

Abstract
A thermophilic and hydrogenogenic carboxydotroph, Carboxydothermus pertinax, performs hydrogenogenic CO metabolism in which CODH-II couples with distally encoded ECH. To enhance our knowledge of its hydrogenogenic CO metabolism, we performed whole transcriptome analysis of C. pertinax grown under 100% CO or 100% N2 using RNA sequencing. Of the 2577 genes, 36 and 64 genes were differentially expressed genes (DEGs) with false discovery rate adjusted P value < 0.05 when grown under 100% CO or 100% N2, respectively. Most of the DEGs were components of 23 gene clusters, suggesting switch between metabolisms via intensive expression changes in a relatively low number of gene clusters. Of the 9 significantly expressed gene clusters under 100% CO, CODH-II and ECH gene clusters were found. Only the ECH gene cluster was regulated by the CO-responsive transcriptional factor CooA, suggesting that others were separately regulated in the same transcriptional cascade as the ECH gene cluster. Of the 14 significantly expressed gene clusters under 100% N2, ferrous iron transport gene cluster involved in anaerobic respiration and prophage region were found. Considering that the expression of the temperate phage was strictly repressed under 100% CO, hydrogenogenic CO metabolism might be stable for C. pertinax.
Electronic supplementary material
The online version of this article (10.1007/s00792-019-01091-x) contains supplementary material, which is available to authorized users.
Keywords: Hydrogenogenic carboxydotroph, Carbon monoxide dehydrogenase, RNA sequencing, Carboxydothermus pertinax
Introduction
Carbon monoxide (CO) is toxic for many organisms. However, some prokaryotes, called CO-utilizing microbes (carboxydotrophs), can use CO as the source of energy and carbon for their growth (CO metabolism) (Mörsdorf et al. 1992). Owing to the low redox potential (− 520 mV) of CO (Grahame and DeMoll 1995), anaerobic carboxydotrophs can couple CO oxidation to various reduction reactions such as sulfate to sulfide (sulfate-reducers), water to H2 (hydrogenogens), Fe3+ to Fe2+ (Fe[III]-reducers), and CO2 to acetate (acetogens) or methane (methanogens) (Oelgeschläger and Rother 2008; Sokolova et al. 2009). Many anaerobic and thermophilic carboxydotrophs have been isolated (Sokolova et al. 2009) from hydrothermal environments where CO is supplied by volcanic gas, photochemical and thermochemical decomposition of organic matter, and as a by-product of certain thermophiles (King and Weber 2007; Techtmann et al. 2009). Among them, 25 strains representing 13 genera (21 species) have been reported as hydrogenogenic carboxydotrophs (Sokolova and Lebedinsky 2013). Recently, in addition to these 25 strains, three thermophilic and hydrogenogenic carboxydotrophs, namely Thermococcus barophilus (Kozhevnikova et al. 2016), Thermoanaerobacter kivui (Weghoff and Müller 2016), and Parageobacillus thermoglucosidasius (Mohr et al. 2018), have been reported. Although a number of strains of Moorella thermoacetica have been isolated, only one strain (M. thermoacetica strain AMP) exhibits hydrogenogenic carboxydotrophy, whereas the other strains are acetogenic carboxydotrophs (Jiang et al. 2009). Because of their ability to use potentially toxic CO and produce H2 as the source of energy for other microbes, hydrogenogenic carboxydotrophs are assumed to be important ‘CO scavengers’ and primary producers in the environment (Sokolova and Lebedinsky 2013; Techtmann et al. 2009; Yoneda et al. 2013, 2015).
In anaerobic carboxydotrophs, CODH, with Ni in its active center, catalyzes the oxidoreductive interconversion between CO and CO2 (Ragsdale 2004). The function of CODH has been principally predicted by the genomic context of each CODH gene (cooS; Techtmann et al. 2012). A model of hydrogenogenic carboxydotrophs, Carboxydothermus hydrogenoformans, possesses five genes (cooS-I to -V) which code for the catalytic subunits of CODHs (CODH-I–V) on its genome (Wu et al. 2005). Based on the genomic context of the gene clusters including each cooS and/or empirical evidence, their functions are predicted as follows: CODH-I, energy conversion conjugated with ECH; CODH-II, NAD(P)H generation; CODH-III, carbon fixation in the Wood–Ljungdahl pathway conjugated with acetyl-CoA synthase (ACS); and CODH-IV, oxidative stress response (Wu et al. 2005; Svetlitchnyi et al. 2001). CODH-V does not conserve sequences responsible for its active center, and hence, its physiological function remains unknown (Inoue et al. 2013). cooS-I is arranged in a gene cluster with genes coding for an electron transfer protein (CooF) (Kerby et al. 1992), Ni insertion protein (CooC) (Kerby et al. 1997), a transcriptional factor (CooA) (Shelver et al. 1995), and ECH-related genes. In most hydrogenogenic carboxydotrophs, cooS-I and ECH-related genes form a gene cluster (CODH−ECH gene cluster) or are closely arranged (Sokolova et al. 2009). The hydrogenogenic CO utilization is performed by a complex of three enzymes; CODH-I, CooF, and ECH complex. CO is oxidized by CODH-I and the generated electron is transferred to CooF. Subsequently, this electron is coupled to proton reduction via the ECH complex, producing H2 and forming a proton gradient (Svetlitchnyi et al. 2001; Soboh et al. 2002).
