

ABSTRACT
DNA methylation is known to be responsive to prenatal exposures, which may be a part of the mechanism linking early developmental exposures to future chronic diseases. Many studies use blood to measure DNA methylation, yet we know that DNA methylation is tissue specific. Placenta is central to fetal growth and development, but it is rarely feasible to collect this tissue in large epidemiological studies; on the other hand, cord blood samples are more accessible. In this study, based on paired samples of both placenta and cord blood tissues from 169 individuals, we investigated the methylation concordance between placenta and cord blood. We then employed a machine-learning-based model to predict locus-specific DNA methylation levels in placenta using DNA methylation levels in cord blood. We found that methylation correlation between placenta and cord blood is lower than other tissue pairs, consistent with existing observations that placenta methylation has a distinct pattern. Nonetheless, there are still a number of CpG sites showing robust association between the two tissues. We built prediction models for placenta methylation based on cord blood data and documented a subset of 1,012 CpG sites with high correlation between measured and predicted placenta methylation levels. The resulting list of CpG sites and prediction models could help to reveal the loci where internal or external influences may affect DNA methylation in both placenta and cord blood, and provide a reference data to predict the effects on placenta in future study even when the tissue is not available in an epidemiological study.
KEYWORDS: DNA methylation, illumina humanmethylation 450, cord blood, placenta, machine learning
Introduction
DNA methylation affects cell differentiation and tissue development especially during the in utero period[1–3]. Many exposures occurring during the prenatal period have been shown to affect DNA methylation of the developing child [4,5]. During this period, the placenta has a key role because it is the organ transferring nutrients from the mother to the fetus, and can adapt to various exposures [6–13]. Nevertheless, placenta is not always easily collectable in large-scale epidemiological studies.
Significant progress in the high-throughput sequencing technology has been achieved over the past decade, and large-scale whole-genome data analyses have identified genetic susceptibility loci in many complex diseases [14–17]. Genome-wide studies have also shown that many DNA methylation patterns are highly conserved across tissues [2,18]. DNA methylation can serve as an important basis for the analysis and prediction of complex diseases. For instance, according to the IlluminaGoldenGate Bead Array platform, which includes 1,505 CpG sites from 807 genes, the mean correlation of the methylation levels in tissues from 11 individuals is 0.85 (range: 0.74 to 0.94)2. Ursini et al. found that methylation in target organs (i.e., the brain prefrontal cortex), which has important biological functions, correlates with the methylation in peripheral blood mononuclear cells [19]. In 2013, Barault et al. reported that leukocyte DNA methylation levels in imprinted genes can serve as substitute markers for DNA methylation in cancer tissue [20]. Ma et al. have proposed statistical models to predict DNA methylation levels in an artery and atrium using DNA methylation levels in peripheral blood because the latter is easily accessible [21]. De Carli et al. performed a locus-specific methylation prediction based on paired cord blood and placenta datasets [22].
In general, the computational methods of DNA methylation can be classified into the following two main types: (1) prediction of the methylation status of whole CpG islands or fragments of CpG islands; and (2) prediction of the methylation status of a single CpG site. The methylation status of a CpG site can be represented as a continuous methylation beta value measured from microarray. Although there are a variety of approaches to DNA methylation prediction, few studies have focused on genome-wide locus-specific methylation status (continuous beta values) across human tissues. In this study, we collected paired samples of human tissues, i.e., cord blood and the placenta, from 169 individuals, and measured methylation levels at individual CpG sites distributed across the human genome using the Illumina HumanMethylation450 BeadChips. Our purposes were to predict the locus-specific methylation status in the placenta based on the DNA methylation in cord blood, to improve prediction accuracy at the CpG sites with variations in the placenta, to determine the enrichment of the set of well-predicted CpGs in functional pathways or disease pathways and to test the utility of the cross-tissue prediction model for potential studies.
Results
The raw methylation pattern across tissues
Most DNA methylation beta values were found to be conserved across the cord blood and placenta when all available probes were assessed (after quality control; QC). The correlation (CpG-wise R2) between the cord blood and placenta in the 169 samples ranged from 0.61 to 0.76 with a mean of 0.66 (Figure 1). Despite the high correlation (CpG-wise R2), there were many CpG sites with methylation levels close to (0,0) and (1,1) in the scatter plot (Figure 2(a)). To visualize the distribution of the measured DNA methylation levels in the cord blood and placenta, we transformed the dot density in the scatter plot of sample #1 into a colour plot using the ggplot2 R package (Supplementary Figure 1). We observed obvious red areas at the two ends of the y = x line; this finding implied that there was a large number of dots at the two ends (CpG sites with extreme values of methylation i.e. close to 0 or close to 1). To evaluate correlations between the tissues at an intermediate level of DNA methylation, we excluded the CpGs with extremely high or low DNA methylation levels in both tissues, i.e. removed the CpG sites meeting two criteria: (1) minimum methylation beta values>0.8 or maximum beta values <0.2 in both tissues and (2) minimum methylation beta values>0.9 or maximum beta values <0.1 in both tissues, and these criteria were the same for both the cord blood and placenta. The correlation calculated with all CpG sites included could be potentially inflated by these almost completely methylated or unmethylated CpG sites. Removing the extreme CpG sites may decrease the artificial between-tissue correlation coefficients and uncover the relation at intermediate CpGs, which would show more tissue specificity. Moreover, the CpGs with intermediate DNA methylation levels could be those showing DNA methtylation variation among individuals: these CpGs are most likely to be influenced by environmental exposures and/or associated with chronic disease development.
Figure 1.
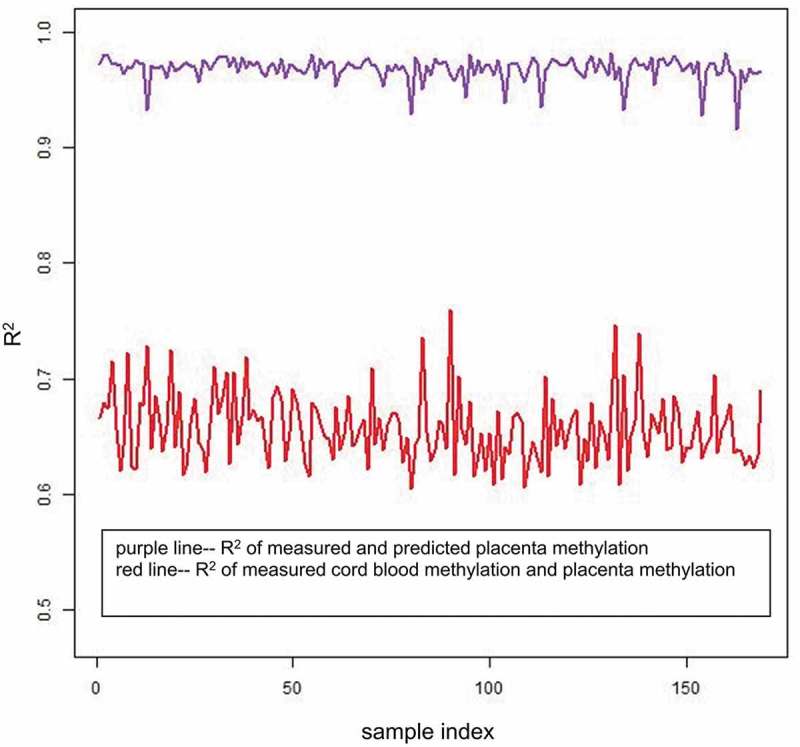
R2 of the 169 samples. The x-axis represents the sample index from 1 to 169, and the y-axis represents the CpG-wise R2 of the 169 samples. The red line represents the CpG-wise R2 of measured methylation beta values in the cord blood and measured methylation beta values in the placenta, and the purple line represents the CpG-wise R2 of measured methylation beta values in the placenta and predicted methylation beta values in the placenta by single-CpG-based SVM and leave-one-out cross-validation.
Figure 2.
