

Abstract
The nosocomial pathogen Acinetobacter baumannii acquired clinical significance due to the rapid development of its multi-drug resistant (MDR) phenotype. A. baumannii strains have the ability to colonize several ecological niches including soil, water, and animals, including humans. They also survive under extremely harsh environmental conditions thriving on rare and recalcitrant carbon compounds. However, the molecular basis behind such extreme adaptability of A. baumannii is unknown. We have therefore determined the complete genome sequence of A. baumannii DS002, which was isolated from agricultural soils, and compared it with 78 complete genome sequences of A. baumannii strains having complete information on the source of their isolation. Interestingly, the genome of A. baumannii DS002 showed high similarity to the genome of A. baumannii SDF isolated from the body louse. The environmental and clinical strains, which do not share a monophyletic origin, showed the existence of a strain-specific unique gene pool that supports niche-specific survival. The strains isolated from infected samples contained a genetic repertoire with a unique gene pool coding for iron acquisition machinery, particularly those required for the biosynthesis of acinetobactin. Interestingly, these strains also contained genes required for biofilm formation. However, such gene sets were either partially or completely missing in the environmental isolates, which instead harbored genes required for alternate carbon catabolism and a TonB-dependent transport system involved in the acquisition of iron via siderophores or xenosiderophores.
Introduction
A surge in the number of multi-drug resistant (MDR) bacteria has increased the severity of many bacterial diseases. The number of infections caused by the MDR strains has almost quadrupled in the last two decades [1, 2]. Of these, the ESKAPE pathogens comprising of Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species form the major proportion of MDR and extremely drug resistance (XDR) strains [3, 4]. The genus Acinetobacter are Gram-negative bacteria belonging to the class Gammaproteobacteria. Nearly 55 different species of Acinetobacter have been isolated from sources as varied as water [5], soil [6], hospitals [7, 8], body fluids [9, 10], and even body lice [11]. Almost all are known to cause human diseases. Acinetobacter baumannii is the predominant species of the genus and accounts for about 80% of reported Acinetobacter infections (https://www.cdc.gov/hai/organisms/acinetobacter.html), which range from pneumonia to serious blood or wound infections, soft tissue infections, and secondary meningitis [12–14]. The symptoms vary depending on the severity of the infection, but strains can also be asymptomatic, residing in tracheostomy sites or open wounds. A. baumannii strains are known for their genome plasticity and the ability to survive on abiotic surfaces [15, 16]. They acquire genes through conventional horizontal gene transfer (HGT), as well as through membrane vesicles [17–22]. Such robust gene acquisition contributes to the evolution of A. baumannii strains exhibiting MDR and XDR.
A total of 3539 A. baumannii genome sequences are available in the NCBI database. All these genomes display resistance traits acquired through HGT as part of the accessory genome. However, the genes that contribute to virulence are found as part of the core genome [12]. Genomes of A. baumannii strains have been compared to understand the molecular basis underlying their rapid acquisition of drug resistance genes. This comparative analysis of genomes revealed a surprisingly high degree of variations in the genomes of various strains, including single nucleotide polymorphisms (SNPs) as well as large DNA fragment variations [23]. Interestingly, in the AYE strain of A. baumannii, an 86 Kb resistance island (RI) was identified carrying 45 resistance genes. However, at the analogous position in the drug-sensitive SDF strain of A. baumannii, a 20 Kb genomic island (GI) devoid of RIs was detected, suggesting site-specific integration of resistance genes at this hotspot in the genome [24]. In addition to the RIs, diverse antibiotic resistance determinants were identified outside of the RIs. Antibiotic resistance gene-bearing integrons and blaOXA-23-containing transposon, Tn2009 were detected in the genome of several A. baumannii isolates. Sequence similarity and phylogenetic analyses have revealed that the resistance genes found in A. baumannii have been acquired relatively recently from bacteria of the genera Pseudomonas, Salmonella, or Escherichia [25].
A. baumannii strains adapt to a variety of ecological niches. They survive in soil and water in a free-living state and can quickly adapt to the infectious mode as a lifestyle. Such robust survival strategies of A. baumannii strains suggest they have acquired the genetic repertoire allowing them to adapt quickly to changing environmental conditions. Unfortunately, few studies have been conducted to identify unique genes that contribute to niche-specific survival of A. baumannii strains [11]. The present study reports a complete genome sequence of A. baumannii DS002, which was isolated from pesticide-polluted agricultural soils. Comparison of its genome sequence with the complete genome sequences of A. baumannii strains available in the NCBI database revealed the existence of unique, niche-specific genes that contribute to the fitness and survival of the bacterium in a variety of different habitats.
Materials and methods
Genome sequencing, assembly, and annotation
A. baumannii DS002 was grown to early stationary phase in LB medium at 30°C and the genomic DNA was isolated using a QIAGEN Genomic-tip Kit. The quality and quantity of the isolated DNA were assessed using agarose gel electrophoresis and Qubit 3.0, which is a fluorometric instrument used for quantification of DNA, RNA, and protein. The isolated DNA was subjected to sequencing using a Pacific Biosciences (PacBio) single-molecule real-time (SMRT) cell RS II instrument (chemistry version P6-C4) at Nucleome Informatics, Hyderabad. A total of 89,614 reads were obtained, out of which, 86,803 reads were more than 500bps in length. The average read length was 5601bp. The genome assembly was done by the Canu 1.8 assembly pipeline using HISEA read overlapper [26, 27]. The Canu+HISEA process involved four basic steps of read error-correction, quality/adapter trimming, contig layout construction and consensus generation. Several iterations of the assembly pipeline were performed to determine the optimal parameters. About 80,184 reads survived the error correction and trimming steps, resulting in eight different assemblies. The assemblies were annotated using Prokka v1.12 [28] and Prodigal 2.6.1 [29] pipelines. Benchmarking Universal Single-Copy Orthologs (BUSCOs) from the gammaproteobacteria set was used as a standard to assess the quality of the assembly and annotation performed [30]. A circular map of the chromosome was generated using Arc with color tool (http://www.ige.tohoku.ac.jp/joho/gmProject/gmdownload.html). The bacterial culture was deposited in the Microbial Type Culture Collection center (MTCC), IMTECH, Chandigarh, India, and is available as Acinetobacter sp. DS002 MTCC11451.
Bacterial pan-genome analysis (BPGA)
In order to identify strain-specific genomic features in a genome and determine the genomic diversity among the A. baumannii strains, the computational pipeline BPGA tool was used [31]. Although a total of 3539 A. baumannii genome sequences are available in the NCBI database most of them are incomplete and exist as scaffolds. In certain cases a complete genome sequence is available but there is an ambiguity with respect to the source of isolation. Therefore, such sequences were not considered for the pan-genome analysis. Only complete genome sequences of A. baumannii having information on the source of isolation were included in the analysis. This list comprises of 78 genome sequences, which includes the complete genome sequence of A. baumannii DS002 (Table 1). The full GenBank files of all 78 genomes downloaded from NCBI served as input for the BPGA analysis. BPGA further processed these files for orthologous cluster analysis and generated an input file containing a total of 281,137 annotated genes. This input file was subsequently used for clustering of genes into families by USEARCH with 50% sequence similarity as a cutoff. To avoid any bias during the sequential addition of genomes, 30 random permutations in the sequence/order of genomes added were carried out. The size of the pan-genome is represented by median values of the total number of distinct gene families and the core genome is represented by the number of shared gene families. Preliminary pan-genome profile analysis was performed to determine the frequency distribution of various gene families among the selected 78 complete genomes. Using the pan-genome sequence extraction module, core, accessory, and unique gene families were extracted. The exclusive gene family analysis module was used to identify homologous gene families that are distinctive for A. baumannii strains (i.e., unique genes or singletons). The pan-genome functional analysis module was used to find the Clusters of Orthologous Groups of proteins (COGs) and KEGG pathway distribution. Evolutionary analysis done by BPGA was based on concatenated core gene alignment using a binary pan-matrix file that depicts the presence or absence of the genes among the genomes. The pan-gene matrix was generated by calculating the contribution of the gene to the orthologous gene clusters. The core genome phylogenetic tree was constructed using protein sequences of 20 random orthologous gene clusters. MUSCLE (MUltiple Sequence Comparison by Log-Expectation) was used for initial multiple sequence alignment and the aligned sequences were then concatenated before constructing a neighbor-joining phylogenetic tree. To assess the genetic diversity among the genomes we performed an average nucleotide identity (ANI) analysis using the FastANI tool [32], which computes a pair-wise ANI value for the sample genome. These ANI values were then used to generate a heatmap using webserver heat mapper (http://www.heatmapper.ca) and to convert into a Euclidean distance matrix. This distance matrix was then uploaded to the web server DendroUPGMA (http://genomes.urv.cat/UPGMA/) [33] to generate a Newick format of the distance matrix. The Newick file is then used in MEGA7 [34] to view the UPGMA (unweighted pair group method with arithmetic mean) Tree based on ANI based Euclidian distance.