Transcription of the genes in the CODH gene cluster in hydrogenogenic CO metabolism is activated by CO-responsive transcriptional factors CooA, RcoM (Kerby et al. 2008), and CorQR (Kim et al. 2015). Of these, CooA is well characterized in the hydrogenogenic carboxydotroph, Rhodospirillum rubrum (Aono et al. 1996; Roberts et al. 2004) and is present in most of the hydrogenogenic carboxydotrophs (Youn et al. 2004). CooA belongs to the cyclic AMP receptor protein family (Shelver et al. 1995). Homodimeric heme protein CooA is inactive in the absence of CO (Shelver et al. 1997). When CooA senses CO, ligand replacement in CooA leads to its conformational change making it active (Shelver et al. 1997). The active form of CooA can bind to the promoter region, thereby activating transcription through contact with RNA polymerase (Leduc et al. 2001). In the R. rubrum genome, CODH gene cluster and ECH gene cluster are closely arranged and the CooA-binding site is found upstream of both cooF in the CODH gene cluster and cooM in the ECH gene cluster (Fox et al. 1996; Rajeev et al. 2012). The previous study shows that CooA homologs are divided into two phylogenetically distinct groups (CooA-1 and CooA-2). CooA-1 is found in the majority of CooA possessing carboxydotrophs, whereas CooA-2 is found in some carboxydotrophs that possess multiple CODH gene clusters in their genomes (Techtmann et al. 2011). Furthermore, CO-binding assay of two CooA groups shows that both CooA-1 and CooA-2 are in their active forms in high CO concentration, whereas only CooA-2 is in active form even in low CO concentration (Techtmann et al. 2011). This difference in CO activation in the two CooA groups enables the bacterium to regulate multiple CODH gene clusters across wide range of CO concentrations (Techtmann et al. 2011). In addition to CooA, recent studies have reported other anaerobic CO-responsive transcriptional factors RcoM and CO-responsive regulatory system CorQR. Similar to CooA, RcoM, whose gene is adjacent to CODH and ECH gene clusters in the hydrogenogenic carboxydotroph Rubrivivax gelatinosus (phylum Proteobacteria) (Wawrousek et al. 2014), possesses a potential CO-sensor domain-containing heme (Kerby et al. 2008). CorQR pair is composed of CorQ with a DNA-binding domain of LysR-type transcriptional regulator (LTTR) family and CorR with 4-vinyl reductase domain instead of heme to sense CO (Kim et al. 2015). Genes encoding CorQR pair also flanked with CODH−ECH gene cluster in the hydrogenogenic carboxydotrophic archaeon, Thermococcus onnurineus (phylum Euryarchaeota) (Kim et al. 2015).
To date, only a few transcriptional studies about hydrogenogenic CO metabolism have been reported. A transcriptomic analysis of the sulfate-reducing carboxydotroph Desulfovibrio vulgaris using microarray reports that the expression of cooS is dependent on active CooA under low CO concentration (Rajeev et al. 2012). In addition, genome-wide primary transcriptomic analysis of T. onnurineus reports that the expression of cooS is significantly upregulated when they utilize CO as the source of energy for H2 production (Cho et al. 2017). However, there are limited data on comprehensive gene expression pattern of hydrogenogenic CO metabolism.
Carboxydothermus pertinax is isolated from an acidic hot spring in Japan (Yoneda et al. 2012). Despite of its hydrogenogenic carboxydotrophy (Yoneda et al. 2012), as per genome analysis, C. pertinax lacks genes encoding CODH-I catalytic subunit (CooS-I) and its transcriptional factor CooA-1 in their CODH-I−ECH gene cluster (Fukuyama et al. 2017). Furthermore, gene expression analysis in C. pertinax has shown that genes encoding CODH-II catalytic subunit (cooS-II) and distantly encoding the ECH catalytic large and small subunits are remarkably upregulated under 100% CO, suggesting that C. pertinax performs hydrogenogenic CO metabolism in which CODH-II couples with distal ECH (Fukuyama et al. 2018). Since C. pertinax possesses one CooA homolog (CooA-2) unlike C. hydrogenoformans possessing two CooA homologs, its transcriptional regulation from CO response is expected to be simpler than that of C. hydrogenoformans. Therefore, study of C. pertinax will further help us to understand the regulation mechanism of hydrogenogenic CO metabolism. In this study, we performed the whole transcriptome analysis of C. pertinax grown on pyruvate under 100% CO or100% N2 by RNA sequencing (RNA-Seq).
Materials and methods
Growth conditions for transcriptome analysis
To prepare RNA for transcriptome analysis, C. pertinax was grown at 65 °C in modified DSM medium 507 under a headspace of 100% CO or 100% N2 gas according to methods previously described (Fukuyama et al. 2018). We added sodium thiosulfate (final concentration, 1 g/L) as the terminal electron acceptor and sodium pyruvate (final concentration, 2 g/L) as the electron donor and carbon source to the medium in both the gas phase conditions to collect enough cells for analysis. Growth was assessed by direct enumeration of SYBR gold-stained cells collected on 0.2 μm black polycarbonate membrane filters (Advantec, Tokyo, Japan) using a fluorescent microscope (Olympus, Tokyo, Japan). The cells exponentially grown in pre-culture were inoculated to fresh medium and cultivated routinely.
Transcriptome analysis of C. pertinax
When C. pertinax reached the late exponential phase as indicated by arrows (Fig. S1), 10 mL of the culture in both the conditions was collected and total RNA was extracted according to the methods previously described (Fukuyama et al. 2018). After removal of contaminating DNA using TURBO DNase (Invitrogen, Carlsbad, CA, USA), total RNA was purified using Agencourt RNAClean XP (Beckman Coulter, Brea, CA, USA) according to the manufacturer’s instructions. Quantification was performed in the Agilent 2100 Bioanalyzer with Agilent RNA6000 pico kit (Agilent Technologies, Santa Clara, CA, USA). Ribosomal RNA (rRNA) was depleted from the purified total RNA using Ribo-ZeroTM Magnetic Kit (Bacteria) (Epicentre, Madison, WI, USA) according to the manufacturer’s instructions. After rRNA removal, the purified total RNA, which was extracted from total 60 mL from the two replicates, was merged to obtain sufficient total RNA for RNA-Seq. Then, the merged total RNA was reverse transcribed to obtain double-strand cDNA (ds cDNA) using PrimeScript Double-Strand cDNA Synthesis Kit (TaKaRa Bio, Shiga, Japan) for RNA-Seq. The ds cDNA library was constructed with 75 bp paired-end libraries prepared by Nextera® XT DNA sample prep kit (Illumina, San Diego, CA, USA) and sequenced using Illumina MiSeq system (Illumina). For reverse transcription quantitative PCR (RT-qPCR), the purified total RNA was reverse transcribed to single-strand cDNA (ss cDNA) using SuperScript III First-Strand Synthesis System (Invitrogen).
RNA-Seq data analysis
Generated high-quality reads of C. pertinax with Q30 (sequence error rate lower than 0.1%) mapped to their draft genome (BDJK01000000) using Tophat version 2.0.13 (Trapnell et al. 2009) with Bowtie2 version 2.2.2 (mismatches 2 bp) (Langmead and Salzberg 2012). We manually removed contaminated rRNA reads which showed high similarity to C. pertinax 5S, 16S, and 23S rRNA. Read count per gene under both conditions was estimated using featureCounts (Liao et al. 2014). Rarefaction curves for reads under both conditions were constructed using PAST ver.3.17 (Hammer et al. 2001). Gene expression levels were compared and normalized using the R statistical package edgeR (Robinson et al. 2010). DEGs were identified as significantly expressed under 100% CO or 100% N2 when their log2 fold change was > 1 or < − 1, respectively, and their FDR adjusted P value (Q value) was < 0.05.
