
Figure 2.
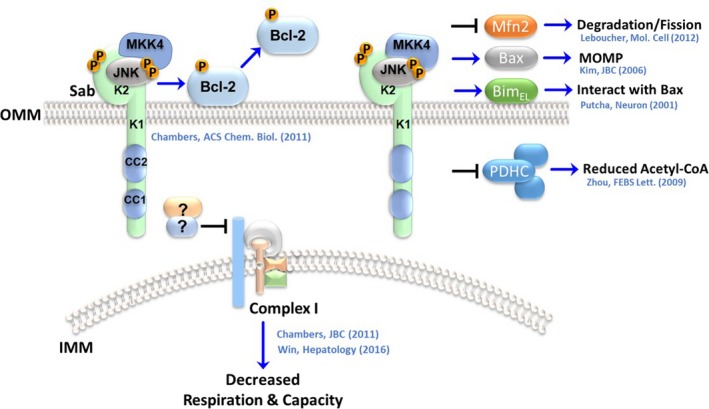
c‐Jun N‐terminal kinase (JNK) on the OMM impacts apoptosis and bioenergetics. JNK signaling has long been linked to cell death and metabolism, and only recently was it realized that mitochondrial JNK signaling on the OMM scaffold protein Sab was critical to the induction of apoptosis through the manipulation of proteins within the Bcl‐2 superfamily. Following activation, JNK translocates to mitochondria. JNK interacts with and phosphorylates the OMM scaffold protein Sab most likely through an interaction with the KIM2 motif (K2) because the KIM1 motif (K1) may be on the inner leaf of the OMM. In cases of neurotoxin exposure, ischemia, and cytotoxic stress, mitochondrial JNK activity leads to amplification of ROS production, organelle dysfunction, and cell death. Mitochondrial JNK can impair complex I by a yet‐to‐be‐described mechanism to impair mitochondrial metabolism and engage ROS production. However, this may be facilitated by signaling on the coiled‐coil motifs (CC1/CC2) of Sab's SH3 domain. Mitochondrial JNK can initiate apoptosis through phosphorylation of Bcl‐2 on Ser70 inducing its emigration from mitochondria. JNK activity on mitochondria has also been demonstrated to impact mitochondrial dynamics through phosphorylation of Mfn2, which leads to its degradation and mitochondrial fission. JNK also influences mitochondrial metabolism directly through the inhibition of PDH via phosphorylation of the Eα1 subunit. In addition to Bcl‐2, JNK can also influence the activities of BH3‐only proteins such as Bim to induce apoptosis, and mitochondrial JNK has been shown to phosphorylate Bax to induce permeabilization of the OMM. The local activities of mitochondrial JNK suggest it could be a significant physiological player in organelle health