Table 1. List of 78 A. baumannii strains and source of isolation.
| Genomic IDs | Strain | Source of isolation | Broader-Isolation Source |
|---|---|---|---|
| AP013357.1 | NCGM-237 | RTI/UTI/Blood | Blood |
| AP014649.1 | IOMTU-433 | Respiratory tracts | Tissue |
| CP000863.1 | ACICU | Cerebrospinal fluid | Body fluids |
| CP024911.1 | AB307_0294_2 | Blood | Blood |
| CP001182.2 | AB0057 | Bloodstream | Blood |
| CP001921.1 | 1656–2 | Hospital Setting- South Korea | Hospital |
| CP001937.1 | MDR-ZJ06 | Bloodstream | Blood |
| CP003500.1 | MDR-TJ | Hospital setting | Hospital |
| CP003846.1 | BJAB07104 | Blood | Blood |
| CP003847.1 | BJAB0715 | Spinal fluid | Body fluids |
| CP003849.1 | BJAB0868 | Ascites | Body fluids |
| CP003856.1 | TYTH-1 | Bacteremia- Taiwan | Bacteremia |
| CP003967.2 | D1279779 | Bacteraemic infection of an indigenous Australian male | Bacteremia |
| CP006768.1 | ZW85-1 | Diarrheal patient feces | Body fluids |
| CP007535.2 | AC29 | Endotracheal Secretion | Body fluids |
| CP007577.1 | AC30 | Endotrachial secretion | Body fluids |
| CP007712.1 | LAC-4 | Outbreak | Outbreak |
| CP008706.1 | AB5075-UW | Tibia/Osteomyelitis | Tissue |
| CP009256.1 | AB031 | Bloodstream | Blood |
| CP009257.1 | AB030 | Bloodstream | Blood |
| CP009534.1 | AbH12O-A2 | Outbreak in Spain | Outbreak |
| CP010397.1 | 6200 | Body fluid | Body fluids |
| CP010779.1 | XH386 | Lower respiratory tract | Tissue |
| CP010781.1 | A1 | Nottingham University Hospital | Hospital |
| CP012006.1 | Ab04-mff | Blood | Blood |
| CP012952.1 | D36 | Wound | Wound |
| CP013924.1 | KBN10P02143 | Pus | Body fluids |
| CP014215.1 | YU-R612 | Sputum | Sputum |
| CP014528.1 | XH858 | Sputum | Sputum |
| CP014539.1 | XH859 | Wound | Wound |
| CP014540.1 | XH857 | Sputum | Sputum |
| CP014541.1 | XH856 | Drainage fluid | Body fluids |
| CP015364.1 | 3207 | Bronchial fluid | Body fluids |
| CP015483.1 | ORAB01 | Body fluid | Body fluids |
| CP016298.1 | CMC-MDR-Ab59 | Sputum | Sputum |
| CP017152.1 | DU202 | Clinical isolate | Hospital |
| CP017642.1 | KAB01 | Bronchial | Body fluids |
| CP017644.1 | KAB02 | Bronchial | Body fluids |
| CP017646.1 | KAB03 | Sputum | Sputum |
| CP017648.1 | KAB04 | Sputum | Sputum |
| CP017650.1 | KAB05 | Blood | Blood |
| CP017652.1 | KAB06 | Wound | Wound |
| CP017654.1 | KAB07 | Sputum | Sputum |
| CP017656.1 | KAB08 | Wound | Wound |
| CP018143.1 | HRAB-85 | Sputum | Sputum |
| CP018254.1 | AF-401 | Small colon | Tissue |
| CP018256.1 | AF-673 | Sputum | Sputum |
| CP018332.1 | A1296 | Sputum | Sputum |
| CP018421.1 | XDR-BJ83 | Human | Human |
| CP018664.1 | ATCC-17978 | Blood | Blood |
| CP020574.1 | 15A5 | Pulmonary | Tissue |
| CP020578.1 | SSA12 | Blood | Blood |
| CP020579.1 | SAA14 | Blood | Blood |
| CP020581.1 | SSMA17 | Bronchial washing fluid | Body fluids |
| CP020584.1 | JBA13 | Sputum | Sputum |
| CP020586.1 | CBA7 | Sputum | Sputum |
| CP020590.1 | 15A34 | Pulmonary | Tissue |
| CP020591.1 | SSA6 | Endotracheal aspirate | Body fluids |
| CP020592.1 | USA2 | Urine | Body fluids |
| CP020595.1 | USA15 | Sputum | Sputum |
| CP020597.1 | HWBA8 | Sputum | Sputum |
| CP020598.1 | WKA02 | Sputum | Sputum |
| CP021342.1 | B8342 | Blood | Blood |
| CP021347.1 | B8300 | Blood | Blood |
| CP021782.1 | A85 | Sputum | Sputum |
| CP024124.1 | AYP-A2 | Wound | Wound |
| CP024576.1 | AbPK1 | Sheep broncho-alveolar lavage | Tissue |
| CP024611.1 | Ab4977 | Sputum | Sputum |
| CP024612.1 | Ab4653 | Sputum | Sputum |
| CP024613.1 | Ab4568 | Sputum | Sputum |
| CP001172.1 | AB307_0294 | Blood | Blood |
| CP025266.1 | SMC_Paed_Ab_BL01 | Blood, Central line | Blood |
| CP027704.1 | DS002 | Soil | Soil |
| CU459141.1 | AYE | Patient with Pneumonia & UTI | Human |
| CU468230.2 | SDF | Body Louse | Environmental |
| LN865143.1 | CIP70.10 | Human skin | Human skin |
| LN868200.1 | R2090 | Tissue | Tissue |
| LN997846.1 | R2091 | Tissue | Tissue |
Comparative genomics
The primary objective of this study is to identify a niche-specific unique gene content(s) in A. baumannii strains. No environmental isolate other than DS002 exists among the 78 genome sequences included in the analysis. Therefore, we have considered the inclusion of the draft genome sequence of DSM30011 to gain insight into the unique gene content in the environmental isolate. The seventy-nine (including DSM30011) sequences of A. baumannii strains were considered for the comparative analysis of virulome, resistome, and genes involved in iron acquisition and metabolic pathways. We compiled a list of genes involved in virulence, resistance, iron acquisition, and carbon metabolism in A. baumannii from different literature studies as well as from the KEGG pathway tools (S1 File). These gene sets were then used as input to perform a Large-Scale BLAST Score Ratio (LS-BSR) analysis [35]. Principal Component Analysis (PCA) plot for LS-BSR output score matrix is generated by using the R package ggplot2 and scatterplot3d. Further, we set a BSR cut-off of 0.6 and above to determine the presence of genes and any value below 0.6 indicated their absence. The matrix file obtained from LS-BSR was used to generate heat maps and was further clustered hierarchically to provide a better understanding of the relationship between the strains using the R platform version 3.4.4 [36].
Resistome and genomic island (GI) prediction
The Resistance Gene Identifier (RGI) software of CARD (The Comprehensive Antibiotic Resistance Database) was used for prediction of the resistome [37]. The presence of GI was predicted by IslandViewer 4 [38], which uses three prediction algorithms such as SIGI-HMM, IslandPath-DIMOB, and IslandPick to calculate codon usage, dinucleotide bias within a genome, and to generate a dataset of GIs and non-GIs from phylogenetically related organisms. The GIs predicted by at least one of the three algorithms is considered for detailed analysis. In order to gain better clarity on the existence of GI and their putative functions, only eleven genome sequences, derived from strains isolated from bacteremia (TYTH-1) (CP003856), hospital (MDR-TJ) (CP003500), body fluid (6200) (CP010397), sputum (HWBA8) (CP020597), wound (AYP-A2) (CP024124), tissue (IOMTU 433) (AP014649), blood (AB031) (CP009256), infectious outbreaks (LAC-4) (CP007712), humans (AYE) (CU459141.1), and environmental (SDF) (CU468230) samples were included along with DS002 genome sequence. These genome sequences were selected by taking one from each source of isolation, and which had the highest number of unique genes.
Quantification of bacterial growth and biofilm formation
A. baumannii strains AYE and DS002 were grown either in LB medium or in minimal salt medium [39] supplemented with phenol (2 mM) as the sole source of carbon at 30°C. The biofilm formation of the strains AYE and DS002 was determined following standard procedures [40].
Results
Genomic size and GC content of A. baumannii DS002
The de novo assembly of the PacBio raw reads using the Canu-HISEA pipeline generated 8 completely circularized contigs (S1 Table). The circularized length reflects the length after removing the overlapping portion. The largest assembled contig corresponds to the bacterial chromosome with a circular length of 3,430,798 bps. The remaining contigs correspond to the plasmid repertoire of the organism. The whole genome sequence of A. baumannii DS002 is available in the NCBI database (Genome Submission-ID: SUB3752749, BioSample submission ID: SUB3749011, BioProject submission ID: SUB3749009, accession number: CP027704.1). The Prokka annotation pipeline identified 18 rRNA, 72 tRNA, 1 tmRNA and a total of 3,569 protein-coding genes on the main chromosome of DS002, with a genomic GC content of 39.6%, which complies with the already reported genomic data of A. baumannii strains. The position of the coding genes on positive and negative strands, the GC skew and dinucleotide bias ratio for DS002 is shown in the circular map of the genome (Fig 1). The single copy orthologous genes (BUSCOs) indicate evolutionarily conserved genes among organisms belonging to a taxonomic class. The BUSCO set of gammaproteobacteria contains a total of 452 conserved orthologous genes. Of these 452 genes, 422 were found in the DS002 genome, suggesting the genome is about 93.4% complete. According to the core and pan-phylogeny analysis, the DS002 strain clustered closely with A. baumannii SDF, a strain isolated from the body louse (Pediculus humanus) (Fig 2 and S1 Fig). Among the sequences of A. baumannii, the sequence of environmental strain SDF showed the lowest ANI at nearly 96% when compared to the rest of the genomes of A. baumannii strains (S2 Fig). Interestingly, the ANI score (~97.5%) of DS002 was close to the genomes of A. baumannii strains isolated from clinical sources. The UPGMA tree constructed using Euclidean distance (obtained from ANI value) showed that strain SDF is most distant and dissimilar when compared to other strains of A. baumannii (S3 Fig). Interestingly, the UPGMA tree analysis revealed that strain DS002 has the highest similarity to A. baumannii strains isolated from sputum. PCA plot generated to gain better visualization on diversity also showed no isolation source specific clustering of A. baumannii strains. Interestingly environmental isolates DS002 and SDF were found as outliers in the PCA plot (S4 Fig).