Analysis to predict the function of DEGs
As a unit of polycistronic transcription, a gene cluster was predicted based on the following criteria: (1) intergenic distance between genes was less than 300 bp, (2) genes were encoded in the same strand, and (3) the visual pattern of read alignment to a locus on C. pertinax genome. The Integrative Genomics Viewer (IGV) software was used to visualize the patterns of read alignment (Thorvaldsdottir et al. 2013). To predict the functions of DEGs, these protein sequences were annotated using BLASTp search (Altschul et al. 1990, 1997) with an e value of 1e−5 at an effective database size of 107 against the COG database (Tatusov 2000). The upstream regions (300 bp) of the predicted gene cluster containing DEGs and solely transcribed DEGs were collected from the C. pertinax draft genome. The primary sigma factor recognition sequences were predicted using Bacterial Promoter Prediction Program (BPROM) with default parameters (Solovyev and Salamov 2011).
The promoter sequences were aligned separately for each motif (− 10 motif and − 35 motif). Logos were prepared using weblogo for visualization (Crooks et al. 2004). To discover motifs in the upstream regions, we used MEME ver5.0.2 program with default parameters (Bailey and Elkan 1994).
Prophage regions encoded in contigs (cut-off < 1500 bp) from the genomes of the three Carboxydothermus species except for the already analyzed C. hydrogenoformans (Wu et al. 2005) were predicted using PHAge Search Tool—Enhanced Release (PHASTER) (Arndt et al. 2016). To classify the predicted prophages in three Carboxydothermus species based on genome-wide similarities, a viral proteomic tree was generated by ViPTree (Nishimura et al. 2017b). The all-against-all distance matrix between viral reference genomes and the predicted phages was calculated on the basis of the normalized bit score of tBLASTx (SG) (Nishimura et al. 2017a), and the proteomic tree was built with BIONJ using the distance matrix in the ViPTree.
RT-qPCR validation for the expression of CO metabolism-related genes
To validate the RNA-Seq expression data, 10 predicted CO metabolism-related genes were selected for RT-qPCR. RT-qPCR primers have been described in the previous study (Fukuyama et al. 2018). PCR amplification was performed according to the previous study (Fukuyama et al. 2018). We served same cDNA samples as RNA-Seq to RT-qPCR. The detection limit of the transcript levels of mRNA for target genes was 1.00 × 102 copies/μL. Relative transcript amounts were calculated using rrsD (16S rRNA) transcripts as an internal standard. All RT-qPCR data represent the mean value of at least triplicate biological determinations.
Accession number of the sequence
RNA-Seq data have been submitted to the DNA Data Bank of Japan Sequence Read Archive under the accession no. DRA007734.
Results
Overview of RNA-Seq
Carboxydothermus pertinax grew under both 100% CO and 100% N2 conditions (Fig. S1). Under 100% CO (carboxydotrophic growth), doubling time was 2.3 h with a final cell density of 8.25 × 107 ± 1.05 × 107 cells/mL. Under 100% N2 (heterotrophic growth), and doubling time was 1.9 h with a final cell density of 4.07 × 108 ± 1.68 × 107 cells/mL. Total RNA was extracted from cells at late exponential phase in both conditions and subjected to RNA-Seq (Fig. S1). A total of approximately 3.54 M and 3.74 M high-quality reads were obtained from 100% CO and 100% N2 conditions, respectively. The total number of the mapped reads to the C. pertinax draft genome was 2.28 M and 1.95 M with 100% CO and 100% N2, respectively (Table S1). Rarefaction curves of the mapped reads under both conditions showed that sequencing data from each condition was exhaustive to describe the transcriptional profile when read counts reached approximately 1.0 M (Fig. S2), suggesting that the count of mapped genes was sufficient for RNA-Seq analysis under both conditions.
In general, central component of bacterial transcription is multi-subunit-DNA-dependent RNA polymerase. When we searched for sigma factors in C. pertinax, 13 sigma factors (one primary sigma factor, 11 alternative sigma factors as sigma70 family, and one sigma54) were found (Table S2). Read count of the primary sigma factor was 6495 and 2855 under 100% CO and 100% N2, respectively. These read counts were an order of magnitude higher among the other sigma70 factors including sigma70 factors for sporulation under both conditions (Table S2). These results clearly indicated that the obtained RNA-Seq reads reflected expression patterns of exponentially growing cells.
Under 100% CO or 100% N2 conditions, 36 genes and 64 genes were identified as DEGs (Q value < 0.05), respectively (Fig. 1). A ratio of the 2577 genes in C. pertinax draft genome to the DEGs was no more than 1.4% under 100% CO and 2.5% under 100% N2 conditions, respectively. Seventy-four percent of the DEGs under 100% CO and 91% of the DEGs under 100% N2 were components of 9 and 14 gene clusters, respectively (Table 1). The remaining 8 DEGs under 100% CO and 6 DEGs under 100% N2 were solely transcribed. As significantly expressed gene clusters are expected to play key roles in both the conditions, we classified them into three categories based on average read counts per kilobase of the genes in the gene clusters containing DEGs (Table 1): very high (average read counts 10,000), high (average read counts 1000), and moderate (average read counts 100).
Fig. 1.
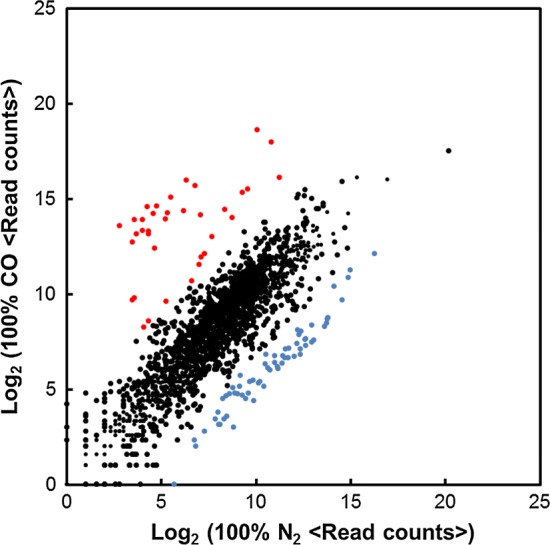
Comparison of read counts between C. pertinax grown under 100% CO and 100% N2. Each plot shows an open-reading frame. Red plots, upregulated DEGs; blue plot, downregulated DEGs; black, unchanged genes
Table 1.