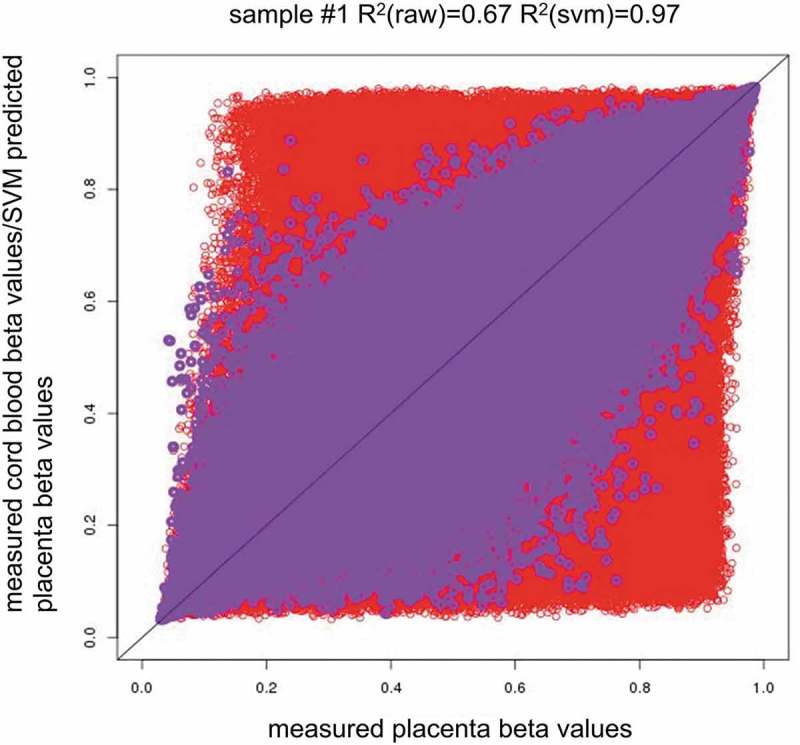
Methylation pattern across tissues and the between-tissue differences across individuals. (2a). Red circles indicate measured placenta beta values vs. measured cord blood beta values (x = measured placenta beta values, y = measured cord blood beta values), and purple circles indicate measured placenta beta values vs. SVM predicted placenta beta values (x = measured placenta beta values, y = predicted placenta beta values by single-CpG-based SVM and leave-one-out cross-validation). R2(raw) = CpG-wise R2 between measured methylation beta values in the placenta and measured methylation beta values in the cord blood. R2(svm) = CpG-wise R2 between measured methylation beta values in the placenta and predicted methylation beta values in the placenta by single-CpG-based SVM and leave-one-out cross-validation.
After removal of the CpG sites with minimum methylation beta values>0.8 or maximum beta values <0.2 in both the cord blood and placenta, some red areas remained in the upper right corner and lower left corner as presented in Supplementary Figure 2, and the maximum density value substantially decreased from 45 to 25. After the exclusion of the CpG sites with minimum DNA methylation beta values>0.9 or maximum beta values<0.1 among all the subjects and tissues, the correlations decreased (CpG-wise R2 ranged from 0.58 to 0.74, mean 0.63). The correlations further decreased (CpG-wise R2 ranged from 0.43 to 0.65, mean 0.51) after removal of the CpG sites with minimum DNA methylation beta values>0.8 or maximum beta values<0.2 among all the subjects and tissues (Table 1, Figure 1, and Supplementary Figures 3 and 4). The magnitude of the difference between the cord blood and placenta was similar across the individuals (Figure 2(b), Supplementary Figures 5–23). The CpG sites manifesting differences in methylation values across tissues determined the tissue-specific patterns of DNA methylation.
Table 1.
CpG-wise R2 in different tissue pairs.
| All CpGs |
Removed CpGs with min methylation beta values >0.9 or max beta values <0.1 |
Removed CpGs with min methylation beta values >0.8 or max beta values <0.2 |
||||||
|---|---|---|---|---|---|---|---|---|
| Tissue pair | Mean R2 | Range | Mean R2 | Range | CpGs removed | Mean R2 | Range | CpGs removed |
| measured Placenta- measured Cord Blood | 0.66 | 0.61, 0.76 | 0.63 | 0.58, 0.74 | 15822 | 0.51 | 0.43, 0.65 | 99076 |
| measured Placenta- SVM predicted Placenta | 0.97 | 0.92, 0.98 | 0.97 | 0.91, 0.98 | 15822 | 0.95 | 0.87, 0.97 | 99076 |
For the ‘All CpGs’ column, we used all CpG sites in each sample to calculate the CpG-wise R2. For the ‘Removed CpGs with min methylation beta values>0.9 or max beta values<0.1’ column, we excluded the extreme CpGs that fell within this range and used the remaining data to calculate the CpG-wise R2, and this column is similar to the ‘Removed CpGs with min methylation beta values >0.8 or max beta values<0.2’ column. For the ‘measured Placenta-measured Cord Blood’ row, the R2 is the squared correlation coefficient of measured methylation in both placenta and cord blood. For the ‘measured Placenta-SVM predicted Placenta’ row, the R2 is the squared correlation coefficient of measured methylation in the placenta and predicted methylation in the placenta by single-CpG-based SVM and leave-one-out cross-validation.
Figure 2b.
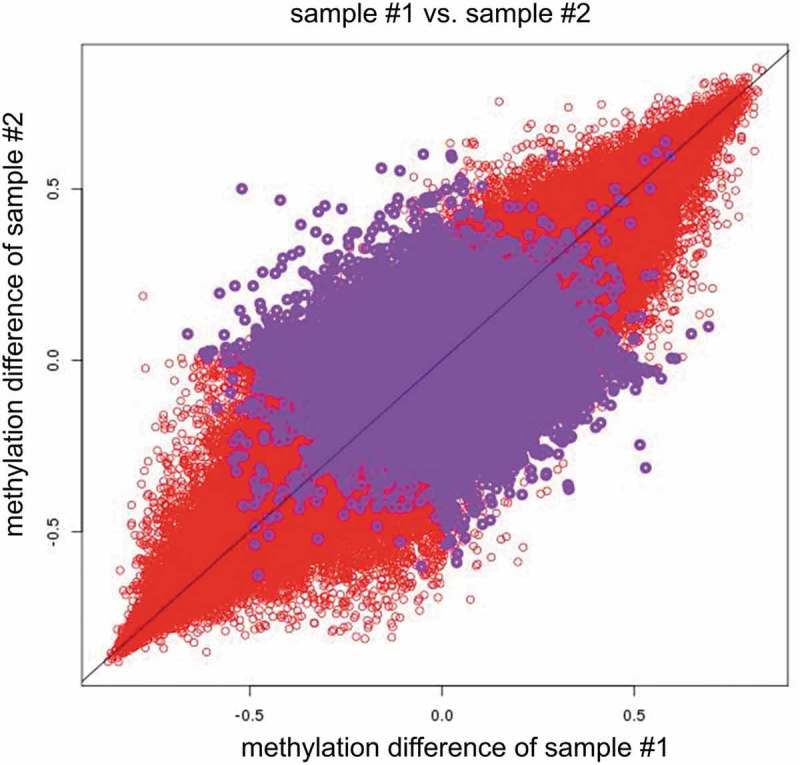
Red circles indicate measured placenta beta values–measured cord blood beta values of sample #1 vs. measured placenta beta values–measured cord blood beta values of sample #2(x = measured placenta beta values–measured cord blood beta values of sample #1, y = measured placenta beta values–measured cord blood beta values of sample #2). Purple circles indicate measured placenta beta values–SVM predicted placenta beta values in sample #1 vs. measured placenta beta values–SVM predicted placenta beta values in sample #2(x = measured placenta beta values–SVM predicted placenta beta values of sample #1, y = measured placenta beta values–SVM predicted placenta beta values of sample #2). Note: ‘–’ represents minus sign.
Locus-specific methylation prediction
We evaluated the utility of the DNA methylation prediction via support vector machine (SVM) and paired tissue samples: the cord blood and placenta. We treated the cord blood as the surrogate tissue and the placenta as the target tissue. A leave-one-out cross-validation procedure was performed to estimate prediction accuracy and avoid overfitting. After iterating through all the samples, we obtained anmatrix of the predicted methylation values in the placenta and a matrix of their observed methylation values as experimentally measured by the Illumina HumanMethylation450 array, where represents the number of samples, and denotes the number of CpG sites. The CpG-wise and sample-wise squared correlation coefficient (R2) and mean absolute error (MAE) between these two matrices were used to evaluate the prediction performance (see Methods).