Fig 1. Circular map of the chromosome.
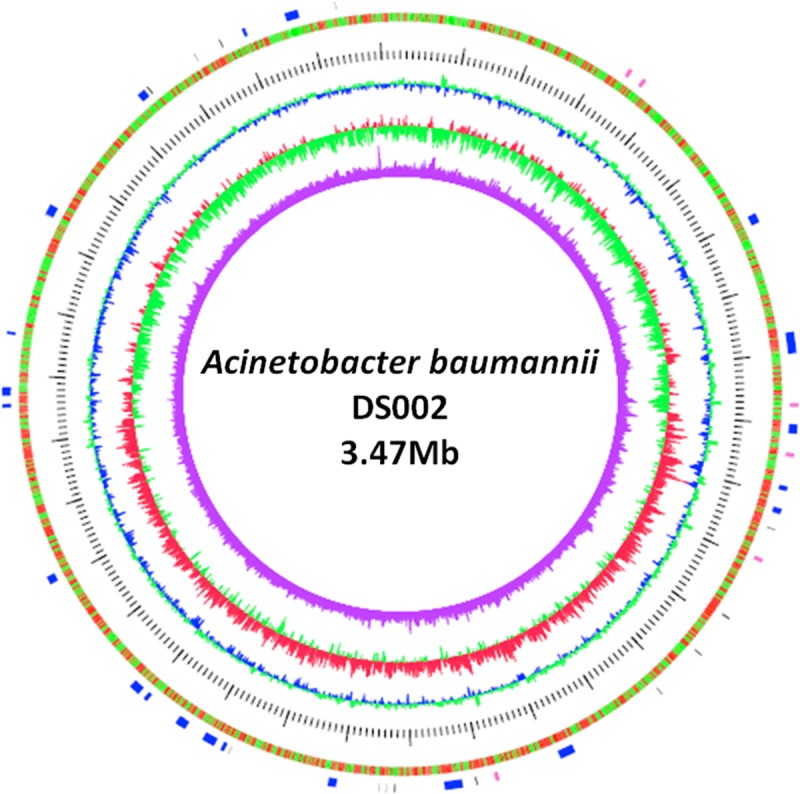
Circle 1 (outer to inner) represents the total 3251 CDS in the genome. Genes present on the positive and negative strands are depicted in red and green colour, respectively. Circles 3, 4 and 5 represent GC content, GC skew, and dinucleotide bias, respectively. The tRNA (black) and rRNA (red) coding genes and the genomic islands (blue) are shown in the outermost discontinuous circle.
Fig 2. Phylogenetic tree.

Core genome-derived phylogenetic tree of 78 A. baumannii strains. The position of DS002 in the phylogenetic tree is highlighted with a blue background and dotted clade line.
Pan-genome analysis
The A. baumannii complex forms the largest species group within the Acinetobacter genus. At present, there are about 135 complete genome sequences for the A. baumannii strains isolated from a variety of ecological niches. The primary aim of the study is to identify unique and accessory genes in the genome of A. baumannii species that might contribute to their exceptional survival strategies under diverse environmental conditions. We have therefore taken only 78 genomes for which the source of isolation is well documented (Table 1). Initially, we created a pan-genome pool by taking the sequences of all genes found in these 78 genomes (281,137 sequences) and segregated them as the core, accessory, and unique gene families. The pan-versus-core gene plot clearly indicates that the pan-genome window is still open for expansion in the case of A. baumannii (Fig 3A) and with the addition of each new genome to the A. baumannii complex a change in the pan-genome profile can result. Distribution of gene families and new genes within the pan-genome of the A. baumannii complex are shown in Fig 3B and 3C. Representative protein sequences of the core (1344), accessory (4644), and unique (1695) genes were identified and their COG and KEGG identities established. Accessory genes, which includes those present in only a few genomes, intriguingly are involved in cellular metabolism, emphasizing an ecological niche-specific change in the genome. Moreover, the genes involved in information storage and processing activities of the cell were all identified in the unique genes of the pan-genome (Fig 3D & 3E). The details of core, accessory and unique genes present in each of the 78 genomes are shown in the S2 Table. After segregation of pan-genome into the core, accessory, and unique genes, we specifically analyzed unique genes from the eleven representative strains to determine whether it is possible to identify a particular feature or features of the genetic composition that might contribute to niche-specific survival of A. baumannii strains.
Fig 3. Pan genome analysis.

Panel A indicates the pan and core genome curves. Panel B shows the frequency distribution of gene families within genomes. The number of new genes added to each genome is depicted in panel C. The COG and KEGG distribution of the representative proteins in the core, accessory, and unique genome are shown in panels D, and E respectively.
Unique genome content
In the unique genome content, we noticed genes that contribute to carbon catabolism, virulence, and drug resistance. As the analysis of the unique genes from all 78 genomes is challenging, we restricted our analysis to eleven representative genomes, taking one representative from each source of isolation with the highest number of unique genes. While analyzing the unique genes we found kgdL in the unique gene pool of A. baumannii 6200, isolated from the body fluid, which codes for the large subunit of membrane-bound gluconate 2-dehydrogenase [41]. Similarly, in A. baumannii HWBA8, a strain isolated from sputum, a membrane-bound PQQ-dependent dehydrogenase was identified as part of its unique genome. These membrane-bound dehydrogenases are linked to the cytoplasmic respiratory chain through their unique N-terminal domains. The PQQ-dependent dehydrogenases play a role in incomplete oxidation of monosaccharides leading to accumulation of breakdown products in the culture medium [41]. The physiological role of these membrane-bound dehydrogenases is still uncertain; however, results of several studies suggest that they provide additional energy-conserving systems, which aids the survival of the strain in environments with high energy demands [42–44].
The hrgA gene exists in the unique genome of AB031
In blood isolate AB031, a hrgA gene was identified as one of the unique genes. The hrgA gene was initially identified in Helicobacter pylori, a well-known pathogen in the human gastrointestinal tract, as part of the restriction and modification system [45]. In H. pylori the hrgA gene has replaced the restriction component hypIIIR locus. The existence of hrgA has been reported in nearly 33% of the sequenced H. pylori strains. Although the physiological or evolutionary significance of such gene replacement is unknown, a recent clinical study associated the hrgA gene with an increased incidence of gastric cancer [46]. The hrgA gene in H.pylori is present upstream of the hypIIIM gene encoding for the methylase component of the HypRM system. Since its existence is unique in AB031, we have analyzed the sequence regions 1000 bps upstream and downstream of the hrgA gene to determine whether hrgA also forms a part of such restriction and modification system in Acinetobacter sp. Interestingly, there was no indication from the DNA sequences of any association of hrgA with a modification system. Instead, we observed a gene coding for glycine tRNA synthetase subunit β and a gene coding for an uncharacterized transporter in the upstream and downstream regions of hrgA, respectively. Since the existence of hrgA is of clinical significance we searched all 3539 A. baumannii genome sequences to identify the hrgA gene. Our BLAST search revealed the presence of hrgA only in two genomes, namely A. baumannii 1297 (perirectal) and A. baumannii AB136 (blood). Both of them are clinical isolates and were isolated from human samples. Several studies have reported the occurrence of horizontal gene transfer between A. baumannii and bacteria belonging to other genera. It is conceivable that if A. baumannii frequently co-infects the host together with these hrgA-bearing pathogens, there is a possibility for A.baumannii to acquire hrgA from, for example, H. pylori, through HGT (41).
The resistome is part of the unique gene pool in AYE
Several studies performed on the human pathogen AYE have established it as an XDR strain of A. baumannii complex. The genes conferring drug resistance, including aadB, encoding a 2-aminoglycoside nucleotidyltransferase, blaOXA-10 encoding beta-lactamase OXA-10 precursor, dhfrI encoding for dihydrofolate reductase type 1, dhfrA10 coding for dihydrofolate reductase type A10, and veb-1 encoding the extended-spectrum beta-lactamase were identified in the unique genome of the AYE strain.