Overview of gene clusters containing DEGs under 100% CO and 100% N2
| Metabolism category | Locus tag | Predicted function | Fold change | No. of genes in gene cluster | No. of DEGs in gene cluster | Average read count | Expression category | |
|---|---|---|---|---|---|---|---|---|
| CO 100% | N2 100% | |||||||
| Significantly expressed gene cluster under CO 100% | ||||||||
| Energy conservation | cpu_03670-03750 | ECH | 480–1760 | 9 | 9 | 35,565 | 48 | Very high |
| cpu_14030-14050 | CODH-II | 70–380 | 3 | 3 | 97,573 | 405 | Very high | |
| cpu_22890-22980 | H2-uptake Ni–Fe-hydrogenase | 20 | 1 | 10 | 471 | 118 | Moderate | |
| Carbohydrate metabolism | cpu_07810-07850 | Anion/Na+ symporter, fumarate hydratase, and succinate dehydrogenase | 460–960 | 5 | 5 | 12,770 | 23 | Very high |
| cpu_15750-15760 | Succinate dehydrogenase | 420–640 | 2 | 2 | 21,045 | 40 | Very high | |
| Transcriptional regulation | cpu_03570-03580 | TetR/AcrR family transcriptional regulator | 20–30 | 1 | 2 | 593 | 46 | Moderate |
| cpu_05930-05940 | LysR-type transcriptional regulator | 60–70 | 2 | 2 | 45,670 | 710 | Very high | |
| Unknown | cpu_03830-03840 | Unknown | 40 | 2 | 2 | 22,067 | 561 | Very high |
| cpu_23290-23300 | Unknown | 10 | 2 | 2 | 4326 | 58 | High | |
| Significantly expressed gene cluster under N2 100% | ||||||||
| Energy conservation | cpu_18560-18580 | Dimethyl sulfoxide reductase | 30–40 | 3 | 3 | 205 | 6819 | High |
| Prophage region | cpu_00830-00840 | Myoviridae like prophage region | 10–50 | 2 | 2 | 971 | 40,766 | Very high |
| cpu_00860-00940 | 9 | 9 | 465 | 15,716 | Very high | |||
| cpu_00950-01010 | 7 | 5 | 32 | 852 | Moderate | |||
| cpu_01060-01070 | 2 | 1 | 163 | 2382 | High | |||
| cpu_01080-01100 | 3 | 3 | 86 | 1504 | High | |||
| cpu_01110-01130 | 3 | 3 | 217 | 5618 | High | |||
| cpu_01140-01200 | 6 | 6 | 139 | 3903 | High | |||
| cpu_01220-01350 | 12 | 10 | 89 | 1428 | High | |||
| Amino acid synthesis | cpu_11010-11030 | Aromatic amino acid biosynthesis | 20–40 | 3 | 3 | 74 | 2021 | High |
| cpu_11630-11700 | Aromatic amino acid biosynthesis | 20–30 | 8 | 4 | 26 | 292 | Moderate | |
| Membrane transport | cpu_18330-18350 | Ferrous iron transport | 10–20 | 3 | 2 | 1174 | 12,640 | Very high |
| Unknown | cpu_13490-13500 | Unknown | 20–50 | 2 | 2 | 35 | 1372 | High |
| cpu_24930-24940 | Unknown | 20 | 2 | 2 | 1862 | 30,424 | Very high | |
Significantly expressed gene clusters under 100% CO or 100% N2
Of the 6 gene clusters in the ‘very high’ category under 100% CO (Table 1), the two gene clusters (cpu_14030-14050 and cpu_03670-03750; 70–380 and 480–1760 fold changes, respectively) were related to energy conservation and encoded CODH-II and ECH, which are essential proteins for hydrogenogenic CO metabolism (Fukuyama et al. 2018) (Fig. 2; Table S3). Furthermore, the two gene clusters were related to carbohydrate metabolism (460–960 and 420–640 fold changes, respectively; Table S3). Of these, the gene cluster (cpu_07810-07850) composed of five genes encoding divalent anion/Na+ symporter, fumarate hydratase α and β subunits, succinate dehydrogenase flavoprotein, and iron–sulfur subunit. The other gene cluster (cpu_15750-15760) composed of two genes encoding succinate dehydrogenase iron–sulfur and cytochrome b subunit. These gene products catalyze the oxidoreductive reaction between succinate and malate in tricarboxylic acid (TCA) cycle. The gene cluster (cpu_05930-05940; 60–70 fold changes) was composed of two genes encoding LTTR, involved in transcriptional regulation, and a hypothetical protein. In addition, a gene encoding another LTTR (cpu_07800) was also identified as a sole DEG (220 fold change). The function of the rest of the gene cluster (cpu_03830-03840) was not predicted from their gene products. Consequently, products of these six gene clusters were considered to play an important and specific role in their hydrogenogenic CO metabolism.
Fig. 2.

Transcriptomic read mapping pattern of the CODH-II gene cluster (a) and the ECH gene cluster (b). Part of genome-wide overview of reads mapped to the C. pertinax draft genome at samples in the CODH-II gene cluster (a) and the ECH gene cluster (b). Read abundance was displayed in a logarithmic scale of 1–30,000 by Integrative Genomics Viewer (Thorvaldsdottir et al. 2013). Red box, DEG under 100% CO; White box, not DEG under 100% CO. Positions of predicted sigma70 promoter sequence was predicted using BPROM with high linear discrimination function score (> 5; Solovyev and Salamov 2011). Black arrows, the position of predicted sigma70 promoter sequence. Red arrow, the position of predicted sigma70 promoter sequence with already known CooA-binding site
Next, we annotated three other categories (high or moderate; Table 1 and S3). These three gene clusters were composed of the following genes: two hypothetical genes (cpu_23290-23300; high), 10 H2-uptake Ni–Fe-hydrogenase genes (cpu_22890-22980; moderate), and TetR/AcrR family transcriptional regulator gene and sodium/phosphate symporter (cpu_03570-03580; moderate).