The mean R2 of all 169 samples increased from 0.66 (CpG-wise R2 between measured DNA methylation in both the cord blood and placenta, then the mean across 169 samples) to 0.97 (mean R2 between measured placenta DNA methylation and predicted placenta DNA methylation according to the single-CpG-based SVM model). The improvement afforded by the proposed method (the single-CpG-based SVM prediction) is illustrated in the scatter plots of sample #1 in Figure 2(a) (measured cord blood vs. measured placenta and predicted placenta vs. measured placenta). The predicted placenta methylation values were much closer to their experimental counterparts (methylation values measured directly in the placenta). The difference between measured and predicted placenta DNA methylation greatly diminished (as compared to the difference between measured cord blood and measured placenta DNA methylation) and was consistent across the samples (Figure 2(b)). A similar improvement was also observed in other samples; we presented examples using samples #1 to #169 (Supplementary Figures 5–42).
After excluding the extreme CpG sites in both the cord blood and placenta, we still noted increases in the correlation (CpG-wise R2). After removal of the CpG sites meeting two criteria (1) minimum methylation beta values>0.8 or maximum beta values <0.2 in both tissues; (2) minimum methylation beta values>0.9 or maximum beta values <0.1 in both tissues, our prediction model enhanced overall CpG-wise R2 (Table 1). These results indicated that we can relatively accurately predict the levels of DNA methylation in the placenta by means of cord blood, including some with DNA methylation levels not at the extremes.
At individual CpG sites, when there were substantial variations (standard deviation[SD] >0.1, 33,314 CpGs) in the placenta, the trends in sample-wise R2 vs. MAE negatively correlated as shown in Figure 3, which indicates that the R2 is larger, and the MAE is smaller, or vice versa. For measured cord blood and placenta methylation, the mean sample-wise R2 for all CpGs was 0.0453, the mean sample-wise R2 for CpGs with SD > 0.1 was 0.0677, and the mean sample-wise R2 for CpGs with SD > 0.2 (786 CpGs) was 0.2064. For measured and predicted placenta methylation, the mean sample-wise R2 for all CpGs was 0.0410, the mean sample-wise R2 for CpGs with SD > 0.1 was 0.0737, and the mean sample-wise R2 for CpGs with SD > 0.2 was 0.2167. The results revealed that the proposed method had higher prediction accuracy evaluated by the mean sample-wise R2 of the CpGs with SD > 0.1 or 0.2 and improved the cross-tissue prediction accuracy at the CpGs with substantial variations in the placenta.
Figure 3.
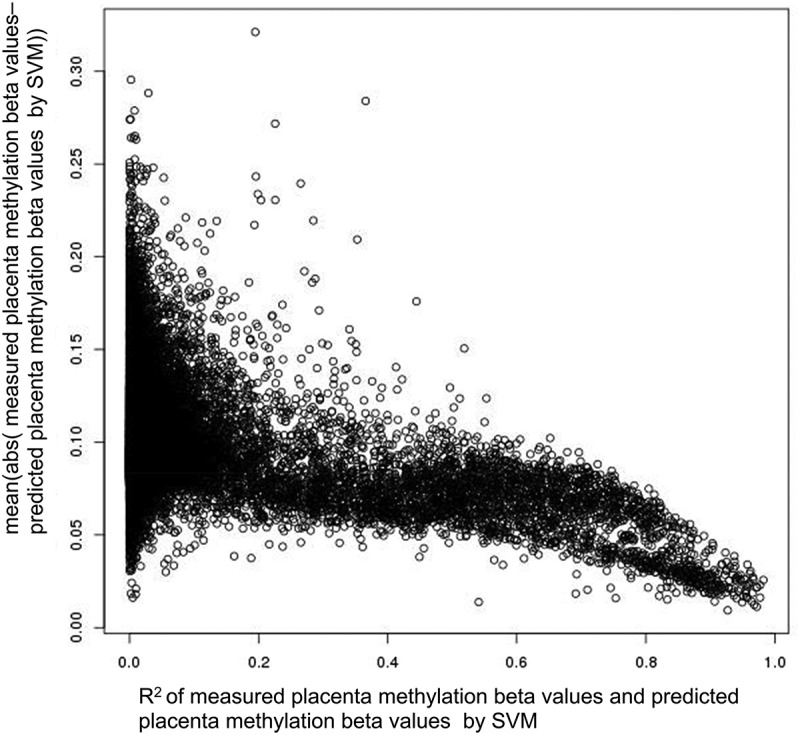
Relationship between R2 and mean absolute error. The y-axis is the sample-wise mean absolute error (MAE) of measured placenta methylation beta values and predicted placenta methylation beta values by single-CpG-based SVM and leave-one-out cross-validation, and the x-axis is the sample-wise R2 of measured placenta methylation beta values and predicted placenta methylation beta values by single-CpG-based SVM and leave-one-out cross-validation (CpGs with SD > 0.1).
The effect of sample size on prediction accuracy
In addition to the leave-one-out cross-validation, we performed evaluation of the impact of the size of the training dataset. We carried out twofold and threefold cross-validation (see Methods) and obtained predictive results listed in Table 2. Additionally, we conducted an experiment to estimate prediction accuracy when changing the size of the training dataset. First, 100 samples were randomly selected from 169 samples to serve as the testing dataset; the training dataset was chosen from the remaining 69 samples; and the sample size of the training dataset was varied from 3 to 69. For each sample size, we used the training dataset to build the single-CpG-based SVM models and utilized the testing dataset to validate the models. The mean CpG-wise R2 of measured placenta methylation and SVM predicted placenta methylation in 100 testing samples were calculated as prediction accuracy and are plotted in Figure 4. We calculated the CpG-wise R2 using all CpG sites and the remaining CpG sites after removing the extreme CpG sites with minimum methylation beta values>0.8 or maximum beta values<0.2, respectively, and found that the mean CpG-wise R2 increased with the sample size both before and after the removal of the extreme CpGs, and the mean CpG-wise R2 reached 0.96 when the sample size was 20 and then rose slowly.
Table 2.
Mean R2 using the twofold and threefold cross-validation methods.
| 2-fold cross-validation |
3-fold cross-validation |
|||||
|---|---|---|---|---|---|---|
| Tissue pair | All CpGs | Removed CpGs with min methylation beta values >0.9 or max beta values <0.1 | Removed CpGs with min methylation beta values >0.8 or max beta values <0.2 | All CpGs | Removed CpGs with min methylation beta values >0.9 or max beta values <0.1 | Removed CpGs with min methylation beta values >0.8 or max beta values <0.2 |
| measured Placenta-measured Cord Blood | 0.6567879 | 0.6333165 | 0.5079859 | 0.6567879 | 0.6333165 | 0.5079859 |
| measured Placenta- SVM predicted Placenta | 0.9672072 | 0.9644117 | 0.9472045 | 0.9676109 | 0.9648506 | 0.9478592 |
For the ‘All CpGs’ column, we used all CpG sites in each sample to calculate the CpG-wise R2, and then obtained the mean R2. For the ‘Removed CpGs with min methylation beta values>0.9 or max beta values<0.1’ column, we excluded the extreme CpGs that fell within this range and used the remaining data to calculate the mean CpG-wise R2, and this column is similar to the ‘Removed CpGs with min methylation beta values>0.8 or max beta values<0.2’ column. For the ‘measured Placenta-measured Cord Blood’ row, the R2 is the squared correlation coefficient of measured methylation in both placenta and cord blood. For the ‘measured Placenta-SVM predicted Placenta’ row, the R2 is the squared correlation coefficient of measured methylation in the placenta and predicted methylation in the placenta by single-CpG-based SVM.
Figure 4.