Rut pathway encoding genes are in the unique genome of B8342
The unique genome of blood isolate B8342 contains genes involved in pyrimidine degradation via the Rut (pyrimidine utilization) pathway. The rutABCDEFG operon gene products facilitate the use of pyrimidines as a sole nitrogen source. In E. coli, the rut operon is part of NtrC regulon [47] and is highly expressed under nitrogen-limiting conditions [48]. The rutA gene codes for pyrimidine oxygenase and is involved in ring cleavage of pyrimidines, thereby producing the toxic ureidoacrylate peracid. The rutF gene is a flavin reductase involved in the regeneration of the flavin mononucleotide cofactor, while rutB encodes an amidohydrolase and is involved in the further breakdown of peracid in vitro. The rutBCD genes are assumed to catalyze the breakdown of ureidoacrylate peracid in vivo, subsequently releasing toxic malonic semialdehyde [49]. In E. coli, two genes, rutE, and ydfG have been reported to convert malonic semialdehyde into 3-hydroxypropionate [50]. Apparently, the Rut pathway is functional only at low, or at room temperature, and not at 37°C [51], as the toxic malonic semialdehyde cannot be converted into 3-hydroxypropionate at 37°C [48]. The physiological concentration of pyrimidines in blood plasma and other body fluids is around 0.4–0.6 μM, which is considerably lower than the intracellular concentrations of 0.5–3 mM [52]. If a pathogen acquires the capacity to utilize pyrimidines as a source of nitrogen, it certainly would be advantageous to blood isolates like A. baumannii B8342. The Rut pathway has also been identified among soil proteobacteria and is suggested to have a role in utilizing pyrimidines generated from decaying plant material as a source of nitrogen [49]. Identification of the Rut pathway in a pathogenic strain signifies its importance in the survival of organisms with pathogenic lifestyles. In contrast to the clinical isolates, the major portion of the unique genome of non-clinical isolate SDF comprises hypothetical genes. Interestingly, its unique genome codes for putative hemaglutinin/hemolysin-related proteins [11]. The presence of such genes in the SDF strain might help its survival as an ectoparasite by feeding on human blood.
Comparison of carbon catabolome
Most of the A. baumannii strains used in the study contained similar carbon metabolic pathways, and show no preference for a specific carbon source (Fig 4). However, the soil isolate includes genes coding for phenol 2-monooxygenase (pheA) and phenol hydroxylase (mphL) enzymes. Interestingly, these genes were absent in most of the clinical isolates. Phenol hydroxylase is a multicomponent monooxygenase that cleaves the aromatic ring and comprises three enzymatic components including a reductase, an oxygenase, and a regulatory component [53]. The oxygenase component has a dinuclear iron center at its active site, in which an oxygen atom is complexed by two iron ions. The reductase component transfers electrons to the dinuclear iron center facilitating the initial hydroxylation and subsequent detoxification of recalcitrant aromatic compounds such as phenol, benzene, toluene, xylene, and methyl or chlorophenols, to generate catechol. Catechol is subsequently degraded either by ortho- (Acinetobacter sps) or meta-cleavage (Pseudomonas spp) pathways [54, 55] enabling the cells to use these rare and recalcitrant carbon compounds as a source of carbon and energy.
Fig 4. Heat map showing LS-BSR analysis of genes involved in carbon catabolism.

Genes extensively discussed in the text as well as the strain DS002 are highlighted in red font.
Monooxygenase-encoding pheA genes have been identified in a number of soil microbes including yeast Trichosporon cutaneum,[56], Pseudomonas pickettii PKO1[57], Bacillus stearothermophilus BR219 [58], and some species of Acinetobacter [59]. A recent study has also shown the presence of a gene coding for phenol hydroxylase in pathogenic strains of A. baumannii. Existence of catabolic potential for phenol in pathogenic A. baumannii strains is a matter of huge concern as phenolic substances are used as disinfectants in hospitals [60]. Since strain DS002 contains the genetic capability for metabolizing phenolic substances we tested if it can grow using phenol as the sole source of carbon. Consistent with the genetic information, DS002 was able to grow in minimal salt medium containing 2mM phenol as the sole source of carbon. However, A. baumannii strain AYE in which no pheA homologues were identified failed to grow under similar culture conditions (S5 Fig). Our study has also revealed the presence of genes involved in phenol degradation in A. baumannii strains isolated from different body fluids (Fig 4) and this feature of these strains poses a major challenge in the use of phenol-based disinfectants in hospital environments.
Virulome of A. baumannii strains
Analysis of the virulome of the A. baumannii strains revealed the presence of similar virulence genes in most of the clinical isolates, suggesting the pathogenic nature of the majority of stains within the A. baumannii complex (Fig 5). Interestingly, a significant variation was observed with respect to the biofilm-associated genes. In the environmental isolates, the operon coding the csu pili chaperone-usher assembly system, which is known to contribute to biofilm formation [61], is only present in pathogenic A. baumannii strains and is absent in the genome of the soil isolate DS002 and the SDF strain (Fig 5). In addition to the csu operon, the biofilm-associated protein (bap) and blue light-sensing (blsA) gene, whose roles in biofilm formation have been established, were found in all pathogenic strains of A. baumannii [62–65]. None of these biofilm-associated virulence genes was observed in soil isolate DS002 and the SDF strain isolated from the body louse [11]. Interestingly, strain DS002 also carries the pgaABCD operon reported to be essential for the production, modification, and export of poly-β-1,6-N-acetyl-D-glucosamine (β-1,6-GlcNAc; PGA). The roles of pgaC and pgaD genes in the biosynthesis of poly-β-1,6-N-acetyl-D-glucosamine (PGA) is well established. The pgaB gene encodes an N-deacetylase that converts 15–20% of GlcNAc to glucosamine and pgaA codes for a membrane porin required for PGA export [66–68]. The absence of pgaA in DS002 might weaken the polysaccharide matrix necessary for biofilm formation. Similarly, the entire pgaABCD operon is absent the genome of the SDF strain (Fig 5). The role of PNAG in biofilm-mediated virulence has been previously reported in almost 30 clinical isolates of Acinetobacter baumannii [69]. In agreement with the genetic evidence, the multidrug-resistant AYE strain, which has the genetic repertoire for biofilm production, was significantly better at forming a biofilm compared with the soil isolate DS002, with no difference observed in their growth (S6 Fig).
Fig 5. Heat map showing LS-BSR analysis of genes involved in virulence.

Genes extensively discussed in the text as well as the strain DS002 are highlighted in red font.
Comparison of the resistome
Strains belonging to the A. baumannii complex show intrinsic β-lactamase activity due to the existence of β-lactamases like the cephalosporinase AmpC [17]. These genes, when associated with the IsAba1-type insertion element, confer resistance to 3rd generation cephalosporins [17]. The weak oxacillinase activity of A. baumannii strains is attributed to the presence of blaOXA-51/69 type oxacillinases [70]. An increased oxacillinase activity, which confers resistance to imipenem, is associated with the presence of IsAba1-type insertion elements [71, 72]. Most of the strains considered in our study, irrespective of their source of isolation, have shown the presence of chromosomally located blaOXA-51-like genes. A subset of oxacillinases is known to exhibit weak carbapenemase activity when compared to the metallo-β-lactamases and hence are grouped in the carbapenem-hydrolyzing class-D β-lactamases [17]. In A. baumannii, different types of Carbapenem-Hydrolyzing Class D β-Lactamases (CHDLs) are observed, which include, OXA-23-like, OXA-40/24-like, OXA-58-like [73], OXA-143-like [74], and OXA-235-like [75]. Significantly, the present study revealed the existence of an OXA-23-like CHDL gene as part of the unique genome in strain IOMTU 433, due to its presence as part of mobile genetic elements [76, 77]. The presence of both intrinsic blaOXA64 (51-like) and acquired blaOXA23 genes was observed in A. baumannii IOMTU 433. Carbapenem-associated outer membrane protein (CarO) is a membrane porin that facilitates uptake of imipenem due to the presence of an imipenem-binding site in the protein (61). Its absence in the genome of DS002 strain, but the presence of ampC, suggests possible weak carbapenemase activity exhibited by the strain (Fig 6).
Fig 6. Heat map showing LS-BSR analysis of genes involved in resistance.

Genes extensively discussed in the text as well as the strain DS002 are highlighted in red font.
Genome sequences of certain strains have shown the presence of genes that code for broad-spectrum β-lactamases, such as cephalosporinase-blaADC, and TEM-1 β-lactamases. Tissue isolate IOMTU 433 also possesses an NDM-1metallo-β- lactamase gene, making the strain resistant to all known classes of β-lactams [78]. The extremely drug-resistant human isolate of A. baumannii AYE was observed to contain veb-1 (Vietnamese extended-spectrum beta-lactamase) genes, which provide resistance towards extended-spectrum cephalosporins and aztreonam.
In addition to β-lactamases, pathogenic strains were also found to express aminoglycoside-modifying enzymes conferring resistance to aminoglycoside antibiotics. All three types of aminoglycoside-modifying enzymes including i) N-acetyltransferases (AAC), which catalyze acetyl CoA-dependent acetylation of an amino group, 2) O-adenyltransferases (ANT) catalyzing ATP-dependent adenylation of hydroxyl group, and 3) O-phosphotransferases (APH) that catalyze ATP-dependent phosphorylation of a hydroxyl group on the aminoglycoside antibiotics [79], are found in the genome of infectious strains, leading to their successful emergence as MDR pathogens (Fig 6). Furthermore, the sul1 gene has been identified in the genome of strains AYP-A2 and AYE, indicating their potential resistance to sulfonamides. However, none of these genes was identified in the genome of soil isolate DS002, probably owing to the strain’s limited exposure to such classes of antibiotics in the soil environment.
Efflux-mediated resistance in A. baumannii strains is well established, and it contributes significantly to MDR. The acinetobacter drug efflux (ade) pumps comprise the first Resistance-Nodulation-Division (RND) efflux pumps to be identified among the clinical isolates of Acinetobacter but is not present in environmental isolates. In accordance with previous reports, the environmental isolate DS002 lacks the adeABC RND efflux pump and the corresponding two-component regulatory system, adeRS, which contribute to resistance towards aminoglycosides, tetracyclines, erythromycin, trimethoprim, fluoroquinolones, some beta-lactams, and tigecycline [80–82]. However, it contains other RND efflux pumps such as adeIJK and adeFGH, which are involved in the efflux of ticarcillin, cephalosporins, aztreonam, fluoroquinolones, tetracyclines, tigecycline, lincosamides, rifampin, chloramphenicol, cotrimoxazole, novobiocin, and fusidic acid (Fig 6). Non-RND efflux pumps, such as abeS and abeM, were also found in the genome of DS002. All the representative pathogenic strains considered in our study were found to code for the adeABC efflux pumps. In addition to these, the AYP-A2 and AYE strains encoded Tet and CmlA efflux pumps, accounting for resistance to tetracycline and chloramphenicol.