Of the four gene clusters in the ‘very high’ category under 100% N2 (Table 1 and S4), the two gene clusters (cpu_00830-00840 and cpu_00860-00940; 40 and 30–50 fold changes, respectively) were a part of the prophage region in C. pertinax (Table S4). From the viral proteomic tree generated using ViPTree, this prophage region (54 genes) was close to P2-like Myoviridae viruses, Vibrio virus X29 (NC_024369), and Vibrio phage phi 2 (KJ545483) (Fig. S4). The gene cluster (cpu_18330-18350; 10–20 fold change) was composed of two genes encoding ferrous iron transport proteins A and B (Lau et al. 2016). In addition, a gene (cpu_17460) encoding ferrous iron permease was identified as sole DEG (10 fold change). The function of the rest of the gene cluster (cpu_24930-24940) could not be predicted from their gene products. Products of these four gene products were also considered to play an important role in their heterotrophic growth.
Of the 10 gene clusters in high or moderate categories under 100% N2 (Table 1 and S4), the gene cluster (cpu_18560-18580; high) was involved in energy conservation and encoded dimethyl sulfoxide reductase catalytic molybdopterin subunit, iron–sulfur cluster protein, and integral membrane protein. The six gene clusters (cpu_01060-01070, cpu_01080-01100, cpu_01110-01130, cpu_01140-01200, and cpu_01220-01350; high, cpu_00950-01010; moderate) were found in the prophage region. Furthermore, the two gene clusters (cpu_11010-11030, high; cpu_11630-11700, moderate) were involved in aromatic amino acid biosynthesis. The function of the rest of the gene cluster (cpu_13490-13500; high) could not be predicted from their gene products.
Expressions of genes involved in hydrogenogenic CO metabolism
Apart from the largely upregulated CODH-II gene cluster, RNA-Seq showed no significant expression changes of the CODH (CODH-III–V). In addition to ECH, C. pertinax possesses the H2-uptake Ni–Fe-hydrogenase gene cluster. In this gene cluster, read count of all genes were relatively low in both the conditions and only one gene (cpu_22950) encoding maturation factor for H2-uptake Ni–Fe-hydrogenase was identified as DEG.
To validate RNA-Seq, we performed gene expression analysis for 10 genes (coos-IIA2F1 in CODH-II gene cluster, cooS-III–V in CODH-III–V gene cluster, cooLH in ECH gene cluster, and hyaAB in H2-uptake Ni–Fe-hydrogenase gene cluster) using RT-qPCR (Fig. S4). The relative transcript levels of five genes (cooS-II, cooA2, cooF1, cooH, and cooL) under 100% CO were 20–640-fold higher than those under 100% N2 (Fig. S4). Relative transcript levels of the three remaining genes (cooS-III–V) were not changed. Of them, relative transcript levels of cooS-IV were below the limit of detection (1.00 × 102 copies/μL). Furthermore, relative transcript levels of the two genes (hyaA and hyaB) were relatively higher under 100% CO. Consequently, RT-qPCR analysis for these genes supported the RNA-Seq data (Fig. S4).
Analysis of the regulation mechanism under hydrogenogenic CO metabolism
To predict the regulation mechanism under hydrogenogenic CO metabolism, we explored the transcription start site (TSS) from 300 bp upstream of 36 transcriptional regions (23 gene clusters containing DEGs and 13 sole DEGs), except for two transcriptional regions, which were significantly transcribed by the polar effect of their upstream. We assigned 36 TSSs from all promoter regions, regardless of their transcriptional categories (very high, high, and moderate). These 5′-untranslated region lengths of transcripts were ranged from 19 to 239 bp. The − 35 motif and − 10 motif (Wösten 1998) for primary sigma factor were strictly conserved among all the promoter regions in both the conditions (Fig. S5).
Among the 16 promoter regions of the significantly expressed transcriptional regions under 100% CO, the CooA-binding site was conserved only in the ECH gene cluster. Expect for the ECH gene cluster, we searched conserved binding motifs for transcriptional factor among other promoter regions. However, there is no consensus motif with a significantly low E value in the three transcriptional categories. Among the 20 promoter regions of the significantly expressed transcriptional regions under 100% N2, there is no consensus motif with a significantly low E value in three transcriptional categories.
Discussion
In this study, we performed the whole transcriptome analysis of C. pertinax possessing only one CO-responsive transcriptional factor (CooA-2) as the first RNA-Seq report in hydrogenogenic carboxydotrophic bacterium. Total RNA from C. pertinax cells which grew under 100% CO and 100% N2 were compared to understand the regulation mechanism between hydrogenogenic CO metabolism and heterotrophic metabolism. RNA-Seq data suggested that C. pertinax switched its metabolism by considerable expression changes in a relatively low number of gene clusters.
Our previous study of C. pertinax shows that electrons from CO oxidation are mainly (62%) consumed to reduce H2O to H2; the remaining electrons are utilized in the reduction of thiosulfate by thiosulfate reductase (cpu_06910-06930) (Fukuyama et al. 2018). Genes involved in the transport of ferrous iron (very high category) were largely upregulated under 100% N2, suggesting that the ferrous irons were positively transported into cells in heterotrophic growth. Considering that ferrous irons were incorporated as hemes or iron–sulfur clusters in various energy-generating and regulatory proteins (Braun and Hantke 2011), C. pertinax could conserve energy via anaerobic respiration in the modified DSM medium 507 containing sodium thiosulfate. In contrast, in hydrogenogenic CO metabolism, transport of ferrous iron (very high category) was significantly and incompletely reduced to low expression (Table 1). Instead, both CODH-II gene cluster and ECH gene cluster were largely upregulated. In addition, expressions of the genes encoding thiosulfate reductase were relatively decreased (average read counts were decreased from 3018 to 1843). Therefore, owing to the sensitivity of heme protein to CO, C. pertinax would switch from anaerobic respiration to hydrogenogenic CO metabolism retaining a part of reducing power to the reduction of thiosulfate. The higher yields of C. pertinax grew under 100% N2 than 100% CO (Fig. S1) would be partly explained by a thermodynamic perspective about anaerobic respiration and hydrogenogenic CO metabolism. In addition, owing to the high sensitivity of CooA-2 to CO (Techtmann et al. 2011), C. pertinax would switch their metabolisms in response to low CO concentration.
In general, carboxydotrophs can fix CO2 or CO to acetyl-CoA via Wood–Ljungdahl pathway (Ragsdale 2004). In fact, transcriptome analysis of the acetogenic carboxydotroph, Clostridium ljungdahlii, shows that a gene cluster containing most of the genes for Wood–Ljungdahl pathway is significantly upregulated in their CO metabolism (Tan et al. 2013). In our RNA-Seq data, however, expressions of genes in CODH-III–ACS gene cluster were not upregulated regardless of CO addition. This result suggested that C. pertinax could fix carbon via other pathways in response to CO. Actually, a frameshift of CODH catalytic subunit gene in two hydrogenogenic carboxydotrophs suggested that carbon fixation via Wood–Ljungdahl pathway is not essential for hydrogenogenic CO metabolism (Wu et al. 2005; Omae et al. 2017).
