
Fig. 1.
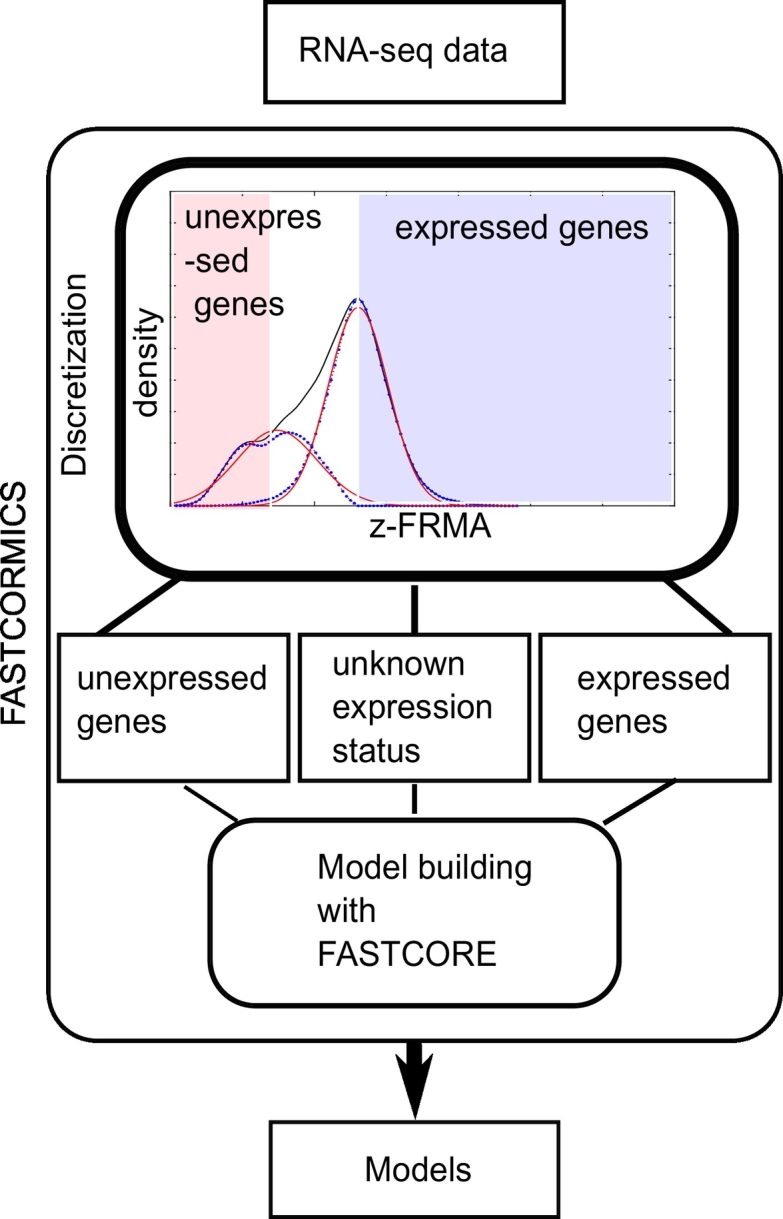
The RNA-seq FASTCORMICS workflow: The workflow uses a discretization step that considers the intensity distribution across all genes to discretize the genes into expressed, undefined expression status, and inexpressed. The discretized values are then mapped to the input model to obtain 3 sets of reactions: core reactions, non-core, and inactive reactions. The bounds of the inactive reactions are set to zero and are removed from the model along with reactions that are no longer able to carry a flux. A modified version of FASTCORE is used to include all the remaining core reactions, with the exception of transporter reactions that are transferred from the core to an unpenalized set (these reactions are not forced in but, as they are not penalized, their inclusion is favoured over non-core reactions). In Recon X models, several hundreds of transporter reactions are controlled by a set of only a few genes. To avoid the unwanted activation of transporter reactions in inactive pathways, transporter reactions are not forced to be active but their activation is not penalized. To obtain consistent models, FASTCORE includes a minimal amount of non-core reactions.