In summary, the genome of strains belonging to A. baumannii complex contains numerous resistance genes and efflux pumps that are either characteristic of A. baumannii species and that have been presumably acquired, resulting in the drug-resistant nature of A. baumannii strains.
Diversity in iron uptake mechanism
Iron acquisition is a major challenge faced by soil bacteria due to the insolubility of Fe (III) in aerobic habitats. Over the period of evolution, bacteria have developed several mechanisms to acquire Fe (III) from the environment. Siderophores are iron-scavenging molecules that are synthesized and released by bacteria for iron uptake. In the genome sequences of Acinetobacter, five major siderophore biosynthetic clusters were reported [83, 84]. Prominent among these is acinetobactin; its biosynthetic genes are found in all genome sequences of A. baumannii except in the non-clinical isolate A. baumannii SDF strain [24, 84]. The genome of soil isolate DS002 lacked most of the acinetobactin biosynthetic genes (basABCDEFGJ) except for basH and basI or the genes involved in acinetobactin uptake (bauAD) except bauB, bauC and bauE, and release (barAB) (Fig 7). Similarly, biosynthesis of the siderophore fimsbactin was specifically identified in A. baumannii ATCC 17978 and ADP1 [85]. This siderophore biosynthetic gene cluster is absent in the DS002 strain. However, the genome of DS002 appears to have all genes involved in the biosynthesis of the siderophore identified in A. baumannii ACICU (gp1672-1683), except the gene that codes for demethylmenaquinone methyltransferase. As this type of iron acquisition mechanism is poorly studied, little information pertaining to the gene products is available in the literature. Some of the genome sequences of A. baumannii complex reveal the presence of genes coding for a catecholate siderophore (A. baumannii ABAYE1888-1889), encoding isochorismatase and a 2, 3-dihydro-2,3-hydroxybenzoate dehydrogenase [86]. These were identified in the genome of DS002. These enzymes produce 2,3-dihydroxybenzoate, an iron-binding molecule, which also serves as a precursor for the synthesis of complex siderophores [86]. The siderophore-mediated uptake of iron by Gram-negative bacteria is facilitated by the presence of TonB-dependent transport systems comprising an outer membrane receptor, a periplasmic binding protein, and an inner membrane permease. Several iron-responsive tonB-dependent receptors involved in cognate siderophore recognition and uptake have been identified in Acinetobacter. Genes coding for fhuA, an outer membrane ferrichrome receptor, fhuB, an inner membrane permease, and fhuD, encoding a periplasmic binding protein, were identified in the genome of DS002, indicating the probable uptake of a ferrichrome-type siderophore. Interestingly, most siderophore-related genes were found on a large GI (771Kb - 800Kb, ~30Kb) predicted in the genome of A. baumannii DS002 (Fig 8). This island contains 30 different ORFs, of which four encode putative transposases, 13 hypothetical genes, and the remaining 13 ORFs appear to be involved in iron acquisition. The genes include fhuB, a permease involved in ferrichrome uptake, feuB, a permease involved in enterochelin uptake and fcuA, a receptor for ferric citrate uptake, along with the fur gene, which is the master transcriptional regulator of the iron-responsive genes. The genes yusV and yclQ, also found in this island, encode putative siderophore-transport system ATP-binding proteins.
Fig 7. Heat map showing LS-BSR analysis of genes involved in iron acquisition.

Genes extensively discussed in the text as well as the strain DS002 are highlighted in red font.
Fig 8. Genomic Island.

The genetic map indicates the genomic island (30Kb) identified in the genome of DS002 with putative iron acquisition machinery. Genes coding a TonB transport system and hypothetical proteins are shown with dark green and blue arrows, respectively. The genetic map also indicates the presence of tnsB, tnsC_1, tnsC_2, tnsE_1 encoding a transposase (red), along with uvrD (grey), racE (light blue) and recF2 (yellow) encoding DNA helicase, racemase, and a DNA replication/repair protein, respectively.
Genomic islands (GIs)
After identifying a GI including genes encoding the iron uptake machinery in DS002, we then looked for GIs in the eleven representative strains. Most of the A. baumannii strains contain on an average 30–40 GIs per genome, indicating a high degree of horizontal mobility of DNA among the strains of the A. baumannii complex. In pathogenic A. baumannii strains, the genes associated with the predicted GIs encode proteins and enzymes involved in either aromatic degradation, formaldehyde detoxification, the type I secretion pathway for protein translocation, restriction-modification systems, amino acid biosynthesis, competence proteins, exoribonuclease VII, or heavy metal resistance (mercury, copper, and arsenic). All predicted GIs can be considered as contributing to general fitness and facilitate survival of the respective strains under unfavorable environmental conditions. The genes contributing to resistance to gentamicin, sulfonamides, β-lactams, streptomycin, and their respective efflux pumps were found on GIs in drug-resistant strains such as AYE, HWBA8, AYP-A2, which were isolated from human, sputum and wound sites, respectively. Interestingly, the majority of GIs in pathogenic strains was found to code for bacteriophage-related genes with a putative function in phage assembly and maintenance (at least 8–59 copies of phage genes). Contrary to the GI landscape in pathogenic strains, the GIs in the soil isolate DS002 did not code for any of these genes, except for copper resistance. Additionally, it contained two copies of prophage integrase genes, which were not seen in other strains, and an island that encoded the ferripyoverdine receptor (fpvA), that is known to be involved in the uptake of the pyoverdine siderophore [87]. In both DS002 and the AYP-A2 strains, one of the islands was found to encode all the NADH: quinone oxidoreductase subunits, which play a key role in respiration.
Discussion
Isolation of MDR strains of A. baumannii has gradually increased ever since they were first identified during an outbreak at a hospital in New York City [88]. In the 1970s, most of the strains were sensitive to well-known antibiotics. Meanwhile, during the span of forty years nearly 70% of the clinical isolates have acquired MDR status [2]. Carbapenems, once considered to be the linchpin against MDR A. baumannii strains were found to be ineffective in controlling MDR strains of A. baumannii. Such a rapid increase in drug resistance requires deeper insights into the correlation between the evolution of A. baumannii and their adaptation to a pathogenic lifestyle.
The comparative genomics performed between the soil isolate DS002 and strains isolated from hospital settings and from body fluids, clearly suggests the existence of unique gene content that contributes to the niche-specific survival of the strains.
The ability of A. baumannii strains to colonize humans depends on the initial establishment of contact with an epithelial cell of the body. The outer membrane porin OmpA is shown to facilitate such contact with the host cells [89]. Once contact is established, the ability of the bacterium to survive in the host depends on its ability to form a protective biofilm. Therefore, the comparative genomic study clearly indicated the existence of the complete genetic machinery for biofilm formation in all strains adapted to a pathogenic lifestyle. However, in strains adapted to survive in soil or water such a genetic machinery is either completely or partially absent. Experimental evidence generated using DS002 and AYE strains have confirmed these findings based on genomic analyses. The AYE strain showed a better biofilm formation ability than DS002 and in contrast, the strains adapted to a pathogenic lifestyle have lost genes required for degradation of rare and/or recalcitrant carbon compounds, such as phenol, as a sole source of carbon and energy. The catabolome required for thriving on phenol or benzoate was only found in environmental isolates. These catabolic traits are non-essential for the strains adapted to a pathogenic life style because they will not be confronted with such chemicals. Strain DS002 was isolated from insecticides-polluted agricultural soils and most commonly used insecticides generate phenolic compounds during the course of their biodegradation [90]. Therefore, they can be a rich carbon source for soil-dwelling bacteria, and hence strain DS002 acquired and retained the catabolome for metabolizing phenol, but the pathogenic strain AYE did not. The ability to degrade phenol as part of a unique genome content in environmental isolates has to be viewed with caution as most of the disinfectants used in hospital settings meanwhile also contain phenolic substances. If A. baumannii strains found in hospitals/intensive care units acquire the capacity to degrade phenol via horizontal gene transfer new disinfectants will need to be developed to remove such strains from nosocomial environments.
Interestingly, when the Rut pathway is considered, certain human isolates retained the corresponding genes. This may be due to the continual replenishment of the nucleotide pool from dead tissue or decaying plant material in the ecological niche and the expression of the Rut pathway is advantageous for the survival of the A. baumannii strains surviving in that habitat.
Irrespective of their lifestyle, acquisition of iron is a major nutritional challenge to all A. baumannii strains. All of them have a well-established transport system for importing iron bound to iron-chelating siderophores. Most of the bacteria possess an endogenous ability to synthesize their own siderophores. However, certain bacteria have efficient siderophore transport systems despite not having an innate ability to synthesize siderophores. In such cases, they presumably scavenge iron bound to siderophores made by other bacteria. The xenosiderophore transport system exists in DS002 on a GI, which certainly helps to compensate for strain’s inability to make its own siderophores (Fig 8).