In addition to Wood–Ljungdahl pathway, C. pertinax is predicted to possess an incomplete TCA cycle and an incomplete 3-hydroxypropionate cycle (Fukuyama et al. 2018). Among the incomplete TCA cycle-related genes, genes for fumarate hydratase (EC: 4.2.1.2; cpu_07820-07830 and cpu_21290-21300) and succinate dehydrogenase (EC: 1.3.5.1; cpu_078240-07850, cpu_15750-15760, and cpu_21260-21280) are multicopy (Fukuyama et al. 2017). Of these gene sets, one encoding fumarate hydratase (cpu_07820-07830) and succinate dehydrogenase (cpu_07840-07850 and cpu_15750-15760; Table S3) were largely expressed under 100% CO (very high category), implying that these gene clusters were upregulated in response to CO and products of these genes enhanced carbon fixation via a part of reductive TCA cycle. Furthermore, C. pertinax can conserve energy via their hydrogenogenic CO metabolism in which CO oxidation is coupled with H2 production and CO2 is generated as a by-product of this reaction (Yoneda et al. 2012; Fukuyama et al. 2018). Therefore, we hypothesized that, in addition to Wood–Ljungdahl pathway, C. pertinax fixed CO2 via reductive incomplete TCA cycle in response to CO.
In bacteria, self-cleaving activity of LexA repressor is stimulated by activated recombinase A (RecA) during response to DNA damage (SOS response) (Butala et al. 2009). As the temperate prophage also utilizes the bacterial SOS response system, host RecA promotes the entry of lytic phase when SOS response occurs in the host. In the prophage region, a gene encoding transcription repressor LexA (cpu_00820) was expressed under both conditions, whereas other genes such as DNA replication (cpu_00950) and many structure proteins were significantly downregulated under hydrogenogenic CO metabolism condition, suggesting that the lysis of the temperate phage was strictly repressed with 100% CO. In addition, these data might imply that hydrogenogenic CO metabolism is more stable than heterotrophic metabolism for C. pertinax.
Of the known anaerobic CO-responsive transcriptional factors, two transcriptional factors (CooA and RcoM for carboxydotrophic bacteria) possess heme domain (Kerby et al. 2008) and the other factor (CorQR for carboxydotrophic archaea) possesses 4-vinyl reductase domain to sense CO (Kim et al. 2015). Of the three transcriptional factors as DEGs under 100% CO, TetR/AcrR family transcriptional factor (cpu_03570) acts as the repressor when it senses cellular environmental dynamics (Deng et al. 2013). On the other hand, LTTRs (cpu_05930 and cpu_07800) is a widespread transcriptional factor in bacteria and acts as either activators or repressors of single or operonic genes (Maddocks and Oyston 2008). However, the LTTRs were not considered to be a CO–responsive transcriptional factor, because these possessed neither heme nor 4-vinyl reductase domains. When we explored transcriptional factor with known CO-sensing domain from C. pertinax using BLASTp search, only CooA-2 with heme domain was found. The CooA-binding site was conserved only upstream of the ECH gene cluster, strongly indicating that the ECH gene cluster was regulated by CO-responsive transcriptional factor CooA regulation. Collectively, it was suggested that only the ECH gene cluster was regulated by active CooA and others were regulated secondarily in the same transcriptional cascade as the ECH gene cluster. Otherwise, the expression of gene clusters, expect for the ECH gene cluster, was regulated by an undiscovered CO-responsive transcriptional factor.
Conclusion
Our RNA-Seq analysis showed that C. pertinax switched its metabolism through intense expression changes of relatively low number of gene clusters. In heterotrophic metabolism, C. pertinax conserved energy via anaerobic respiration. On the other hand, owing to the inactivation of heme protein by CO, C. pertinax performed hydrogenogenic CO metabolism under 100% CO instead of anaerobic respiration retaining a part of reducing power from CO oxidation to the reduction of thiosulfate. Notably, genes in the temperate phage were strictly expressed under heterotrophic growth. This result might imply that hydrogenogenic CO metabolism might be more stable for C. pertinax. When C. pertinax switches heterotrophic metabolism to CO metabolism, of the significantly expressed gene clusters under 100% CO, only the ECH gene cluster was regulated by CooA regulation. In addition, no potential CO-responsive transcriptional factors were conserved in C. pertinax. These results suggested that only the ECH gene cluster was regulated by active CooA and others were separately regulated in the same transcriptional cascade as the ECH gene cluster.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
This work was supported by Grant-in-Aid for Scientific Research (A) (25252038), (A) (16H02569), and (S) (16H06381) from The Ministry of Education, Culture, Sports, Science and Technology (MEXT).