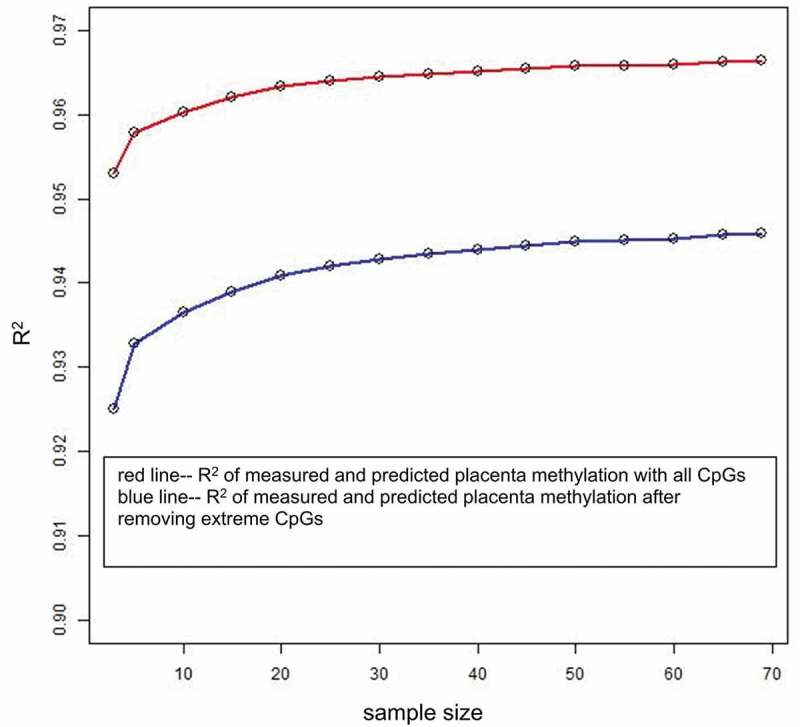
Effect of sample size on prediction accuracy. The x-axis is the sample size of the training dataset, and the y-axis is the mean of CpG-wise R2 for measured placenta methylation beta values and predicted placenta methylation beta values by single-CpG-based SVM for 100 testing samples. For the blue line, the extreme CpG sites with a minimum methylation beta value >0.8 or a maximum beta value <0.2 were removed.
The single-CpG-based model vs. multiple-CpG-based model
Our cross-tissue prediction model based on a single CpG only chose DNA methylation at the same CpG site as a predictor; however, the DNA methylation at a CpG site correlated with certain other CpG sites in the genome, and those other sites may contribute to further improvement of prediction accuracy. We tested the utility of SVM, which can leverage information from multiple correlating CpGs as predictors to enhance cross-tissue prediction accuracy (see Methods). For one sample, the computational complex is M×(M–1), where M is the number of all CpG sites available. We randomly selected 20 CpGs with substantial SD in the placenta but small R2 based on the single-CpG prediction to assess the usefulness of this approach. The CpG sites were selected with SD > 0.1 and sample-wise R2 < 0.1. The latter is the squared correlation coefficient of measured placenta DNA methylation and the placenta DNA methylation predicted by the single-CpG-based SVM. For each selected CpG site, in each round of the leave-one-out cross-validation procedure, we used the training dataset to select 30 CpG sites in the cord blood that highly correlated with the target CpG in the placenta, and we included these correlating CpG sites in the SVM models as predictors.
A neighbouring-CpG-based model has been proposed elsewhere to predict DNA methylation levels in the placenta by means of DNA methylation levels in cord blood and includes two predictors: (1) the same CpG site and (2) the mean DNA methylation levels of neighbouring CpG sites [22]. To examine the utility of stand-alone and multiple CpG sites in prediction models, the single-CpG-based SVM model, 30-correlating-CpG-based SVM model and neighbouring-CpG-based SVM model [22] were compared. After randomly choosing 20 CpG sites that satisfied the above criteria, we applied the three prediction models to these CpG sites and calculated the mean sample-wise R2 of measured and predicted DNA methylation among the 20 CpG sites (Supplementary Table 1). The results suggested that the multiple-correlating-CpG-based method can improve prediction accuracy (mean sample-wise R2) from 0.00366 in the single-CpG-based model to 0.04063 in the 30-correlating-CpG-based model, whereas the mean sample-wise R2 between measured cord blood methylation and placenta methylation at these 20 CpG sites was 0.01791, and the mean sample-wise R2 yielded by the neighbouring-CpG-based model was 0.00713. We hypothesized that a larger sample size may further improve the prediction model by incorporating multiple correlating CpGs because the increased number of degrees of freedom from a greater number of samples can afford more accurate estimates of the model parameters.
A comparison of well-predicted CpGs in datasets cord blood–placenta, blood–artery, and blood–atrium
To determine how many common CpG sites could be well predicted among different human tissues, we chose the well-predicted CpGs with R2 > 0.8 from the following three datasets: Cord Blood–Placenta, Blood–Artery [21] and Blood–Atrium [21], where sample-wise R2 is the squared correlation coefficient of measured DNA methylation values in the placenta, artery or atrium and predicted DNA methylation values in the placenta, artery or atrium (on the basis of the single-CpG-based SVM prediction). The number of all CpGs available after QC in datasets Blood–Artery and Blood–Atrium was 435,605, and the number of all CpGs available after QC in the Cord Blood–Placenta dataset was 437,882. A Venn diagram was plotted to show the overlaps among the sets of well-predicted CpGs from the three datasets (Figure 5). The number of well-predicted CpGs in the Blood–Atrium dataset was 3,684, that in the Blood–Artery dataset 3,751, and in the Cord Blood–Placenta dataset 1,012. The number of well-predicted CpG in the overlap between datasets Blood–Atrium and Cord Blood–Placenta was 204, that in the overlap between datasets Blood–Artery and Cord Blood–Placenta was 198, between the Blood–Artery and Blood–Atrium datasets 2,149, and among the three datasets: 152. The Venn diagram illustrates that 152 common CpGs can be well predicted across the three datasets. The results indicate that 15% of well-predicted CpGs in the Cord Blood–Placenta dataset are consistent with 4% of well-predicted CpGs in the Blood–Atrium and Blood–Artery datasets. Moreover, the number of well-predicted CpGs in the overlap between the Blood–Artery dataset and Blood–Atrium dataset is more than 10 times that between the Cord Blood–Placenta dataset and one of the other two datasets, even though sample size in the Cord Blood–Placenta dataset is much larger (n = 169 vs. n = 14 in the Blood–Artery or – Atrium dataset), because the latter two tissues (atrium and artery) are more similar. These results imply that closer tissue types could show relatively higher prediction performance.
Figure 5.
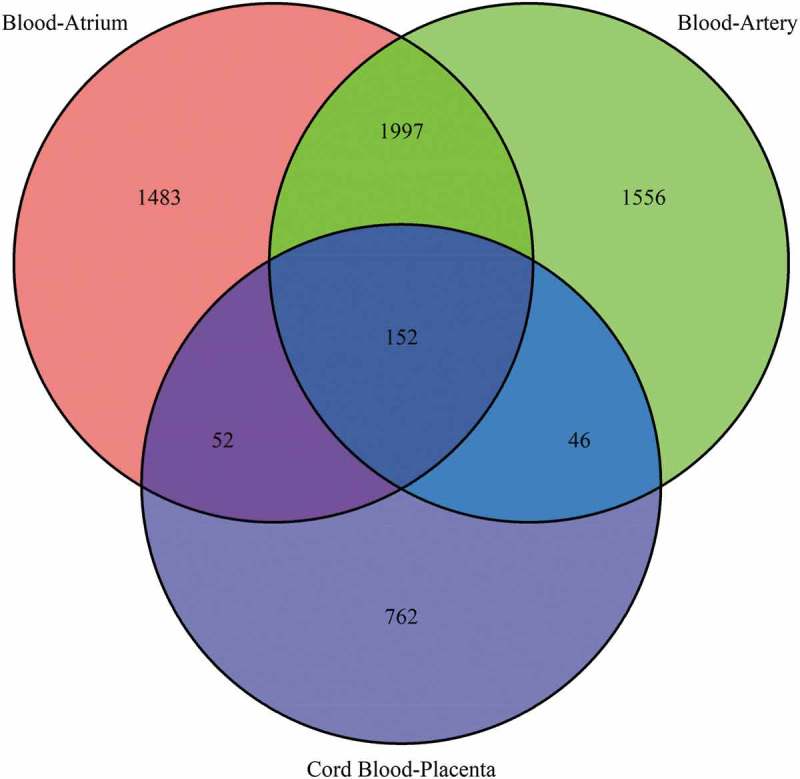
Venn diagram of well-predicted CpGs across the three datasets. The numbers in the circle represent the well-predicted CpGs (sample-wise R2 > 0.8) in the three datasets, and the Venn diagram shows the intersection of the well-predicted CpGs across the following three datasets: cord blood–placenta, blood–artery and blood–atrium.