Likewise, significant niche-specific genetic differences are seen with respect to drug resistance. The genes coding for all three types of aminoglycoside-modifying enzymes is absent in the genome of both DS002 and SDF strains. Similarly, the adeABC operon encoding RND efflux pumps and the corresponding two-component regulatory system, adeRS, are absent in soil isolates. Therefore, strain DS002 is sensitive to aminoglycosides, tetracyclines, erythromycin, trimethoprim, and fluoroquinolones (data not shown). However, the genome of DS002 indicates the presence of adeIJK and adeFGHRND efflux pumps along with other non-RND efflux pumps like abeS and abeM, suggesting that these potentially contribute to the resistance of the respective strain towards certain antibiotics like chloramphenicol, cotrimoxazole, novobiocin, and fusidic acid.
Interestingly, the virulome of A. baumannii is conserved in all A. baumannii strains. The presence of virulence factors, such as the BfmRS system, OmpA, phospholipase D in DS002, suggests a latently infectious property of the strain, irrespective of the source of its isolation. However, the soil isolates have lost the potential to infect humans due to a lack of biofilm formation ability. As the virulence factors are available in all A. baumannii strains, the soil-dwelling strains could, therefore, acquire the potential to colonize humans if they picked up genes encoding biofilm formation through HGT. Thus, the comparative genome analysis highlights not only apparent genetic differences found among A. baumannii strains but also the survival strategies being adopted by these A. baumannii strains.
Conclusion
The comparative genome analysis highlighted the selective expansion of unique, niche-specific genome content in A. baumannii. The expanded unique genome content contribute to the strain’s adaptability to different ecological habitats. Our study clearly revealed the expansion of drug-resistance genes only in clinical isolates, as it confers a selective advantage for the survival of clinical isolates in an ecological niche that is frequently exposed to all kinds of antibiotics. Interestingly, no difference in the virolome of the different A. baumannii strains exists. However, the genetic makeup required for biofilm formation, an essential feature for colonization of a host, is only seen in clinical isolates. Because biofilm formation is not essential for the survival of soil isolates, the loss of critical genes involved in biofilm formation is frequently observed. Niche-specific genome expansion has also revealed an impact on carbon metabolism. The pheA found in unique genome content enables the survival of cells using phenol as the sole source of carbon, is only seen in strains that are frequently exposed to phenolic substances.
Supporting information
(XLSX)
The position of DS002 in the phylogenetic tree is highlighted with a blue background and dotted clade line.
(TIF)
(TIF)
Strains with similar isolation source are shown with identical colors. The dotted blue line indicates the clade of soil isolate DS002 highlighted in blue.
(TIF)
Strains with similar isolation source are shown with identical colors.
(TIF)
(TIF)
OD590 values obtained for the ethanol extracts prepared from these two tubes indicating the extent of biofilm formation is shown in panel B. Panel C represents the bacterial growth as measured by OD600.
(TIF)
(DOCX)
(DOCX)
Data Availability
Complete genomes gbk file is available from the NCBI database. GenBank: CP027704.1.
Funding Statement
HY is the recipient of women scientist grant from Department of Science and Technology (DST)(SR/WOS-A/LS-1225/2015(G)), New Delhi (http://www.dst.gov.in/scientific-programmes/scientific-engineering-research/women-scientists-programs). CSIR, New Delhi and DST, New Delhi supported research work in DS* laboratory. Dept. of Animal Biology received special assistance from DST, New Delhi under FIST-level-II. School of Life Sciences received financial assistance under DBT-CREBB programme (Department of Biotechnology)(http://www.dbtindia.nic.in/). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Basak S, Singh P, Rajurkar M. Multidrug Resistant and Extensively Drug Resistant Bacteria: A Study. Journal of pathogens. 2016;2016:4065603 10.1155/2016/4065603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cohen ML. Changing patterns of infectious disease. Nature. 2000;406(6797):762–7. 10.1038/35021206 . [DOI] [PubMed] [Google Scholar]
- 3.Boucher HW, Talbot GH, Bradley JS, Edwards JE, Gilbert D, Rice LB, et al. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clinical infectious diseases: an official publication of the Infectious Diseases Society of America. 2009;48(1):1–12. 10.1086/595011 . [DOI] [PubMed] [Google Scholar]
- 4.Rice LB. Federal funding for the study of antimicrobial resistance in nosocomial pathogens: no ESKAPE. The Journal of infectious diseases. 2008;197(8):1079–81. 10.1086/533452 . [DOI] [PubMed] [Google Scholar]
- 5.Huys G, Bartie K, Cnockaert M, Hoang Oanh DT, Phuong NT, Somsiri T, et al. Biodiversity of chloramphenicol-resistant mesophilic heterotrophs from Southeast Asian aquaculture environments. Research in microbiology. 2007;158(3):228–35. 10.1016/j.resmic.2006.12.011 . [DOI] [PubMed] [Google Scholar]
- 6.Repizo GD, Viale AM, Borges V, Cameranesi MM, Taib N, Espariz M, et al. The Environmental Acinetobacter baumannii Isolate DSM30011 Reveals Clues into the Preantibiotic Era Genome Diversity, Virulence Potential, and Niche Range of a Predominant Nosocomial Pathogen. Genome biology and evolution. 2017;9(9):2292–307. 10.1093/gbe/evx162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Park JY, Kim S, Kim SM, Cha SH, Lim SK, Kim J. Complete genome sequence of multidrug-resistant Acinetobacter baumannii strain 1656–2, which forms sturdy biofilm. Journal of bacteriology. 2011;193(22):6393–4. 10.1128/JB.06109-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gao F, Wang Y, Liu YJ, Wu XM, Lv X, Gan YR, et al. Genome sequence of Acinetobacter baumannii MDR-TJ. Journal of bacteriology. 2011;193(9):2365–6. 10.1128/JB.00226-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lean SS, Yeo CC, Suhaili Z, Thong KL. Comparative Genomics of Two ST 195 Carbapenem-Resistant Acinetobacter baumannii with Different Susceptibility to Polymyxin Revealed Underlying Resistance Mechanism. Frontiers in microbiology. 2015;6:1445 10.3389/fmicb.2015.01445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Iacono M, Villa L, Fortini D, Bordoni R, Imperi F, Bonnal RJ, et al. Whole-genome pyrosequencing of an epidemic multidrug-resistant Acinetobacter baumannii strain belonging to the European clone II group. Antimicrobial agents and chemotherapy. 2008;52(7):2616–25. 10.1128/AAC.01643-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vallenet D, Nordmann P, Barbe V, Poirel L, Mangenot S, Bataille E, et al. Comparative analysis of Acinetobacters: three genomes for three lifestyles. PloS one. 2008;3(3):e1805 10.1371/journal.pone.0001805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Antunes LC, Visca P, Towner KJ. Acinetobacter baumannii: evolution of a global pathogen. Pathogens and disease. 2014;71(3):292–301. 10.1111/2049-632X.12125 . [DOI] [PubMed] [Google Scholar]
- 13.Hiraki Y, Yoshida M, Masuda Y, Inoue D, Tsuji Y, Kamimura H, et al. Successful treatment of skin and soft tissue infection due to carbapenem-resistant Acinetobacter baumannii by ampicillin-sulbactam and meropenem combination therapy. International journal of infectious diseases: IJID: official publication of the International Society for Infectious Diseases. 2013;17(12):e1234–6. 10.1016/j.ijid.2013.05.002 . [DOI] [PubMed] [Google Scholar]
- 14.Michalopoulos A, Falagas ME. Treatment of Acinetobacter infections. Expert opinion on pharmacotherapy. 2010;11(5):779–88. 10.1517/14656561003596350 . [DOI] [PubMed] [Google Scholar]
- 15.Pendleton JN, Gorman SP, Gilmore BF. Clinical relevance of the ESKAPE pathogens. Expert review of anti-infective therapy. 2013;11(3):297–308. 10.1586/eri.13.12 . [DOI] [PubMed] [Google Scholar]
- 16.Houang ET, Sormunen RT, Lai L, Chan CY, Leong AS. Effect of desiccation on the ultrastructural appearances of Acinetobacter baumannii and Acinetobacter lwoffii. Journal of clinical pathology. 1998;51(10):786–8. 10.1136/jcp.51.10.786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Da Silva GJ, Domingues S. Insights on the Horizontal Gene Transfer of Carbapenemase Determinants in the Opportunistic Pathogen Acinetobacter baumannii. Microorganisms. 2016;4(3). 10.3390/microorganisms4030029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thomas CM, Nielsen KM. Mechanisms of, and barriers to, horizontal gene transfer between bacteria. Nature reviews Microbiology. 2005;3(9):711–21. 10.1038/nrmicro1234 . [DOI] [PubMed] [Google Scholar]