Abbreviations
- CODH
Carbon monoxide dehydrogenase
- ECH
Energy-converting hydrogenase
- DEG
Differential expression gene
- LTTR
LysR-type transcriptional regulator
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Altschul SF, Gish W, Miller W, et al. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schäffer AA, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucl Acids Res. 1997;25:3389–3402. doi: 10.1093/NAR/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aono S, Nakajima H, Saito K, Okada M. A novel heme protein that acts as a carbon monoxide-dependent transcriptional activator in Rhodospirillum rubrum. Biochem Biophys Res Commun. 1996;228:752–756. doi: 10.1006/bbrc.1996.1727. [DOI] [PubMed] [Google Scholar]
- Arndt D, Grant JR, Marcu A, et al. PHASTER: a better, faster version of the PHAST phage search tool. Nucl Acids Res. 2016;44:W16–W21. doi: 10.1093/nar/gkw387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Elkan C (1994) Fitting a mixture model by expectation maximization to discover motifs in biopolymers. In: Altman R, Brutlag D, Karp P, Lathrop R and Searls D (eds) Proceedings of the 2nd international conference on intelligent systems for molecular biology. AAAI Press, Menlo Park, pp 226–233 [PubMed]
- Braun V, Hantke K. Recent insights into iron import by bacteria. Curr Opin Chem Biol. 2011;15:328–334. doi: 10.1016/J.CBPA.2011.01.005. [DOI] [PubMed] [Google Scholar]
- Butala M, Žgur-Bertok D, Busby SJW. The bacterial LexA transcriptional repressor. Cell Mol Life Sci. 2009;66:82–93. doi: 10.1007/s00018-008-8378-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho S, Kim MS, Jeong Y, et al. Genome-wide primary transcriptome analysis of H2-producing archaeon Thermococcus onnurineus NA1. Sci Rep. 2017;7:43044. doi: 10.1038/srep43044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng W, Li C, Xie J. The underling mechanism of bacterial TetR/AcrR family transcriptional repressors. Cell Signal. 2013;25:1608–1613. doi: 10.1016/J.CELLSIG.2013.04.003. [DOI] [PubMed] [Google Scholar]
- Fox J, He Y, Shelver D, et al. Characterization of the region encoding the CO-induced hydrogenase of Rhodospirillum rubrum. J Bacteriol. 1996;178:6200–6208. doi: 10.1128/jb.178.21.6200-6208.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuyama Y, Omae K, Yoneda Y, et al. Draft genome sequences of Carboxydothermus pertinax and C. islandicus, hydrogenogenic carboxydotrophic bacteria. Genome Announc. 2017;5:e01648-16. doi: 10.1128/genomeA.01648-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuyama Y, Omae K, Yoneda Y, et al. Insight into energy conservation via alternative carbon monoxide metabolism in Carboxydothermus pertinax revealed by comparative genome analysis. Appl Environ Microbiol. 2018;84:e00458-18. doi: 10.1128/AEM.00458-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grahame DA, DeMoll E. Substrate and accessory protein requirements and thermodynamics of acetyl-CoA synthesis and cleavage in Methanosarcina barkeri. Biochemistry. 1995;34:4617–4624. doi: 10.1021/BI00014A015. [DOI] [PubMed] [Google Scholar]
- Hammer Ø, Harper DAT, Ryan P. PAST: Paleontological statistics software package for education and data analysis. Palaeontol Electron. 2001;4:9. [Google Scholar]
- Inoue T, Takao K, Yoshida T, et al. Cysteine 295 indirectly affects Ni coordination of carbon monoxide dehydrogenase-II C-cluster. Biochem Biophys Res Commun. 2013;441:13–17. doi: 10.1016/j.bbrc.2013.09.143. [DOI] [PubMed] [Google Scholar]
- Jiang B, Henstra AM, Paulo PL, et al. Atypical one-carbon metabolism of an acetogenic and hydrogenogenic Moorella thermoacetica strain. Arch Microbiol. 2009;191:123–131. doi: 10.1007/s00203-008-0435-x. [DOI] [PubMed] [Google Scholar]
- Kerby RL, Hong SS, Ensign SA, et al. Genetic and physiological characterization of the Rhodospirillum rubrum carbon monoxide dehydrogenase system. J Bacteriol. 1992;174:5284–5294. doi: 10.1128/jb.174.16.5284-5294.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerby RL, Ludden PW, Roberts GP. In vivo nickel insertion into the carbon monoxide dehydrogenase of Rhodospirillum rubrum: molecular and physiological characterization of cooCTJ. J Bacteriol. 1997;179:2259–2566. doi: 10.1128/JB.179.7.2259-2266.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerby RL, Youn H, Roberts GP. RcoM: a new single-component transcriptional regulator of CO metabolism in bacteria. J Bacteriol. 2008;190:3336–3343. doi: 10.1128/JB.00033-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MS, Choi AR, Lee SH, et al. A novel CO-responsive transcriptional regulator and enhanced H2 production by an engineered Thermococcus onnurineus NA1 strain. Appl Environ Microbiol. 2015;81:1708–1714. doi: 10.1128/AEM.03019-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King GM, Weber CF. Distribution, diversity and ecology of aerobic CO-oxidizing bacteria. Nat Rev Microbiol. 2007;5:107–118. doi: 10.1038/nrmicro1595. [DOI] [PubMed] [Google Scholar]
- Kozhevnikova DA, Taranov EA, Lebedinsky AV, et al. Hydrogenogenic and sulfidogenic growth of Thermococcus archaea on carbon monoxide and formate. Microbiology. 2016;85:400–410. doi: 10.1134/S0026261716040135. [DOI] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau CKY, Krewulak KD, Vogel HJ. Bacterial ferrous iron transport: the Feo system. FEMS Microbiol Rev. 2016;40:273–298. doi: 10.1093/femsre/fuv049. [DOI] [PubMed] [Google Scholar]
- Leduc J, Thorsteinsson MV, Gaal T, Roberts GP. Mapping CooA. RNA polymerase interactions. Identification of activating regions 2 and 3 in CooA, the co-sensing transcriptional activator. J Biol Chem. 2001;276:39968–39973. doi: 10.1074/jbc.M105758200. [DOI] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- Maddocks SE, Oyston PCF. Structure and function of the LysR-type transcriptional regulator (LTTR) family proteins. Microbiology. 2008;154:3609–3623. doi: 10.1099/mic.0.2008/022772-0. [DOI] [PubMed] [Google Scholar]
- Mohr T, Aliyu H, Küchlin R, et al. CO-dependent hydrogen production by the facultative anaerobe Parageobacillus thermoglucosidasius. Microb Cell Fact. 2018;17:108. doi: 10.1186/s12934-018-0954-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mörsdorf G, Frunzke K, Gadkari D, Meyer O. Microbial growth on carbon monoxide. Biodegradation. 1992;3:61–82. doi: 10.1007/BF00189635. [DOI] [Google Scholar]
- Nishimura Y, Watai H, Honda T, et al (2017a) Environmental viral genomes shed new light on virus-host interactions in the ocean. mSphere 2:e00359–16. 10.1128/msphere.00359-16 [DOI] [PMC free article] [PubMed]