Distribution of genomic annotation categories
The correlation of cord blood and the placenta may vary in different regions of the genome, and this situation may affect the prediction results. Accordingly, we calculated the genomic distribution stratified by Illumina annotation category for the dataset of all the CpGs and the dataset of 1,012 well-predicted CpGs. Three genomic categories: ‘CpG island’, ‘promoter’ and ‘enhancer’ were chosen to determine the differences between the ‘all-CpGs’ dataset and the ‘1,012 well-predicted CpGs’ dataset. The proportions of the three above-mentioned genomic categories were 0.32, 0.21, and 0.22, respectively, for the all-CpGs dataset and 0.32, 0.21, and 0.17 for the well-predicted CpGs dataset. The CpG island-related distributions are listed in Table 3. The proportions for the all-CpGs dataset were larger than those for the well-predicted CpGs dataset at the ‘Shore’, and the proportions for the all-CpGs dataset were smaller than those for the well-predicted CpGs dataset at the ‘Shelf’. The results showed that the proportions of ‘CpG island’ and ‘promoter’ in the well-predicted CpGs dataset are equal to those in the all-CpGs dataset, but the proportion of ‘enhancer’ is smaller in the former than in the latter.
Table 3.
CpG island-related distributions.
| Dataset | Island | N_Shelf | N_Shore | S_Shelf | S_Shore | Other |
|---|---|---|---|---|---|---|
| All CpGs | 0.3199 | 0.0501 | 0.1318 | 0.0445 | 0.1031 | 0.3506 |
| Well-predicted CpGs | 0.3192 | 0.0642 | 0.1196 | 0.0583 | 0.0672 | 0.3715 |
The categories were designated by Illumina annotation. Five genomic annotation categories including island, northern shelf (N_shelf), southern shelf (S_shelf), northern shore (N_shore) and southern shore (S_shore) are listed. Each proportion is the number of the CpGs belonging to the same category divided by the sum of CpGs in the dataset. The ‘All CpGs’ row is the proportion for the all CpGs dataset and the ‘Well-predicted CpGs’ row is the proportion for the well-predicted CpGs dataset.
Cross-tissue prediction improves the utility of cord blood
We performed clustering analyses using measured placenta methylation, predicted placenta methylation and measured cord blood methylation (see Supplementary Figure 43). Supplementary Figure 43(a) depicts the clustering of measured placenta methylation, 43B presents the clustering of predicted placenta methylation, and 43C depicts the clustering of measured cord blood methylation. We subdivided the 169 samples into four groups according to the four most distinct sub-branches from top to bottom of the clustering tree, and labelled each group with a color rectangle in each clustering figure. The overall matched proportion is defined as the total number of matching samples in the four groups divided by the number of all samples. After running through the four groups in every possible combination, we finally hit on the four matching groups in (1) measured placenta methylation and predicted placenta methylation (A-B), and (2) measured placenta methylation and measured cord blood methylation (A-C), according to the maximum overall matched proportion. The same colour rectangle indicates the matching groups. The overall matched proportion of the former was 48.5%, and that of the latter was 43.2%. The proportion of matching samples for each group was defined as the number of the matching samples in one group divided by the number of samples in the matching group in measured placenta methylation. The results of clustering comparisons are listed in Supplementary Table 2. The proportion of matching samples of the first three columns in plot 43B is greater than or equal to that in plot 43C, but the proportion of matching samples of the fourth column is smaller in plot 43B than in plot 43C. This finding is expected since all proportions should add up to 100%. This result was obtained in the largest group is reassuring because the larger group contains samples that are not so close to one another as compared to samples in the smaller group. The results show that the clustering based on predicted placenta methylation is more similar to the clustering based on measured placenta methylation. Thus, our prediction models improve the utility of cord blood as a surrogate of the placenta. This finding suggests that the predicted placenta methylation could be helpful for any analysis that involves array-wide CpG sites, such as clustering analysis, multi-dimensional scaling (similar to principal component analysis but more robust to outliers), and epigenetic similarity matrix construction for mixed model association analysis.
Pathway analysis of well-predicted CpG sites
We report a list of CpGs (Supplementary Table 3) that can be well predicted with R2 > 0.8 (sample-wise R2 is the squared correlation coefficient between measured placenta DNA methylation and predicted placenta DNA methylation using the single-CpG-based SVM). To identify the potential gene pathways associated with these well-predicted CpGs, we performed a pathway analysis using the ‘missMethyl’ R package [23] (see Methods). The set of 1,012 well-predicted CpGs turned out to be enriched in 143 pathways associated with biological processes in the KEGG pathway database (False Discovery Rate [FDR] < 0.05), but was not found to be enriched in any pathway in the Gene Ontology (GO) database (FDR < 0.05). We provide Supplementary Table 4 with rows for each KEGG category tested and various statistics of interest, such as FDR. The hsa04930, hsa04940, and hsa04750 are related to type II diabetes mellitus, type I diabetes mellitus, and inflammatory mediator regulation of transient receptor potential channels, respectively.
Discussion
In this study, first we examined the methylation correlation between the placenta and cord blood, and then a machine-learning-based method was proposed to predict the DNA methylation values in the placenta based on the DNA methylation values in the cord blood. Our results indicate that the cross-tissue prediction method can improve the utility of cord blood at a certain number of loci, and a large size of a training sample can enhance prediction accuracy. Furthermore, the set of 1,012 well-predicted CpG sites is enriched in 143 KEGG pathways, and we also provide the corresponding SVM models established by our dataset for potential studies.
A comparison with the existing DNA methylation prediction models
In their early stage, most computational methods and tools were developed to predict the methylation status of CpG island fragments [24–29]. When high-throughput microarray and sequencing data became widely and publicly available, the methods proposed later were able to predict DNA methylation status at a CpG site in the whole genome [30–32]. Fan et al. developed a computational model to predict DNA methylation levels and discover more rheumatoid-arthritis-related genes on the basis of 14 human tissues with both whole-genome bisulfite sequencing and Illumina HumanMethylation450 array data [33]. This model integrates cell type-specific Illumina HumanMethylation450 array data and common DNA sequence features, and then predicts the methylation levels of CpGs outside the Illumina HumanMethylation450 covered sites. The correlation coefficient between the measured and predicted methylation values is 0.9 in leave-one-tissue-out cross-validation procedures. Zhang et al. developed a random forest classifier to predict the DNA methylation status in whole blood and identified the features that contribute to prediction accuracy [34]. A deep-learning model named ‘CpGenie’ can predict the DNA methylation status of a CpG site in immortalized cell lines and the impact of non-coding variants on DNA methylation [35]. The two above-mentioned methods are both based on sequence context information and output predicted DNA methylation status as ‘methylated’ or ‘unmethylated’.
There are two studies focused on cross-tissue prediction [21,22]. Ma et al. proposed a linear regression model (LM) and SVM model to predict DNA methylation levels in an artery and atrium using the DNA methylation levels in peripheral blood [21]. De Carli et al. predicted DNA methylation levels in the placenta using DNA methylation levels in cord blood [22], and this statistical model integrates the same CpG site and the mean DNA methylation levels of neighbouring CpG sites identified by the Aclust R package [36]. We compared the cross-tissue prediction models: the 30-correlating-CpG-based SVM model and neighbouring-CpG-based SVM [22] model and reported the mean sample-wise R2 of measured and predicted DNA methylation (Supplementary Table 1). The results show that the 30-correlating-CpG-based SVM model has higher prediction accuracy than does the neighbouring-CpG-based SVM model at the CpG sites with substantial variations in the placenta.
