].Shanker G, Hampson RE, Aschner M. Methylmercury stimulates arachidonic acid release and cytosolic phospholipase A2 expression in primary neuronal cultures. Neurotoxicology 2004; 25: 399–406. [DOI] [PubMed] [Google Scholar]
- [103].Bolanos JP, Heales SJR, Land JM, Clark JB. Effect of peroxynitrite on the mitochondrial respiratory chain: differential susceptibility of neurones and astrocytes in primary culture. J Neurochem 1995; 64: 1965–1972. [DOI] [PubMed] [Google Scholar]
- [104].Dringen R, Pfeiffer B, Hamprecht B. Synthesis of the antioxidant glutathione in neurons: supply by astrocytes of CysGly as precursor for neuronal glutathione. J Neurosci 1999; 19: 562–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Makar TK, Nedergaard M, Preuss A, Gelbard AS, Perumal AS, Cooper AJ. Vitamin E, ascorbate, glutathione, glutathione disulfide, enzymes of glutathione metabolism in cultures of chick astrocytes and neurons: evidence that astrocytes play an important role in antioxidative processes in the brain. J Neurochem 1994; 62: 45–53. [DOI] [PubMed] [Google Scholar]
- [106].Barker JE, Bolanos JP, Land JM, Clark JB, Heales SJR. Glutathione protects astrocytes from peroxynitrite mediated mitochondrial damage: implications for neuronal/astrocytic trafficking and neurodegeneration. Dev Neurosci 1996; 18: 391–396. [DOI] [PubMed] [Google Scholar]
- [107].Clarkson TW. The toxicology of mercury. Crit Rev Clin Lab Sci 1997; 34: 369–403. [DOI] [PubMed] [Google Scholar]
- [108].Murakami M Naraba H, Tanioka T, et al. Regulation of prostaglandin E2 biosynthesis by inducible membrane-associated prostaglandin E2 synthase that acts in concert with cyclooxygenase-2. J Biol Chem 2000; 275: 32783–32792. [DOI] [PubMed] [Google Scholar]
- [109].Tanioka T, Nakatani Y, Semmyo N, Murakami M, Kudo I. Molecular identification of cytosolic prostaglandin E2 synthase that is functionally coupled with cyclooxygenase-1 in immediate prostaglandin E2 biosynthesis. J Biol Chem 2000; 42: 32775–32782. [DOI] [PubMed] [Google Scholar]
- [110].Tanikawa N, Ohmiya Y, Ohkubo H, et al. Identification and characterization of a novel type of membrane-associated prostaglandin E synthase. Biochem Biophys Res Comm 2002; 291: 884–889. [DOI] [PubMed] [Google Scholar]
- [111].Bezzi P, Carmignoto G, Pasti L, et al. Prostaglandins stimulate calcium-dependent glutamate release in astrocytes. Nature 1998; 391: 281–285. [DOI] [PubMed] [Google Scholar]
- [112].Nishihara I, Minami T, Watanabe Y, Ito S, Hayaishi O. Prostaglandin E2 stimulates glutamate release from synaptosomes of rat spinal cord. Neurosci Lett 1995; 196: 57–60. [DOI] [PubMed] [Google Scholar]