- 19.Rumbo C, Fernandez-Moreira E, Merino M, Poza M, Mendez JA, Soares NC, et al. Horizontal transfer of the OXA-24 carbapenemase gene via outer membrane vesicles: a new mechanism of dissemination of carbapenem resistance genes in Acinetobacter baumannii. Antimicrobial agents and chemotherapy. 2011;55(7):3084–90. 10.1128/AAC.00929-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fulsundar S, Harms K, Flaten GE, Johnsen PJ, Chopade BA, Nielsen KM. Gene transfer potential of outer membrane vesicles of Acinetobacter baylyi and effects of stress on vesiculation. Applied and environmental microbiology. 2014;80(11):3469–83. 10.1128/AEM.04248-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Traglia GM, Place K, Dotto C, Fernandez JS, Montana S, Bahiense CDS, et al. Interspecies DNA acquisition by a naturally competent Acinetobacter baumannii strain. International journal of antimicrobial agents. 2019. 10.1016/j.ijantimicag.2018.12.013 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krahn T, Wibberg D, Maus I, Winkler A, Bontron S, Sczyrba A, et al. Intraspecies Transfer of the Chromosomal Acinetobacter baumannii blaNDM-1 Carbapenemase Gene. Antimicrobial agents and chemotherapy. 2016;60(5):3032–40. 10.1128/AAC.00124-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu F, Zhu Y, Yi Y, Lu N, Zhu B, Hu Y. Comparative genomic analysis of Acinetobacter baumannii clinical isolates reveals extensive genomic variation and diverse antibiotic resistance determinants. BMC genomics. 2014;15:1163 10.1186/1471-2164-15-1163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Adams MD, Goglin K, Molyneaux N, Hujer KM, Lavender H, Jamison JJ, et al. Comparative genome sequence analysis of multidrug-resistant Acinetobacter baumannii. Journal of bacteriology. 2008;190(24):8053–64. 10.1128/JB.00834-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fournier PE, Vallenet D, Barbe V, Audic S, Ogata H, Poirel L, et al. Comparative genomics of multidrug resistance in Acinetobacter baumannii. PLoS genetics. 2006;2(1):e7 10.1371/journal.pgen.0020007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Khiste N, Ilie L. HISEA: HIerarchical SEed Aligner for PacBio data. BMC bioinformatics. 2017;18(1):564 10.1186/s12859-017-1953-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, Phillippy AM. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome research. 2017;27(5):722–36. 10.1101/gr.215087.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30(14):2068–9. 10.1093/bioinformatics/btu153 . [DOI] [PubMed] [Google Scholar]
- 29.Hyatt D, Chen GL, Locascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC bioinformatics. 2010;11:119 10.1186/1471-2105-11-119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Simao FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31(19):3210–2. 10.1093/bioinformatics/btv351 . [DOI] [PubMed] [Google Scholar]
- 31.Chaudhari NM, Gupta VK, Dutta C. BPGA- an ultra-fast pan-genome analysis pipeline. Scientific reports. 2016;6:24373 10.1038/srep24373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jain C, Rodriguez RL, Phillippy AM, Konstantinidis KT, Aluru S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nature communications. 2018;9(1):5114 10.1038/s41467-018-07641-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Garcia-Vallve S, Palau J, Romeu A. Horizontal gene transfer in glycosyl hydrolases inferred from codon usage in Escherichia coli and Bacillus subtilis. Molecular biology and evolution. 1999;16(9):1125–34. 10.1093/oxfordjournals.molbev.a026203 . [DOI] [PubMed] [Google Scholar]
- 34.Kumar S, Stecher G, Tamura K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Molecular biology and evolution. 2016;33(7):1870–4. 10.1093/molbev/msw054 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sahl JW, Caporaso JG, Rasko DA, Keim P. The large-scale blast score ratio (LS-BSR) pipeline: a method to rapidly compare genetic content between bacterial genomes. PeerJ. 2014;2:e332 10.7717/peerj.332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Team RC. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: URL http://wwwR-projectorg/. 2013. [Google Scholar]
- 37.Jia B, Raphenya AR, Alcock B, Waglechner N, Guo P, Tsang KK, et al. CARD 2017: expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic acids research. 2017;45(D1):D566–D73. 10.1093/nar/gkw1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.LeVay S. The patchy intrinsic projections of visual cortex. Progress in brain research. 1988;75:147–61. . [DOI] [PubMed] [Google Scholar]
- 39.Pandeeti EV, Siddavattam D. Purification and Characterization of Catechol 1,2-Dioxygenase from Acinetobacter sp. DS002 and Cloning, Sequencing of Partial catA Gene. Indian journal of microbiology. 2011;51(3):312–8. 10.1007/s12088-011-0123-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li C, Zhu L, Pan D, Li S, Xiao H, Zhang Z, et al. Siderophore-Mediated Iron Acquisition Enhances Resistance to Oxidative and Aromatic Compound Stress in Cupriavidus necator JMP134. Applied and environmental microbiology. 2019;85(1). 10.1128/AEM.01938-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Toyama H, Furuya N, Saichana I, Ano Y, Adachi O, Matsushita K. Membrane-bound, 2-keto-D-gluconate-yielding D-gluconate dehydrogenase from "Gluconobacter dioxyacetonicus" IFO 3271: molecular properties and gene disruption. Applied and environmental microbiology. 2007;73(20):6551–6. 10.1128/AEM.00493-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hommes RW, van Hell B, Postma PW, Neijssel OM, Tempest DW. The functional significance of glucose dehydrogenase in Klebsiella aerogenes. Archives of microbiology. 1985;143(2):163–8. . [DOI] [PubMed] [Google Scholar]
- 43.van Schie BJ, Hellingwerf KJ, van Dijken JP, Elferink MG, van Dijl JM, Kuenen JG, et al. Energy transduction by electron transfer via a pyrrolo-quinoline quinone-dependent glucose dehydrogenase in Escherichia coli, Pseudomonas aeruginosa, and Acinetobacter calcoaceticus (var. lwoffi). Journal of bacteriology. 1985;163(2):493–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.De Bont JAM DP, Van Schie BJ, Van Dijken JP, Frank JJzn and Kuenen JG. Role of quinoprotein glucose-dehydrogenase in gluconic acid production by Acinetobacter calcoaceticus. Antonie van Leeuwenhoek. 1984;50:76–7. [Google Scholar]
- 45.Ando T, Aras RA, Kusugami K, Blaser MJ, Wassenaar TM. Evolutionary history of hrgA, which replaces the restriction gene hpyIIIR in the hpyIII locus of Helicobacter pylori. Journal of bacteriology. 2003;185(1):295–301. 10.1128/JB.185.1.295-301.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Loewen PC, Alsaadi Y, Fernando D, Kumar A. Genome Sequence of a Tigecycline-Resistant Clinical Isolate of Acinetobacter baumannii Strain AB031 Obtained from a Bloodstream Infection. Genome announcements. 2014;2(5). 10.1128/genomeA.01036-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zimmer DP, Soupene E, Lee HL, Wendisch VF, Khodursky AB, Peter BJ, et al. Nitrogen regulatory protein C-controlled genes of Escherichia coli: scavenging as a defense against nitrogen limitation. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(26):14674–9. 10.1073/pnas.97.26.14674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim KS, Pelton JG, Inwood WB, Andersen U, Kustu S, Wemmer DE. The Rut pathway for pyrimidine degradation: novel chemistry and toxicity problems. Journal of bacteriology. 2010;192(16):4089–102. 10.1128/JB.00201-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Parales RE, Ingraham JL. The surprising Rut pathway: an unexpected way to derive nitrogen from pyrimidines. Journal of bacteriology. 2010;192(16):4086–8. 10.1128/JB.00573-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fujisawa H, Nagata S, Misono H. Characterization of short-chain dehydrogenase/reductase homologues of Escherichia coli (YdfG) and Saccharomyces cerevisiae (YMR226C). Biochimica et biophysica acta. 2003;1645(1):89–94. . [DOI] [PubMed] [Google Scholar]
- 51.Loh KD, Gyaneshwar P, Markenscoff Papadimitriou E, Fong R, Kim KS, Parales R, et al. A previously undescribed pathway for pyrimidine catabolism. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(13):5114–9. 10.1073/pnas.0600521103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Traut TW. Physiological concentrations of purines and pyrimidines. Molecular and cellular biochemistry. 1994;140(1):1–22. . [DOI] [PubMed] [Google Scholar]
- 53.Divari S, Valetti F, Caposio P, Pessione E, Cavaletto M, Griva E, et al. The oxygenase component of phenol hydroxylase from Acinetobacter radioresistens S13. European journal of biochemistry. 2003;270(10):2244–53. . [DOI] [PubMed] [Google Scholar]
- 54.Pessione E, Divari S, Griva E, Cavaletto M, Rossi GL, Gilardi G, et al. Phenol hydroxylase from Acinetobacter radioresistens is a multicomponent enzyme. Purification and characterization of the reductase moiety. European journal of biochemistry. 1999;265(2):549–55. . [DOI] [PubMed] [Google Scholar]
- 55.Powlowski J, Shingler V. Genetics and biochemistry of phenol degradation by Pseudomonas sp. CF600. Biodegradation. 1994;5(3–4):219–36. . [DOI] [PubMed] [Google Scholar]
- 56.Neujahr HY, Gaal A. Phenol hydroxylase from yeast. Purification and properties of the enzyme from Trichosporon cutaneum. European journal of biochemistry. 1973;35(2):386–400. . [DOI] [PubMed] [Google Scholar]
- 57.Kukor JJ, Olsen RH. Molecular cloning, characterization, and regulation of a Pseudomonas pickettii PKO1 gene encoding phenol hydroxylase and expression of the gene in Pseudomonas aeruginosa PAO1c. Journal of bacteriology. 1990;172(8):4624–30. 10.1128/jb.172.8.4624-4630.1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kim IC, Oriel PJ. Characterization of the Bacillus stearothermophilus BR219 phenol hydroxylase gene. Applied and environmental microbiology. 1995;61(4):1252–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xu Y, Chen M, Zhang W, Lin M. Genetic organization of genes encoding phenol hydroxylase, benzoate 1,2-dioxygenase alpha subunit and its regulatory proteins in Acinetobacter calcoaceticus PHEA-2. Current microbiology. 2003;46(4):235–40. 10.1007/s00284-002-3840-4 . [DOI] [PubMed] [Google Scholar]