The impact of partially methylated domains on the cross-tissue prediction
Although most of human tissues have high levels of DNA methylation genome-wide, the placenta manifests lower methylation levels [37,38]. Studies have showed that partially methylated domains (PMDs) are extended genomic regions manifesting a reduced average level of DNA methylation and may play a crucial role in special placental methylation patterns [39–43]. Schroeder et al. carried out genome-wide sequencing of bisulfate-treated DNA (MethylC-seq) and Illumina Infinium450K array in the placenta and found that partially methylated domains are stable across placental samples and lead to global hypomethylation and distinct methylation patterns as compared with other human tissues, including cord blood [44]. The latter contains only the subset of cells (nucleated red blood cells, nRBC) that has similarly low array-wide DNA methylation relative to the placenta [45].
Subsequently, we calculated the means of CpG-wise R2 of methylation values between paired tissues, and they turned out to be 0.66, 0.83, and 0.81 for cord blood and placenta, blood and atrium [21], blood and artery [21], respectively; means of sample-wise R2 are 0.0453, 0.1345, and 0.1280, respectively; means of sample-wise R2 of measured and predicted placenta, atrium, or artery methylation values are 0.0410, 0.1767, and 0.1783, respectively. The results indicate that the correlation between the cord blood and placenta is lower than that between blood and atrium and between blood and artery. The methylation pattern causes a decrease in prediction accuracy between the cord blood and placenta compared to blood–atrium and blood–artery.
Epidemiological utility
We provided two examples to demonstrate how our results might be useful for potential studies. Salihu et al. used cord blood DNA methylation to investigate differential methylation levels in candidate genes for preterm birth between black and non-black individuals [46]. They found that DNA methylation at CpG site cg07404485 (PON1gene) had a lower methylation level in black individuals. By examining our database, we found that cord blood DNA methylation at this CpG site was positively associated (R = 0.508, p value = 1.835e-12) with that in the placenta, suggesting that DNA methylation in the placenta might have a lower methylation level in black individuals than in non-black individuals. Joubert et al. conducted a meta-analysis of the association between newborn cord blood DNA methylation and maternal smoking in pregnancy across 13 cohorts, and identified 6,073 statistically differentially methylated CpGs in relation to maternal smoking [47]. By examining our database, we found that cord blood DNA methylation at six CpG sites was positively associated with that in the placenta: R = 0.896 (cg16909109), R = 0.804 (cg07573717), R = 0.925 (cg16309518), R = 0.902 (cg20544437), R = 0.805 (cg00453258), and R = 0.878 (cg02948944), all p-values < 2.2e-16.
We collected and classified the well-predicted CpG sites in Supplementary Table 3, where the CpG sites (1,012) with R2 > 0.8 are listed in data sheet 1, the CpG sites (5,380) with R2 > 0.5 are listed in data sheet 2 and the CpG sites (20,493) with R2 > 0.2 are listed in data sheet 3. The prospective investigators who have only cord blood methylation data and a disease or trait – who are interested in the association of the significant loci in the placenta with the disease or trait – may predict the methylation levels at these significant loci in the placenta with the methylation levels in cord blood based on our newly developed cross-tissue model (see Supplementary files) and then analyse the association of the predicted methylation in the placenta with the disease or trait. To implement a convenient application for potential studies, we built 1,012 SVM models (single-CpG prediction models) at well-predicted CpG sites (Supplementary Table 3, data sheet 1) using 169 paired cord blood and placenta samples and ultimately saved them as an R data file (Supplementary files). We hope additional epigenome-wide association studies and publicly available data will facilitate application of the cross-tissue model for exploring the mechanism underlying the association between the placenta and a disease or trait in the future because the placenta is central to fetal growth and development.
Strength and limitation of cross-tissue prediction
In this investigation, the proposed method was able to improve the utility of methylation in cord blood for predicting methylation in the placenta at a certain number of loci, and the mean sample-wise R2 slightly increased from 0.21 to 0.22 (SD > 0.2). Moreover, we built SVM models based on multiple correlating CpG sites to enhance the prediction performance at some CpG sites with substantial SD and small sample-wise R2. The reason for choosing the CpGs with substantial SD is that the CpGs with small SD do not reveal differences in methylation levels across samples, while the CpGs with substantial SD may provide sufficient resolution for cross-tissue prediction.
By comparing leave-one-out cross-validation (Table 1) with twofold and threefold cross-validation (Table 2), we observed that the mean CpG-wise R2 produced by the former is greater than that yielded by the latter, and the mean CpG-wise R2 of threefold cross-validation was slightly greater than that of twofold cross-validation. The training sample size was 168 for leave-one-out cross-validation, 112 for 3-fold cross-validation and 84 for 2-fold cross-validation. The results suggest that a larger training sample size can improve prediction accuracy. Furthermore, on the basis of the experiments on training sample size (Figure 4), we recommend that the sample size of the training dataset is greater than 20.
At the CpG sites with substantial variation, the accuracy of the cross-tissue prediction could be improved further by incorporation of correlating CpG sites. The single-CpG-based SVM model, 30-correlating-CpGs-based SVM model, and neighbouring-CpG-based SVM model are compared in Supplementary Table 1. The SVM model based on multiple correlating CpGs performed better at a small number of CpG sites (cg22844669 and cg06085683) but did not work well with all the CpG sites included.
In summary, our results have shown that a machine-learning-based method can predict DNA methylation in the placenta using DNA methylation in cord blood at a limited number of loci and 1,012 prediction models at well-predicted CpG sites were established for potential future studies. Nonetheless, it is still challenging to develop a cross-tissue prediction model with satisfactory accuracy at all CpG sites; therefore, investigators need to be careful when designing future potential applications.
Methods
Study population and tissue collection
The cord blood and placenta tissues were obtained from participants in Genetics of Glucose Regulation in Gestation and Growth (Gen3G), which is a prospective population-based cohort study of pregnant women and their newborns receiving care at the Centre Hospitalier Universitaire de Sherbrooke (CHUS) in Canada [48–50]. Expecting mothers were recruited during the first trimester of pregnancy if they were older than 18 years of age, had a singleton pregnancy and did not receive a diagnosis of pre-pregnancy diabetes or gestational diabetes during the first trimester. In total, 169 paired cord blood and placenta samples were included in this analysis with information regarding epigenome-wide DNA methylation. The study protocols were approved by the CHUS ethics review board and written informed consents were obtained from all the women before their enrolment in the study in accordance with the Declaration of Helsinki.
DNA methylation normalization and quality control
Among our overall sample of women and newborns, we obtained 192 paired cord blood–placenta dyads. After extracting the DNA from the cord blood and placenta, we used HumanMethylation450 BeadChips (Illumina, Inc., San Diego, CA, USA) to measure the DNA methylation levels at 485,512 CpG sites across the genome. The samples were imported into the R software as a raw data object of the red-green channel set (rgSet) via the minfi package [51] in Bioconductor. We applied the ‘dasen’ function in the R WateRmelon package [52] to normalize the probes. Eight controls were removed, and seven outliers were further excluded according to principal component analysis plots. Individual data points with detection p > 0.01 were treated as missing data. We removed each sample with more than 20% of missing values across the epigenome, excluded each CpG with more than 5% of missing values across all samples, and filtered the CpGs that overlapped with single-nucleotide polymorphisms (SNPs) and/or cross-hybridized to other loci [53,54]. In total, 169 cord blood–placenta samples with 437,882 CpG sites met the quality control standards for both tissues and were included in the analysis.
Statistical models for methylation prediction
Cross-tissue prediction models based on single CpG sites
The cross-tissue prediction models were built using a training dataset to predict the methylation value (level) in a testing dataset. Suppose that the values of the DNA methylation in the training dataset were split into two matrices, i.e., and , where corresponds to the surrogate tissue, and denotes the target tissue. Each sample is a row, and each CpG is a column in the matrix that contains samples and CpG sites. Let and ( and ) be the elements of matrices and , respectively. Then, is the ith row, and is the j-th column of experimentally measured methylation matrix . A similar definition was used for and. For a given CpG site, we chose andas the training dataset to build an SVM model, designated as. For a new sample, predicted methylation value in the target tissue could be calculated by applyingto the newly developed model; i.e.,, where is the methylation value of the surrogate tissue from the sample being predicted.