- Nishimura Y, Yoshida T, Kuronishi M, et al. ViPTree: the viral proteomic tree server. Bioinformatics. 2017;106:12826–12831. doi: 10.1093/bioinformatics/btx157. [DOI] [PubMed] [Google Scholar]
- Oelgeschläger E, Rother M. Carbon monoxide-dependent energy metabolism in anaerobic bacteria and archaea. Arch Microbiol. 2008;190:257–269. doi: 10.1007/s00203-008-0382-6. [DOI] [PubMed] [Google Scholar]
- Omae K, Yoneda Y, Fukuyama Y, et al. Genomic analysis of Calderihabitans maritimus KKC1, a thermophilic, hydrogenogenic, carboxydotrophic bacterium isolated from marine sediment. Appl Environ Microbiol. 2017;83:e00832-17. doi: 10.1128/AEM.00832-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragsdale SW. Life with carbon monoxide. Crit Rev Biochem Mol Biol. 2004;39:165–195. doi: 10.1080/10409230490496577. [DOI] [PubMed] [Google Scholar]
- Rajeev L, Hillesland KL, Zane GM, et al. Deletion of the Desulfovibrio vulgaris carbon monoxide sensor invokes global changes in transcription. J Bacteriol. 2012;194:5783–5793. doi: 10.1128/JB.00749-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts GP, Youn H, Kerby RL. CO-sensing mechanisms. Microbiol Mol Biol Rev. 2004;68:453–473. doi: 10.1128/MMBR.68.3.453-473.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelver D, Kerby RL, He Y, Roberts GP. Carbon monoxide-induced activation of gene expression in Rhodospirillum rubrum requires the product of cooA, a member of the cyclic AMP receptor protein family of transcriptional regulators. J Bacteriol. 1995;177:2157–2163. doi: 10.1128/JB.177.8.2157-2163.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelver D, Kerby RL, He Y, Roberts GP. CooA, a CO-sensing transcription factor from Rhodospirillum rubrum, is a CO-binding heme protein. Proc Natl Acad Sci. 1997;94:11216–11220. doi: 10.1073/pnas.94.21.11216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soboh B, Linder D, Hedderich R. Purification and catalytic properties of a CO-oxidizing:H2-evolving enzyme complex from Carboxydothermus hydrogenoformans. Eur J Biochem. 2002;269:5712–5721. doi: 10.1046/j.1432-1033.2002.03282.x. [DOI] [PubMed] [Google Scholar]
- Sokolova T, Lebedinsky A. CO-oxidizing anaerobic thermophilic prokaryotes. In: Satyanarayana T, Littlechild JK, editors. Thermophilic microbes in environmental and industrial biotechnology. 2. Dordrecht: Springer; 2013. pp. 203–231. [Google Scholar]
- Sokolova TG, Henstra AM, Sipma J, et al. Diversity and ecophysiological features of thermophilic carboxydotrophic anaerobes. FEMS Microbiol Ecol. 2009;68:131–141. doi: 10.1111/j.1574-6941.2009.00663.x. [DOI] [PubMed] [Google Scholar]
- Solovyev V, Salamov A. Automatic annotation of microbial genomes and metagenomic sequences. In: Li RW, editor. Metagenomics and its applications in agriculture, biomedicine and environmental studies. Hauppauge: Nova Science Publishers; 2011. pp. 61–78. [Google Scholar]
- Svetlitchnyi V, Peschel C, Acker G, Meyer O. Two membrane-associated NiFeS-carbon monoxide dehydrogenases from the anaerobic carbon-monoxide-utilizing eubacterium Carboxydothermus hydrogenoformans. J Bacteriol. 2001;183:5134–5144. doi: 10.1128/JB.183.17.5134-5144.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Y, Liu J, Chen X, et al. RNA-seq-based comparative transcriptome analysis of the syngas-utilizing bacterium Clostridium ljungdahlii DSM 13528 grown autotrophically and heterotrophically. Mol BioSyst. 2013;9:2775–2784. doi: 10.1039/c3mb70232d. [DOI] [PubMed] [Google Scholar]
- Tatusov RL. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucl Acids Res. 2000;28:33–36. doi: 10.1093/nar/28.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Techtmann SM, Colman AS, Robb FT. “That which does not kill us only makes us stronger”: the role of carbon monoxide in thermophilic microbial consortia. Environ Microbiol. 2009;11:1027–1037. doi: 10.1111/j.1462-2920.2009.01865.x. [DOI] [PubMed] [Google Scholar]
- Techtmann SM, Colman AS, Murphy MB, et al. Regulation of multiple carbon monoxide consumption pathways in anaerobic bacteria. Front Microbiol. 2011;2:147. doi: 10.3389/fmicb.2011.00147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Techtmann SM, Lebedinsky AV, Colman AS, et al. Evidence for horizontal gene transfer of anaerobic carbon monoxide dehydrogenases. Front Microbiol. 2012;3:132. doi: 10.3389/fmicb.2012.00132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative genomics viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2013;14:178–192. doi: 10.1093/bib/bbs01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wawrousek K, Noble S, Korlach J, et al. Genome annotation provides insight into carbon monoxide and hydrogen metabolism in Rubrivivax gelatinosus. PLoS One. 2014;9:e114551. doi: 10.1371/journal.pone.0114551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weghoff MC, Müller V. CO metabolism in the thermophilic acetogen Thermoanaerobacter kivui. Appl Environ Microbiol. 2016;82:2312–2319. doi: 10.1128/AEM.00122-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wösten MM. Eubacterial sigma-factors. FEMS Microbiol Rev. 1998;22:127–150. doi: 10.1111/j.1574-6976.1998.tb00364.x. [DOI] [PubMed] [Google Scholar]
- Wu M, Ren Q, Durkin AS, et al. Life in hot carbon monoxide: the complete genome sequence of Carboxydothermus hydrogenoformans Z-2901. PLoS Genet. 2005;1:e65. doi: 10.1371/journal.pgen.0010065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneda Y, Yoshida T, Kawaichi S, et al. Carboxydothermus pertinax sp. nov., a thermophilic, hydrogenogenic, Fe(III)-reducing, sulfur-reducing carboxydotrophic bacterium from an acidic hot spring. Int J Syst Evol Microbiol. 2012;62:1692–1697. doi: 10.1099/ijs.0.031583-0. [DOI] [PubMed] [Google Scholar]
- Yoneda Y, Yoshida T, Daifuku T, et al. Quantitative detection of carboxydotrophic bacteria Carboxydothermus in a hot aquatic environment. Fundam Appl Limnol/Arch für Hydrobiol. 2013;182:161–170. doi: 10.1127/1863-9135/2013/0374. [DOI] [Google Scholar]
- Yoneda Y, Kano SI, Yoshida T, et al. Detection of anaerobic carbon monoxide-oxidizing thermophiles in hydrothermal environments. FEMS Microbiol Ecol. 2015;91:fiv093. doi: 10.1093/femsec/fiv093. [DOI] [PubMed] [Google Scholar]
- Youn H, Kerby RL, Conrad M, Roberts GP. Functionally critical elements of CooA-related CO sensors. J Bacteriol. 2004;186:1320–1329. doi: 10.1128/JB.186.5.1320-1329.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.