
Fig. 1.
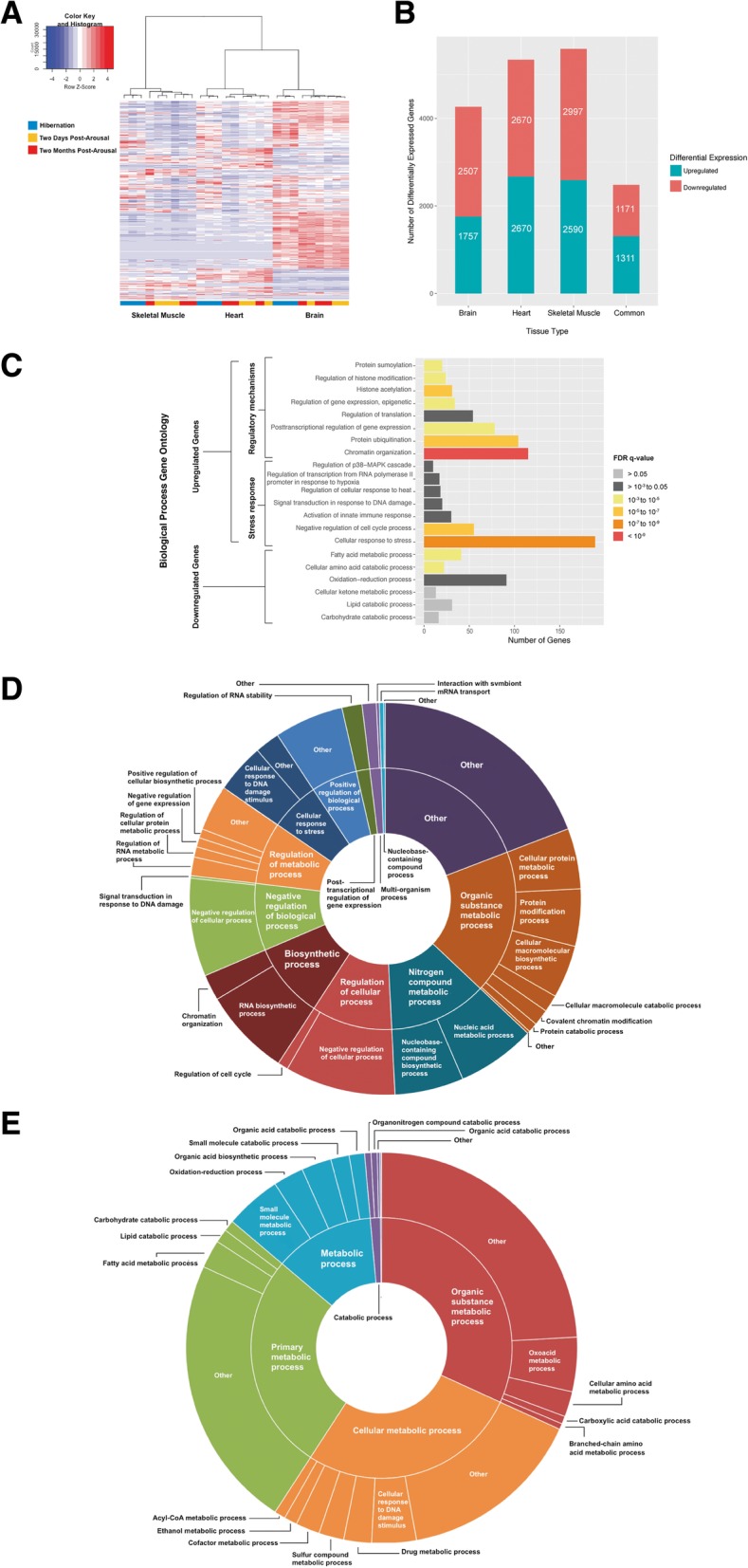
Differential gene expression and gene ontology enrichment analysis. a Heatmap of the 3000 most highly expressed genes in all 27 samples with hierarchical clustering of samples. Each column represents a sample, and each row represents a gene. Each tile in the heatmap shows the normalized expression of a gene (Z-score), which was calculated by subtracting the mean expression value (counts per million) of a gene across all samples from the sample specific expression value, then divided by the standard deviation of the mean expression value of the gene. Hierarchical clustering and the dendogram were calculated using Ward’s method. Colour key shows Z-score, with blue indicating lower expression and red indicating higher expression compared to the mean across all samples. b Bar plot of the number of differentially expressed genes during hibernation as calculated in EdgeR (Log2 fold change > 0.585 – i.e. 1.5-fold change – and FDR < 0.05). The number of differentially expressed genes are overlaid on the bars. c Bar plot of selected enriched gene ontology (GO) terms of upregulated and downregulated genes common to all tissues during hibernation, with color indicating FDR (q-value) of the GO term. d Donut plot of significantly enriched (FDR < 0.05) biological process GO terms for upregulated genes common to all tissues in hibernating individuals. e Donut plot of significantly enriched biological process GO terms for downregulated genes common to all tissues in hibernating individuals. The size of each segment is relative to the number of genes that fall within the specific gene ontology in our dataset