Cross-tissue prediction models based on multiple correlating CpG sites
We proposed a framework to predict the methylation levels in a target tissue using multiple correlating CpG sites. We used the training surrogate data matrix () and target CpG dataset () to build the linear regression models and calculated the p-values. The correlating CpG sites ranked by the p-values were then selected as good predictors to build the SVM model. For a given CpG site j, we used and as the training dataset and then obtained the correlating CpG sites (where ) according to the p-values of regression models, where ; , is the j-th column; and is the number of the selected correlating CpG sites. The prediction model was denoted as, where and . For a new sample, predicted value in the target tissue could be calculated by applying to the newly developed model, i.e., , where is the methylation value of the surrogate tissue from the sample subjected to the prediction.
Assessment of the prediction models
We applied the leave-one-out cross-validation method to evaluate the prediction performance of the proposed models. The steps were as follows:
(1) Constructing datasets
One sample () is left out and used as the testing dataset, and the other samples serve as the training dataset.
(2) Building the models
Statistical prediction models (i.e., SVM) are built using the training dataset.
(3) Prediction
The predicted values are obtained for the target tissue samples in the testing dataset by substituting the methylation values of the surrogate tissue in the testing dataset into the newly developed models.
(4) Repeat steps (1)–(3) for all samples
(5) Evaluation parameters
After predicted methylation values were obtained for all samples and all CpG sites (where ), prediction accuracy was evaluated by squared correlation coefficient (R2) and mean absolute error (MAE) for each specific sample (CpG-wise , MAE = ) or each specific CpG site (sample-wise , MAE = ).
Twofold cross-validation
(1) Randomly divide the 169 samples into two sets with one containing 84 samples, and the other containing 85 samples.
(2) Use one set as the training set and the other as the testing set and then switch them.
(3) Predict the methylation values for all 169 samples and calculate R2.
Threefold cross-validation
(1) Randomly divide the 169 samples into three subsets, with two containing 56 samples and the third containing 57 samples.
(2) Use one as the testing set and the other two as the training set and then switch the training set and testing set.
(3) Predict methylation values for all the 169 samples and calculate R2.
Pathway enrichment analysis
The ‘gometh’ function in the ‘missMethyl’ R package was employed for the enrichment analysis [23]. This function takes a vector of significant CpG sites, and maps the CpG sites to Entrez Gene IDs, then tests for GO term or KEGG pathway enrichment by a hypergeometric test, taking into account the number of CpG sites per gene on the Illumina HumanMethylation450 or EPIC array. The main parameters were set as follows:
sig.cpg = the 1,012 well-predicted CpG sites,
all.cpg = all CpG sites on the Illumina HumanMethylation450 array,
collection = ‘KEGG’,
prior.prob = TRUE.
Implementation of the cross-tissue prediction in the r packages
We proposed the following two statistical models: the single-CpG-based SVM model and multiple-correlating-CpG-based SVM model. The SVM model was implemented using the e1071 R packages [55]. We also provide R scripts, which are available for downloading from our laboratory website (http://lianglab.rc.fas.harvard.edu/CordBloodPlacentaMethylation/), based on these models to perform cross-tissue methylation prediction. The R functions provided in this package can be utilized to construct prediction model for the methylation values within a pair of surrogate and target tissues. The established prediction model can then be applied to a new dataset where only the methylation value in the surrogate tissue is available and outputs a predicted methylation value of the target tissue. Furthermore, for the 1,012 well-predicted CpG sites, we built single-CpG-based SVM models using our 169 cord blood and placenta samples. The users may apply the methylation values in cord blood to calculate the methylation values in the placenta on the basis of our SVM models.
Funding Statement
This work was supported by the National Natural Science Foundation of China (61471078), Program for Liaoning Excellent Talents in University (LJQ2015011), visiting scholar grant of the China Scholarship Council (201806575028), Fundamental Research Funds for the Central Universities [3132014306, 3132015213, 3132017075], Natural Science Foundation of Liaoning Province of China, Scientific Research Foundation for the Returned Overseas Chinese Scholars, State Education Ministry. Gen3G was supported by a Fonds de la recherche du Québec en santé (FRQ-S) operating grant [to M.-F.H.]; a Canadian Institute of Health Research [CIHR] Operating grant (to [M.-F.H.]); a Diabète Québec grant (to P.P.) and a Canadian Diabetes Association operating grant. MFH is the recipient of an ADA Pathways to Stop Diabetes Early Investigator Award [1-15-ACE-26]. LB is a research scholar from the Fonds de recherche du Québec en santé (FRQS) and a member of the FRQS-funded Centre de recherche du CHUS (affiliated to the Centre hospitalier universitaire de Sherbrooke). Funding was also obtained from NIEHS [P30 ES000002].
Acknowledgments
First, we would like to acknowledge all women for their participation and their willingness to let their newborns be part of the study. We acknowledge the Blood Sampling Clinic in Pregnancy at Centre HospitalierUniversitaire de Sherbrooke (CHUS), which supports research activities integrated to the Blood Sampling in Pregnancy clinic; the assistance of clinical research nurses and research assistants for recruiting women and obtaining consent for the study; the CHUS biomedical laboratory for performing biochemical assays. We also acknowledge CHUS Research Obstetric Services for their help in collecting delivery samples.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary Material
Supplemental data for the article can be accessed here.
References
- [1].Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6–21. [DOI] [PubMed] [Google Scholar]
- [2].Byun HM, Siegmund KD, Pan F, et al. Epigenetic profiling of somatic tissues from human autopsy specimens identifies tissue- and individual-specific DNA methylation patterns. Hum Mol Genet. 2009;18:4808–4817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Fleisch AF, Wright RO, Baccarelli AA.. Environmental epigenetics: a role in endocrine disease? J Mol Endocrinol. 2012;49:R61–R7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Laufer BI, Kapalanga J, Castellani CA, et al. Associative DNA methylation changes in children with prenatal alcohol exposure. Epigenomics-Uk. 2015;7:1259–1274. [DOI] [PubMed] [Google Scholar]
- [5].Wang IJ, Chen SL, Lu TP, et al. Prenatal smoke exposure, DNA methylation, and childhood atopic dermatitis. Clin Exp Allergy. 2013;43:535–543. [DOI] [PubMed] [Google Scholar]
- [6].Suzuki M, Maekawa R, Patterson NE, et al. Amnion as a surrogate tissue reporter of the effects of maternal preeclampsia on the fetus. Clin Epigenetics. 2016;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Zhang XJ, Pei LJ, Li RT, et al. Spina bifida in fetus is associated with an altered pattern of DNA methylation in placenta. J Hum Genet. 2015;60:605–611. [DOI] [PubMed] [Google Scholar]
- [8].Nelissen ECM, Dumoulin JCM, Daunay A, et al. Placentas from pregnancies conceived by IVF/ICSI have a reduced DNA methylation level at the H19 and MEST differentially methylated regions. Hum Reprod. 2013;28:1117–1126. [DOI] [PubMed] [Google Scholar]
- [9].Turan N, Ghalwash MF, Katari S, et al. DNA methylation differences at growth related genes correlate with birth weight: a molecular signature linked to developmental origins of adult disease? Bmc Med Genomics. 2012. 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Turan N, Katari S, Gerson LF, et al. Inter- and intra-individual variation in allele-specific DNA methylation and gene expression in children conceived using assisted reproductive technology. Plos Genet. 2010;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Wong EC, Hatakeyama C, Robinson WP, et al. DNA methylation at H19/IGF2 ICR1 in the placenta of pregnancies conceived by in vitro fertilization and intracytoplasmic sperm injection. Fertil Steril. 2011;95:2524–U544. [DOI] [PubMed] [Google Scholar]