- 60.Sahl JW, Johnson JK, Harris AD, Phillippy AM, Hsiao WW, Thom KA, et al. Genomic comparison of multi-drug resistant invasive and colonizing Acinetobacter baumannii isolated from diverse human body sites reveals genomic plasticity. BMC genomics. 2011;12:291 10.1186/1471-2164-12-291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tomaras AP, Dorsey CW, Edelmann RE, Actis LA. Attachment to and biofilm formation on abiotic surfaces by Acinetobacter baumannii: involvement of a novel chaperone-usher pili assembly system. Microbiology. 2003;149(Pt 12):3473–84. 10.1099/mic.0.26541-0 . [DOI] [PubMed] [Google Scholar]
- 62.Longo F, Vuotto C, Donelli G. Biofilm formation in Acinetobacter baumannii. The new microbiologica. 2014;37(2):119–27. . [PubMed] [Google Scholar]
- 63.Brossard KA, Campagnari AA. The Acinetobacter baumannii biofilm-associated protein plays a role in adherence to human epithelial cells. Infection and immunity. 2012;80(1):228–33. 10.1128/IAI.05913-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mussi MA, Gaddy JA, Cabruja M, Arivett BA, Viale AM, Rasia R, et al. The opportunistic human pathogen Acinetobacter baumannii senses and responds to light. Journal of bacteriology. 2010;192(24):6336–45. 10.1128/JB.00917-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gaddy JA, Actis LA. Regulation of Acinetobacter baumannii biofilm formation. Future microbiology. 2009;4(3):273–8. 10.2217/fmb.09.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Itoh Y, Rice JD, Goller C, Pannuri A, Taylor J, Meisner J, et al. Roles of pgaABCD genes in synthesis, modification, and export of the Escherichia coli biofilm adhesin poly-beta-1,6-N-acetyl-D-glucosamine. Journal of bacteriology. 2008;190(10):3670–80. 10.1128/JB.01920-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Friedman L, Kolter R. Two genetic loci produce distinct carbohydrate-rich structural components of the Pseudomonas aeruginosa biofilm matrix. Journal of bacteriology. 2004;186(14):4457–65. 10.1128/JB.186.14.4457-4465.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mack D, Fischer W, Krokotsch A, Leopold K, Hartmann R, Egge H, et al. The intercellular adhesin involved in biofilm accumulation of Staphylococcus epidermidis is a linear beta-1,6-linked glucosaminoglycan: purification and structural analysis. Journal of bacteriology. 1996;178(1):175–83. 10.1128/jb.178.1.175-183.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Choi AH, Slamti L, Avci FY, Pier GB, Maira-Litran T. The pgaABCD locus of Acinetobacter baumannii encodes the production of poly-beta-1-6-N-acetylglucosamine, which is critical for biofilm formation. Journal of bacteriology. 2009;191(19):5953–63. 10.1128/JB.00647-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Heritier C, Poirel L, Fournier PE, Claverie JM, Raoult D, Nordmann P. Characterization of the naturally occurring oxacillinase of Acinetobacter baumannii. Antimicrobial agents and chemotherapy. 2005;49(10):4174–9. 10.1128/AAC.49.10.4174-4179.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chen TL, Lee YT, Kuo SC, Hsueh PR, Chang FY, Siu LK, et al. Emergence and Distribution of Plasmids Bearing the blaOXA-51-like gene with an upstream ISAba1 in carbapenem-resistant Acinetobacter baumannii isolates in Taiwan. Antimicrobial agents and chemotherapy. 2010;54(11):4575–81. 10.1128/AAC.00764-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Turton JF, Ward ME, Woodford N, Kaufmann ME, Pike R, Livermore DM, et al. The role of ISAba1 in expression of OXA carbapenemase genes in Acinetobacter baumannii. FEMS microbiology letters. 2006;258(1):72–7. 10.1111/j.1574-6968.2006.00195.x . [DOI] [PubMed] [Google Scholar]
- 73.Evans BA, Amyes SG. OXA beta-lactamases. Clinical microbiology reviews. 2014;27(2):241–63. 10.1128/CMR.00117-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Higgins PG, Poirel L, Lehmann M, Nordmann P, Seifert H. OXA-143, a novel carbapenem-hydrolyzing class D beta-lactamase in Acinetobacter baumannii. Antimicrobial agents and chemotherapy. 2009;53(12):5035–8. 10.1128/AAC.00856-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Higgins PG, Perez-Llarena FJ, Zander E, Fernandez A, Bou G, Seifert H. OXA-235, a novel class D beta-lactamase involved in resistance to carbapenems in Acinetobacter baumannii. Antimicrobial agents and chemotherapy. 2013;57(5):2121–6. 10.1128/AAC.02413-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Turton JF, Baddal B, Perry C. Use of the accessory genome for characterization and typing of Acinetobacter baumannii. Journal of clinical microbiology. 2011;49(4):1260–6. 10.1128/JCM.02335-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mugnier PD, Poirel L, Naas T, Nordmann P. Worldwide dissemination of the blaOXA-23 carbapenemase gene of Acinetobacter baumannii. Emerging infectious diseases. 2010;16(1):35–40. 10.3201/eid1601.090852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Raghunath D. New metallo beta-lactamase NDM-1. The Indian journal of medical research. 2010;132:478–81. [PMC free article] [PubMed] [Google Scholar]
- 79.Shaw KJ, Rather PN, Hare RS, Miller GH. Molecular genetics of aminoglycoside resistance genes and familial relationships of the aminoglycoside-modifying enzymes. Microbiological reviews. 1993;57(1):138–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Coyne S, Courvalin P, Perichon B. Efflux-mediated antibiotic resistance in Acinetobacter spp. Antimicrobial agents and chemotherapy. 2011;55(3):947–53. 10.1128/AAC.01388-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wieczorek P, Sacha P, Hauschild T, Zorawski M, Krawczyk M, Tryniszewska E. Multidrug resistant Acinetobacter baumannii—the role of AdeABC (RND family) efflux pump in resistance to antibiotics. Folia histochemica et cytobiologica. 2008;46(3):257–67. 10.2478/v10042-008-0056-x . [DOI] [PubMed] [Google Scholar]
- 82.Huys G, Cnockaert M, Nemec A, Swings J. Sequence-based typing of ade B as a potential tool to identify intraspecific groups among clinical strains of multidrug-resistant Acinetobacter baumannii. Journal of clinical microbiology. 2005;43(10):5327–31. 10.1128/JCM.43.10.5327-5331.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mortensen BL, Skaar EP. Host-microbe interactions that shape the pathogenesis of Acinetobacter baumannii infection. Cellular microbiology. 2012;14(9):1336–44. 10.1111/j.1462-5822.2012.01817.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Antunes LC, Imperi F, Towner KJ, Visca P. Genome-assisted identification of putative iron-utilization genes in Acinetobacter baumannii and their distribution among a genotypically diverse collection of clinical isolates. Research in microbiology. 2011;162(3):279–84. 10.1016/j.resmic.2010.10.010 . [DOI] [PubMed] [Google Scholar]
- 85.Proschak A, Lubuta P, Grun P, Lohr F, Wilharm G, De Berardinis V, et al. Structure and biosynthesis of fimsbactins A-F, siderophores from Acinetobacter baumannii and Acinetobacter baylyi. Chembiochem: a European journal of chemical biology. 2013;14(5):633–8. 10.1002/cbic.201200764 . [DOI] [PubMed] [Google Scholar]
- 86.Eijkelkamp BA, Hassan KA, Paulsen IT, Brown MH. Investigation of the human pathogen Acinetobacter baumannii under iron limiting conditions. BMC genomics. 2011;12:126 10.1186/1471-2164-12-126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Meyer JM, Stintzi A, Poole K. The ferripyoverdine receptor FpvA of Pseudomonas aeruginosa PAO1 recognizes the ferripyoverdines of P. aeruginosa PAO1 and P. fluorescens ATCC 13525. FEMS microbiology letters. 1999;170(1):145–50. 10.1111/j.1574-6968.1999.tb13367.x . [DOI] [PubMed] [Google Scholar]
- 88.Alsan M, Klompas M. Acinetobacter baumannii: An Emerging and Important Pathogen. Journal of clinical outcomes management: JCOM. 2010;17(8):363–9. [PMC free article] [PubMed] [Google Scholar]
- 89.Choi CH, Lee JS, Lee YC, Park TI, Lee JC. Acinetobacter baumannii invades epithelial cells and outer membrane protein A mediates interactions with epithelial cells. BMC microbiology. 2008;8:216 10.1186/1471-2180-8-216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lal R, Pandey G, Sharma P, Kumari K, Malhotra S, Pandey R, et al. Biochemistry of microbial degradation of hexachlorocyclohexane and prospects for bioremediation. Microbiology and molecular biology reviews: MMBR. 2010;74(1):58–80. 10.1128/MMBR.00029-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(XLSX)
The position of DS002 in the phylogenetic tree is highlighted with a blue background and dotted clade line.
(TIF)
(TIF)
Strains with similar isolation source are shown with identical colors. The dotted blue line indicates the clade of soil isolate DS002 highlighted in blue.
(TIF)
Strains with similar isolation source are shown with identical colors.
(TIF)
(TIF)
OD590 values obtained for the ethanol extracts prepared from these two tubes indicating the extent of biofilm formation is shown in panel B. Panel C represents the bacterial growth as measured by OD600.
(TIF)
(DOCX)
(DOCX)
Data Availability Statement
Complete genomes gbk file is available from the NCBI database. GenBank: CP027704.1.