- [12].Litzky JF, Deyssenroth MA, Everson TM, et al. Placental imprinting variation associated with assisted reproductive technologies and subfertility. Epigenetics-Us. 2017;12:653–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Xu N, Barlow GM, Cui J, et al. Comparison of genome-wide and gene-specific DNA methylation profiling in first-trimester chorionic villi from pregnancies conceived with infertility treatments. Reprod Sci. 2017;24:996–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Ikram MK, Sim X, Jensen RA, et al. Four novel Loci (19q13, 6q24, 12q24, and 5q14) influence the microcirculation in vivo. Plos Genet. 2010;6:e1001184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hardy J, Singleton A. Genomewide association studies and human disease. N Engl J Med. 2009;360:1759–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Scott LJ, Mohlke KL, Bonnycastle LL, et al. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science. 2007;316:1341–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Dixon AL, Liang L, Moffatt MF, et al. A genome-wide association study of global gene expression. Nat Genet. 2007;39:1202–1207. [DOI] [PubMed] [Google Scholar]
- [18].Caliskan M, Cusanovich DA, Ober C, et al. The effects of EBV transformation on gene expression levels and methylation profiles. Hum Mol Genet. 2011;20:1643–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ursini G, Bollati V, Fazio L, et al. Stress-related methylation of the catechol-O-methyltransferase Val 158 allele predicts human prefrontal cognition and activity. J Neurosci. 2011;31:6692–6698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Barault L, Ellsworth RE, Harris HR, et al. Leukocyte DNA as surrogate for the evaluation of imprinted Loci methylation in mammary tissue DNA. Plos One. 2013;8:e55896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Ma BS, Wilker EH, Willis-Owen SAG, et al. Predicting DNA methylation level across human tissues. Nucleic Acids Res. 2014;42:3515–3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].De Carli MM, Baccarelli AA, Trevisi L, et al. Epigenome-wide cross-tissue predictive modeling and comparison of cord blood and placental methylation in a birth cohort. Epigenomics-Uk. 2017;9:231–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Phipson B, Maksimovic J, Oshlack A. missMethyl: an R package for analyzing data from illumina’s humanMethylation450 platform. Bioinformatics. 2016;32:286–288. [DOI] [PubMed] [Google Scholar]
- [24].Bock C, Paulsen M, Tierling S, et al. CpG island methylation in human lymphocytes is highly correlated with DNA sequence, repeats, and predicted DNA structure. Plos Genet. 2006;2:243–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Das R, Dimitrova N, Xuan Z, et al. Computational prediction of methylation status in human genomic sequences. Proc Natl Acad Sci U S A. 2006;103:10713–10716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Fang F, Fan S, Zhang X, et al. Predicting methylation status of CpG islands in the human brain. Bioinformatics. 2006;22:2204–2209. [DOI] [PubMed] [Google Scholar]
- [27].Feltus FA, Lee EK, Costello JF, et al. Predicting aberrant CpG island methylation. Proc Natl Acad Sci U S A. 2003;100:12253–12258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Previti C, Harari O, Zwir I, et al. Profile analysis and prediction of tissue-specific CpG island methylation classes. BMC Bioinformatics. 2009;10:116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Zheng H, Wu HW, Li JP, et al. CpGIMethPred: computational model for predicting methylation status of CpG islands in human genome. Bmc Med Genomics. 2013;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].He J, Sun MA, Wang Z, et al. Characterization and machine learning prediction of allele-specific DNA methylation. Genomics. 2015;106:331–339. [DOI] [PubMed] [Google Scholar]
- [31].Shi Y, Guo Y, Hu Y, et al. Position-specific prediction of methylation sites from sequence conservation based on information theory. Sci Rep. 2015;5:12403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Zhou X, Li ZC, Dai Z, et al. Prediction of methylation CpGs and their methylation degrees in human DNA sequences. Comput Biol Med. 2012;42:408–413. [DOI] [PubMed] [Google Scholar]
- [33].Fan S, Li C, Ai R, et al. Computationally expanding infinium HumanMethylation450 BeadChip array data to reveal distinct DNA methylation patterns of rheumatoid arthritis. Bioinformatics. 2016;32:1773–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Zhang WW, Spector TD, Deloukas P, et al. Predicting genome-wide DNA methylation using methylation marks, genomic position, and DNA regulatory elements. Genome Biol. 2015;16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Zeng H, Gifford DK. Predicting the impact of non-coding variants on DNA methylation. Nucleic Acids Res. 2017;45:e99–e99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Sofer T, Schifano ED, Hoppin JA, et al. A-clustering: a novel method for the detection of co-regulated methylation regions, and regions associated with exposure. Bioinformatics. 2013;29:2884–2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Ehrlich M, Gama-Sosa MA, Huang LH, et al. Amount and distribution of 5-methylcytosine in human DNA from different types of tissues of cells. Nucleic Acids Res. 1982;10:2709–2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Gama-Sosa MA, Midgett RM, Slagel VA, et al. Tissue-specific differences in DNA methylation in various mammals. Biochim Biophys Acta. 1983;740:212–219. [DOI] [PubMed] [Google Scholar]
- [39].Aran D, Toperoff G, Rosenberg M, et al. Replication timing-related and gene body-specific methylation of active human genes. Hum Mol Genet. 2011;20:670–680. [DOI] [PubMed] [Google Scholar]
- [40].Chu T, Handley D, Bunce K, et al. Structural and regulatory characterization of the placental epigenome at its maternal interface. Plos One. 2011;6:e14723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Lister R, Pelizzola M, Dowen RH, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Novakovic B, Saffery R. DNA methylation profiling highlights the unique nature of the human placental epigenome. Epigenomics-Uk. 2010;2:627–638. [DOI] [PubMed] [Google Scholar]
- [43].Salhab A, Nordstrom K, Gasparoni G, et al. A comprehensive analysis of 195 DNA methylomes reveals shared and cell-specific features of partially methylated domains. Genome Biol. 2018;19:150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Schroeder DI, Blair JD, Lott P, et al. The human placenta methylome. Proc Natl Acad Sci U S A. 2013;110:6037–6042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].de Goede OM, Lavoie PM, Robinson WP. Characterizing the hypomethylated DNA methylation profile of nucleated red blood cells from cord blood. Epigenomics-Uk. 2016;8:1481–1494. [DOI] [PubMed] [Google Scholar]
- [46].Salihu HM, Das R, Morton L, et al. Racial differences in DNA-methylation of CpG sites within preterm-promoting genes and gene variants. Matern Child Health J. 2016;20:1680–1687. [DOI] [PubMed] [Google Scholar]
- [47].Joubert BR, Felix JF, Yousefi P, et al. DNA methylation in newborns and maternal smoking in pregnancy: genome-wide consortium meta-analysis. Am J Hum Genet. 2016;98:680–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Guillemette L, Allard C, Lacroix M, et al. Genetics of glucose regulation in gestation and growth (Gen3G): a prospective prebirth cohort of mother-child pairs in Sherbrooke, Canada. Bmj Open. 2016;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Cardenas A, Allard C, Doyon M, et al. Validation of a DNA methylation reference panel for the estimation of nucleated cells types in cord blood. Epigenetics-Us. 2016;11:773–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Allard C, Desgagne V, Patenaude J, et al. Mendelian randomization supports causality between maternal hyperglycemia and epigenetic regulation of leptin gene in newborns. Epigenetics-Us. 2015;10:342–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Aryee MJ, Jaffe AE, Corrada-Bravo H, et al. Minfi: a flexible and comprehensive bioconductor package for the analysis of infinium DNA methylation microarrays. Bioinformatics. 2014;30:1363–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Pidsley R, Wong CCY, Volta M, et al. A data-driven approach to preprocessing Illumina 450K methylation array data. Bmc Genomics. 2013;14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Chen YA, Lemire M, Choufani S, et al. Discovery of cross-reactive probes and polymorphic CpGs in the illumina infinium humanmethylation450 microarray. Epigenetics-Us. 2013;8:203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Li J, Zhu X, Yu K, et al. Genome-wide analysis of DNA methylation and acute coronary syndrome. Circ Res. 2017;120:1754–1767. [DOI] [PubMed] [Google Scholar]
- [55].Karatzoglou A, Meyer D, Hornik K. Support vector machines in R. J Stat Softw. 2006;15. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.